

Background: Friedreich ataxia (FRDA) is caused by an expanded GAA triplet repeat (GAA-TR) mutation that results in FXN transcriptional deficiency.
Results: Repressive chromatin spreads from the expanded GAA-TR mutation, switching off the FXN gene promoter.
Conclusion: Reduced transcriptional initiation is the major cause of FXN transcriptional deficiency in FRDA.
Significance: Reactivation of FXN transcriptional initiation is a potential therapeutic strategy in FRDA.
Keywords: Gene Transcription, Heterochromatin, Neurological Diseases, Nucleotide Repeat Disease, Transcription Promoter, Friedreich Ataxia, Expanded Triplet Repeat, Nucleosome Positioning, Transcription Start Site
Abstract
Most individuals with Friedreich ataxia (FRDA) are homozygous for an expanded GAA triplet repeat (GAA-TR) mutation in intron 1 of the FXN gene, which results in deficiency of FXN transcript. Consistent with the expanded GAA-TR sequence as a cause of variegated gene silencing, evidence for heterochromatin has been detected in intron 1 in the immediate vicinity of the expanded GAA-TR mutation in FRDA. Transcriptional deficiency in FRDA is thought to result from deficient elongation through the expanded GAA-TR sequence because of repeat-proximal heterochromatin and abnormal DNA structures adopted by the expanded repeat. There is also evidence for deficient transcriptional initiation in FRDA, but its relationship to the expanded GAA-TR mutation remains unclear. We show that repressive chromatin extends from the expanded GAA-TR in intron 1 to the upstream regions of the FXN gene, involving the FXN transcriptional start site. Using a chromatin accessibility assay and a high-resolution nucleosome occupancy assay, we found that the major FXN transcriptional start site, which is normally in a nucleosome-depleted region, is rendered inaccessible by altered nucleosome positioning in FRDA. Consistent with the altered epigenetic landscape the FXN gene promoter, a typical CpG island promoter, was found to be in a transcriptionally non-permissive state in FRDA. Both metabolic labeling of nascent transcripts and an unbiased whole transcriptome analysis revealed a severe deficiency of transcriptional initiation in FRDA. Deficient transcriptional initiation, and not elongation, is the major cause of FXN transcriptional deficiency in FRDA, and it is related to the spread of repressive chromatin from the expanded GAA-TR mutation.
Introduction
Friedreich ataxia (FRDA)2 is the most common inherited ataxia, and it is characterized clinically by progressive sensory ataxia, cardiomyopathy, and a predisposition to diabetes (1). The disease is variably progressive, and there are no effective therapies currently available to slow the inevitable phenotypic deterioration. FRDA is inherited as an autosomal recessive condition, and the vast majority of patients are homozygous for an abnormally expanded GAA triplet repeat (GAA-TR) sequence in intron 1 of the FXN gene (2). Whereas normal alleles contain <30 triplets, disease-causing expanded alleles typically contain 100–1700 triplets. Cells and tissues from patients who are homozygous for the expanded GAA-TR sequence show a severe deficiency of FXN transcript (3). This ultimately leads to a deficiency of frataxin, a mitochondrial protein that plays an important role in Fe-S cluster biogenesis and in mitochondrial iron metabolism (4). The precise delineation of the mechanism(s) by which the expanded GAA-TR results in transcriptional deficiency will be crucial for the development of rationally designed therapies for FRDA.
Saveliev et al. (5) made the seminal discovery that expanded triplet repeats, including the expanded GAA-TR sequence, can result in epigenetic silencing of a closely linked transgene via a mechanism resembling position effect variegation. They showed that the expanded GAA-TR served as a source of HP-1-mediated heterochromatin, which was sufficient to silence a nearby reporter gene. Several groups have since identified molecular signatures of heterochromatin in the vicinity of the expanded GAA-TR in intron 1 of the FXN gene in cells and tissues from FRDA patients (6–13), including histone deacetylation (H3K9Ac, H3K14Ac, and H4K5Ac), histone trimethylation (H3K9me3 and H3K27me3), and CpG DNA methylation. Although these changes were seen both upstream and downstream of the expanded GAA-TR, evidence of heterochromatin was most pronounced immediately upstream of the GAA-TR but still within intron 1. Importantly, Herman et al. (6) showed that derivatives of a 2-aminobenzamide histone deacetylase inhibitor were able, albeit partially, to reverse both the epigenetic defect and the transcriptional silencing in lymphoid cells from FRDA patients. Indeed, a lead compound based on this histone deacetylase inhibitor is currently in clinical development as a rational therapeutic agent for Friedreich ataxia.
The expanded GAA-TR sequence is also known to adopt abnormal structures such as the triplex-based “sticky” DNA (14) and R-loops (15), both of which have the ability to interfere with transcriptional elongation. Indeed, destabilization of triplex-based structures via polyamides resulted in partial reversal of the transcriptional deficiency in lymphoblastoid cells from FRDA patients (16). The expanded GAA-TR in intron 1 does not result in abnormal splicing (3), resulting instead in lower quantities of a normally spliced FXN transcript in FRDA with a normal half-life (10). Taken together, these observations support the model that FXN transcriptional deficiency in FRDA is due to deficient transcriptional elongation, mediated via heterochromatin in intron 1 in the immediate vicinity of the expanded GAA-TR and possibly also one or more abnormal structures adopted by the expanded GAA-TR.
Interestingly, there is also evidence of transcriptional deficiency and of markers of repressive chromatin located further upstream from the changes noted in the immediate vicinity of the expanded GAA-TR in intron 1. For instance, in fibroblast cell lines from FRDA patients, markers of repressive chromatin were detected at the FXN transcriptional start site (FXN-TSS) (17). Furthermore, reduced RNAPII occupancy (12) and deficiency of FXN transcript (11) were noted in exon 1, i.e. considerably upstream of the expanded GAA-TR in intron 1. Indeed, Kumari et al. (11) also demonstrated reduced levels of serine 5-phosphorylated RNAPII, i.e. the initiating form of RNAPII, near the FXN promoter in patient-derived cell lines. These findings clearly support the existence of an additional transcriptional defect in FRDA: one that involves transcriptional initiation (or immediate post-initiation) of the FXN gene. However, the relative magnitude and the mechanism of this initiation defect in FRDA remain unclear, and its relationship to the expanded GAA-TR in intron 1 also remains unknown.
Heterochromatin, once formed, tends to spread to contiguous DNA sequence with some facility. Repressive chromatin associated with variegated silencing caused by expanded triplet repeats was found to spread over at least a few thousand bases (5). Given that the distance between the expanded GAA-TR in intron 1 and the FXN-TSS (and FXN promoter) is <2 kb, we decided to characterize the distance over which repressive chromatin spreads upstream of the expanded GAA-TR in FRDA. We further sought to determine whether the FXN gene promoter had been functionally and structurally altered so as to result in deficiency of transcriptional initiation.
We found that in FRDA repressive chromatin extends from the expanded GAA-TR in intron 1 toward the promoter region of the FXN gene and encompasses the FXN-TSS. The FXN-TSS is thus rendered inaccessible via altered nucleosome positioning that obliterates the nucleosome-depleted region (NDR) within which FXN transcriptional initiation is known to occur. The FXN promoter in FRDA is thus switched to a transcriptionally non-permissive state, which results in a severe deficiency of transcriptional initiation. Indeed, deficient transcriptional initiation, and not elongation, seems to be the major cause of FXN transcriptional deficiency in FRDA. These data have important implications for the pathogenesis and treatment of FRDA and perhaps also for other diseases caused by expanded triplet repeats.
EXPERIMENTAL PROCEDURES
Cell Lines
Most experiments involved the comparison of three lymphoblast cell lines from individuals with FRDA versus three cell lines from healthy subjects. Lymphoblast cell lines from three healthy subjects, GM15851, GM22671, GM22647, and two individuals with FRDA, GM15850 (650 and 1030 repeats) and GM16209 (800 and 800 repeats), were obtained from Coriell Cell Repositories. An additional FRDA lymphoblast cell line, OK22 (400 and 860 repeats), was established in the laboratory. However, in NOMe-Seq and RNA-Seq assays, we compared two FRDA versus two non-FRDA lymphoblast cell lines; in these assays, FRDA-1 and FRDA-2 refer to GM15850 and OK22, and CNTR-1 and CNTR-2 refer to GM22647 and GM22671, respectively.
Chromatin Immunoprecipitation (ChIP)
Chromatin for conventional ChIP assays was prepared according to ChIP-IT® Express Sonication Shearing Kit (Active Motif) with some modifications. Briefly, 6 million cells were cross-linked with 1% freshly prepared formaldehyde. Cross-linked nuclei were sheared with the EpiShearTM sonicator (Active Motif) to yield a chromatin size range of 200 to 800 bp. Immunoprecipitation was performed using anti-RNAPII (Millipore, 17-620), anti-H2A.Z (Millipore, 17-10048), and anti-histone H3 (Millipore, 17-10254). Immunoprecipitated chromatin-bead complexes were washed and transferred to new tubes for elution to minimize contamination with non-specifically bound chromatin to the tube surface. Immunoprecipitated chromatin was eluted from the beads, reverse cross-linked, treated with proteinase K, and extracted using phenol-chloroform. The immunoprecipitated DNA and the input DNA were analyzed by real-time PCR using the ΔΔCt method using an Eppendorf RealPlex-4 Mastercycler with Power SYBR Green PCR Mastermix (Applied Biosystems).
Chromatin for mononucleosomal ChIP was prepared using the ChIP-IT® Express Enzymatic Shearing Kit (Active Motif). Mononucleosomal preparations (size ∼ 150 bp) were obtained by optimizing the digestion reaction with enzymatic shearing mixture. Immunoprecipitation was performed using anti-H3K27me3 (Millipore, 17-622) and anti-H4K5Ac (Millipore, 07-327).
Each value of immunoprecipitated DNA was normalized to the corresponding input DNA. For conventional ChIP, relative recovery was calculated over histone H3 enrichment corresponding to the region being analyzed. All quantitative PCRs were performed in a two-step cycle with an annealing/extension temperature of 62 °C (see Table 1 for primer sequences).
TABLE 1.
Primer sequences used in this study
*, primers were also used for strand-specific reverse transcription.
| Primer | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| Primers used for chromatin immunoprecipitation | ||
| P4 (−2632 to −2513) | CCAAGGAAGTGGAGAAAGACAAG | TCCAGCTCCCTGCAAAGTTG |
| P3 (−1719 to −1602) | CCCTGGACCACCTCTTTGATAG | GAGCTAGGGATGGGGGAATC |
| P2 (−1261 to −1134) | CGGCTCACATTTGACATCCTC | GTGCCTTGCCTATTGTTGATAGAT |
| P1 (−358 to −232) | CCCCACATACCCAACTGCTG | GCCCGCCGCTTCTAAAATTC |
| U1 (−204 to −77) | GGGGTCCTGGTTGCACTCC | GCACTCTTCTGTGGGGGAGC |
| E1 (−46 to +59) | AGCACCCAGCGCTGGAGG | TGGGCTGGGCTGGGTGACGCCAGG |
| I1 (+277 to +396) | ACTAGCTCACCCCGCTCCT | AAATGTCCCCTTTTCCTTCG |
| I2 (+666 to +778) | TGAACGAAATGCTTTCCTGG | GGGCAGAATCTGGAATAAAGG |
| I3 (+1006 to +1121) (Ref. 6) | GAAACCCAAAGAATGGCTGTG | TTCCCTCCTCGTGAAACACC |
| Primers used for RT-PCR | ||
| Ex1 (Ref. 11) | AGCAGCATGTGGACTCTC | TGGGCTGGGCTGGGTGACGCCAGG* |
| Ex1-In1 | CCGACATCGATGCGACCTGC | GTTCCCGGCGCGGATACTTACT |
| Ex2-Ex4 | CCGCGCAAGTTCGAACCAA | TTCCTAGATCTCCACCCAGT |
| RPS29 | GCACTGCTGAGAGCAAGATG | ATAGGCAGTGCCAAGGAAGA |
| Primers used for NOMe-Seq | ||
| Amplicon 1 | CCTATCCAAAAAAAACTTTACAAAAC | GGTTAGATTTTAGGAAGAATTTTTT |
| Amplicon 2 | AATCTCCCTTAAATCAAAAATCC | GATTTGGGTGAGGGTTTGGGTTTGG |
| Primers used for chromatin accessibility assay | ||
| FXN-TSS2 | TCCTGAGGTCTAACCTCTAGCTGC | TGGGCTGGGCTGGGTGACGCCAGG |
Quantitative RT-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen). Reverse transcription was performed either with a mixture of random hexamers and oligo(dT) primers or strand-specific primers using the QuantiTect® Reverse Transcription kit (Qiagen). mRNA levels were quantified by real-time PCR relative to RPS29 using the ΔΔCt method using an Eppendorf RealPlex-4 Mastercycler with SsoAdvancedTM SYBR® Green supermix (Bio-Rad). Quantitative PCRs were performed in a two-step cycle with an annealing/extension temperature of 70 °C (see Table 1 for primer sequences).
Chromatin Accessibility Assay
The EpiQTM chromatin analysis kit (Bio-Rad) was used with minor modifications. For each cell line, a set of two 200,000 cell aliquots were analyzed; one aliquot was treated with nuclease (37 °C for 30 min), and the other was used as an undigested control. 5 ng of DNA isolated from each sample was used for quantitative PCR, done in triplicate to determine % accessibility, which was calculated using the data analysis tool available from Bio-Rad. The β hemoglobin (HBB) gene was used as the inaccessible reference for measuring relative chromatin accessibility at various gene loci. The GAPDH gene was used as a constitutively accessible control, and the RHO gene was used as the inaccessible control. PCR primer sequences for HBB, GAPDH, and RHO are as provided in the EpiQTM chromatin analysis kit. The region of the FXN gene analyzed is shown in Fig. 2A, and the PCR primer sequences are listed in Table 1.
FIGURE 2.

Reduced chromatin accessibility at the major FXN-TSS in FRDA. A, the major FXN-TSS (FXN-TSS2 at −59), which maps within an NDR (DNase I-hypersensitive cluster in ENCODE), is depicted with an arrow in boldface type. The region analyzed for chromatin accessibility, which includes FXN-TSS2, is indicated by a horizontal line. B, relative chromatin accessibility (%) is significantly reduced in FRDA versus non-FRDA (CNTR) cells. Two control regions are shown that do not show any difference in chromatin accessibility in FRDA versus non-FRDA cells; a constitutively accessible region of the GAPDH gene, and a region of the RHO gene that is known to be inaccessible. These data represent the cumulative results from two complete experiments using three FRDA and three non-FRDA (CNTR) lymphoblast cell lines, with each assayed in triplicate. Error bars represent ± S.E. **, p < 0.01.
Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq)
NOMe-Seq was performed according to the manufacturer's instructions (Active Motif) with a few modifications. Briefly, 750,000 cells were cross-linked as described above. Chromatin was sheared by sonication on ice with 25% amplitude for 3 min of 30 s “on” and 30 s “off” cycle. The GpC methyltransferase reaction was performed on sheared chromatin from ∼250,000 cells. 1–2 μg of the GpC methyltransferase-treated DNA was subjected to bisulfite conversion. 20–50 ng of each bisulfite-treated DNA was used for PCR amplification (see Table 1 for primer sequences of the two overlapping amplicons analyzed) using AmpliTaq® Gold DNA polymerase (Applied Biosystems). PCR products were TA-cloned using the TOPO TA Cloning® Kit (Invitrogen) and transformed into competent Escherichia coli (DH5α), and individual clones were sequenced. Cytosines at GpC sites that are accessible to GpC methyltransferase (i.e. not within a nucleosome) are methylated and therefore not converted by bisulfite treatment (i.e. read as cytosine) and are denoted as a white box in Fig. 3. Conversely, inaccessible cytosines, which remain unmethylated after GpC methyltransferase treatment, are converted via bisulfite treatment to uracil (i.e. read as thymine) and are denoted as a black box in Fig. 3. The discriminatory capacity of the assay relies on the efficiency of bisulfite conversion; therefore, only clones that demonstrated complete conversion efficiency at all non-GpC (and non-CpG) sites were used for NOMe-Seq analysis (>98% conversion of all non-GpC sites was attained for the clones shown in Fig. 3).
FIGURE 3.

Altered nucleosome positioning in the vicinity of the major FXN-TSS and obliteration of the naturally occurring NDR in FRDA. NOMe-Seq data, measuring the nucleosomal footprint in vivo, for two non-FRDA (CNTR) and two FRDA lymphoblast cell lines are shown above and below the line depicting the region in the vicinity of FXN-TSS2 (the major FXN-TSS). The boxed region corresponds to the DNase I-hypersensitive cluster or NDR per ENCODE, within which TSS2 maps, consistent with its location in a region of relatively open chromatin configuration. NOMe-Seq data from eight clones each for the four cell lines analyzed is depicted in the form of white and black boxes, each of which represents accessible and inaccessible GpC sites, respectively (see “Experimental Procedures”). The region around TSS2 in the two non-FRDA cell lines shows many accessible GpC sites, indicating that it is in a NDR. In both FRDA cell lines, the region around TSS2 is largely inaccessible, indicating nucleosomal repositioning and obliteration of the naturally occurring NDR. Interestingly, TSS1, which maps just outside the NDR, is equally inaccessible in non-FRDA and FRDA cell lines.
RNA-Seq/Transcriptome Analysis
Total RNA was extracted from two non-FRDA (GM22647 and GM22671) and two FRDA (GM15850 and OK22) lymphoblast cell lines. Strand-specific libraries were generated from poly-A-selected RNA. Multiplexed libraries were paired end-sequenced (100-bp reads) on the Illumina HiSeq 2500 system for a total of 2 × 20–30 million reads per sample. The sequence quality as calculated by % of ≥Q30 bases (PF) was 92.45, 92.66, 92.09, and 92.49 for GM22647 (CNTR-1), GM22671 (CNTR-2), GM15850 (FRDA-1), and OK22 (FRDA-2), respectively.
Metabolic Labeling of Nascent RNA in Live Cells
Metabolic labeling of nascent RNA was performed using the Click-iT® Nascent RNA Capture Kit (Invitrogen). Briefly, lymphoblastoid cells at a density of 1 million/ml were incubated with 0.2 mm 5-ethynyl uridine. 2 million cells were removed after 0.5, 1, 2, and 4 h of 5-ethynyl uridine treatment. RNA was extracted using the TriPure Isolation Reagent (Roche Applied Science). 1 μg of 5-ethynyl uridine RNA was used for biotinylation by Click reaction. After purification, 250 ng of biotinylated RNA was used for pulldown with streptavidin beads. Biotinylated RNA bound to beads were serially washed, and the bound RNA was reverse-transcribed using the SuperScript® VILOTM cDNA synthesis kit (Invitrogen) following the manufacturer's instructions. The mRNA levels were quantified by real-time PCR relative to RPS29 from total biotinylated RNA using the ΔΔCt method on the Eppendorf RealPlex-4 Mastercycler with SsoAdvanced SYBR Green Supermix (Bio-Rad). All quantitative PCRs were performed in a two-step cycle with an annealing-extension temperature of 70 °C (see Table 1 for primer sequences).
Statistical Methods
Statistical significance was calculated using paired, two-tailed Student's t test. p < 0.05 was considered significant.
RESULTS
Repressive Chromatin Extends from the Expanded GAA Repeat in Intron 1 toward the FXN Gene Promoter and Encompasses the FXN-TSS in FRDA
The 5′ region of the FXN gene spanning the putative promoter, the FXN-TSS, 5′-UTR, exon 1, and the sequence upstream of the expanded GAA-TR in intron 1 (Fig. 1A) were analyzed by chromatin immunoprecipitation for markers of repressive chromatin. Lymphoblast cell lines from three FRDA patients, each homozygous for the expanded GAA-TR sequence (range, 400–1030 triplets), and three non-FRDA controls, were analyzed for enrichment of H3K27me3 and hypoacetylation of H4K5, i.e. known markers of repressive chromatin. FRDA patients showed both enrichment of H3K27me3 and hypoacetylation of H4K5 for regions I3 through P1 (Fig. 1, B and C), indicating the presence of repressive chromatin spanning the region of the FXN gene from the expanded GAA-TR in intron 1 to the FXN-TSS. The extended FXN promoter region, spanning up to 2.6 kb upstream of the FXN-TSS, seemed to be spared in FRDA. These data support the model that in FRDA repressive chromatin spreads upstream from the expanded GAA-TR in intron 1 to encompass the FXN-TSS.
FIGURE 1.
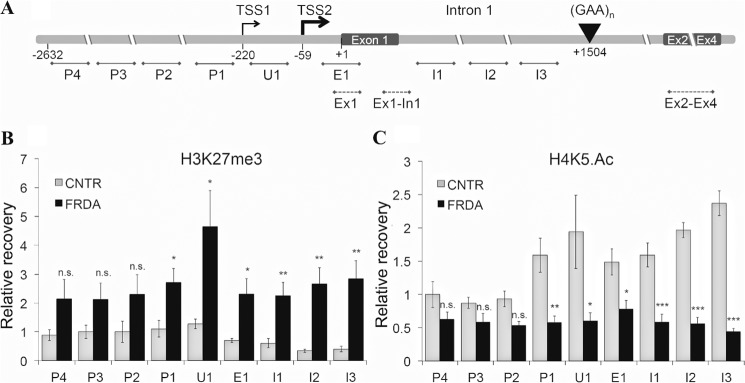
Repressive chromatin extends from the expanded GAA-TR in intron 1 to the FXN-TSS in FRDA. A, the relevant portion of the FXN gene is shown, with the GAA-TR sequence in intron 1 and the positions of the major and minor FXN transcriptional start sites (TSS2 at −59 and TSS1 at −220, respectively). Regions depicted with solid lines were evaluated by ChIP. Of these, three amplicons spanned the relevant portions of intron 1 (I1–3), one sampled the 5′-UTR/exon 1 (E1; immediately downstream of FXN-TSS2), one sampled the 5′-UTR (U1; in between the two reported FXN-TSS) and four amplicons covered the putative FXN promoter up to 2.6 kb upstream of the FXN-TSS (P1–4). Regions depicted with dotted lines were analyzed by quantitative RT-PCR. All location numbers are relative to the A (+1) in the initiation codon. B and C, mononucleosomal ChIP showing enrichment for H3K27me3 (B), and hypoacetylation of H4K5 (C) extending from intron 1 (I3) up to and including the FXN-TSS. These data represent the cumulative results from two complete experiments using three FRDA and three non-FRDA control (CNTR) lymphoblast cell lines, each assayed in triplicate. The x axis labels refer to similarly labeled regions depicted in A. Error bars represent ±S.E. *, p < 0.5; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Reduced Accessibility of the FXN-TSS in FRDA via Altered Nucleosome Positioning and Obliteration of the Nucleosome-depleted Region of the FXN Gene Promoter
To test whether the repressive chromatin in FRDA results in reduced accessibility of the FXN-TSS, a quantitative chromatin accessibility assay was performed based on measurement of nuclease sensitivity in live cells. As is typical for most human CpG island promoters (18), the major TSS of the FXN gene, FXN-TSS2 (position −59; Fig. 2A) is known to map within an NDR (DNase I-hypersensitive cluster in ENCODE; Fig. 2A). Indeed, FXN-TSS2 is likely the only active FXN-TSS in lymphoblasts (11); also see transcriptome analysis below). Chromatin accessibility at FXN-TSS2 was measured in three FRDA and three non-FRDA lymphoblast cell lines. Although it was highly accessible in non-FRDA cells, FXN-TSS2 showed significantly reduced chromatin accessibility in FRDA patients (Fig. 2B). In contrast, a constitutively accessible site in the GAPDH gene was found to be highly accessible, and a region of the gene encoding rhodopsin that is inaccessible in all cells except photoreceptors was found to be highly inaccessible, with no noticeable difference in accessibility between FRDA and non-FRDA cell lines (Fig. 2B). This indicates that in FRDA, repressive chromatin results in reduced chromatin accessibility at the FXN-TSS.
To determine the mechanism of reduced chromatin accessibility at the FXN-TSS in FRDA, the region spanning the NDR was analyzed using NOMe-Seq, a high-resolution in vivo footprint assay for nucleosome occupancy (19). In the NOMe-Seq assay, inaccessible GpC dinucleotides indicate protection by nucleosomes (and vice versa), and sequencing of individual cloned PCR products thus reveals the pattern of nucleosome occupancy in individual chromatin molecules. The NOMe-Seq assay was especially suitable because of the high density of GpC dinucleotides in the vicinity of the two reported FXN-TSS (positions −59 and −220). Eight individual clones were thus analyzed from each of two FRDA and two non-FRDA cell lines (Fig. 3; white and black boxes indicate accessible and inaccessible GpC dinucleotides, respectively). As expected, FXN-TSS2 mapped within an NDR in non-FRDA control cell lines (Fig. 3; upper panels). In contrast, FRDA patients showed a dramatic loss of accessibility at FXN-TSS2, with almost no accessible GpC dinucleotides (Fig. 3; lower panels). This indicates that inaccessibility of FXN-TSS2 in FRDA is caused by altered nucleosome positioning at the NDR. FXN-TSS1, which is not active in lymphoblasts (11), was found to map just outside the NDR despite its close physical proximity to FXN-TSS2 and was equally inaccessible in non-FRDA and FRDA cell lines (Fig. 3). Taken together, our data indicate that FXN-TSS2, the major FXN-TSS, is much less accessible in FRDA due to altered nucleosome positioning and obliteration of the naturally occurring NDR.
Because the NOMe-Seq protocol uses bisulfite conversion, it is also possible to simultaneously detect naturally occurring CpG methylation. However, no endogenous CpG methylation was detected in either the two FRDA or two non-FRDA cell lines in the vicinity of the two FXN-TSS.
Reduced Transcriptional Permissiveness of the FXN Promoter in FRDA
The transcriptional permissiveness of typical human CpG island promoters is characterized by a well positioned +1 nucleosome preceded by a NDR (18) that is preassociated with RNAPII and the chromatin insulator CTCF (20, 21). Active genes are typically enriched for the transcriptionally permissive histone modification, H3K4me3, and the histone variant, H2A.Z, both of which are highly pronounced at the +1 nucleosome (21–23). We sought to determine whether the repressive chromatin and reduced accessibility of the FXN-TSS in FRDA had resulted in switching of the FXN promoter to a transcriptionally non-permissive state. Markers of transcriptional permissiveness at the FXN-TSS and the +1 nucleosome were assessed by chromatin immunoprecipitation in three FRDA and three non-FRDA cell lines. Indeed, the FXN-TSS in FRDA was found to be in a state of reduced transcriptional permissiveness as evidenced by reduced occupancy of RNAPII and CTCF (Fig. 4, A and B), and depletion of H2A.Z and H3K4me3 (Fig. 4, C and D). These data are consistent with observations previously made by us (17) and others (11, 24).
FIGURE 4.
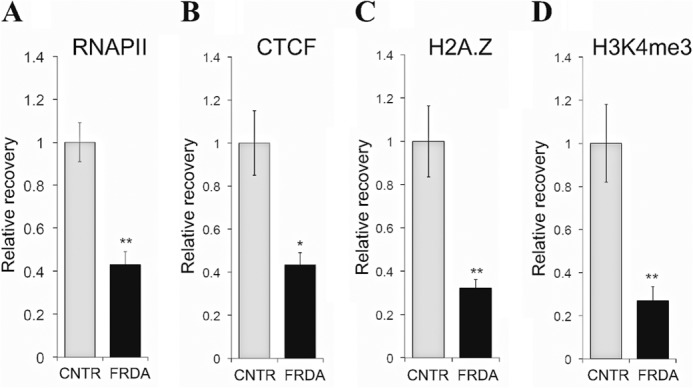
Reduced transcriptional permissiveness of the FXN promoter in FRDA. ChIP showing reduced occupancy of RNAPII (A) and CTCF (B), and depletion of H2A.Z (C) and H3K4me3 (D) in FRDA versus non-FRDA (CNTR) cells, indicating a state of reduced transcriptional permissiveness. RNAPII, H2A.Z and H3K4me3 ChIP was performed at E1 (Fig. 1A) and CTCF ChiP was performed in a region spanning FXN-TSS2 (between E1 and U1 in Fig. 1A) as described previously (17). These data represent the cumulative results from two complete experiments using three FRDA and three non-FRDA (CNTR) lymphoblast cell lines, with each assayed in triplicate. Error bars represent ± S.E. *, p < 0.05; **, p < 0.01.
Deficient FXN Transcriptional Initiation in FRDA
Given the repressive chromatin at FXN-TSS and its switch to a non-permissive transcriptional state in FRDA, we reasoned that this should lead to transcriptional deficiency even upstream of the expanded GAA-TR. To test this, quantitative RT-PCR was performed to measure steady-state levels of FXN transcript both upstream (exon 1 (Ex1 in Fig. 1A) and the exon 1-intron 1 junction (Ex1-In1 in Fig. 1A)) and downstream (spliced product of exons 2–4 (Ex2-Ex4 in Fig. 1A)) of the GAA-TR in three FRDA and three non-FRDA cell lines. FRDA patients showed a deficiency of steady-state levels of FXN transcript, both upstream (Fig. 5, A and B) and downstream (Fig. 5C) of the expanded GAA-TR. The reduction of transcriptional activity upstream of the expanded GAA-TR suggested that the FXN promoter is less active in FRDA and further suggested that transcriptional deficiency was not simply due to a defect in transcriptional elongation.
FIGURE 5.
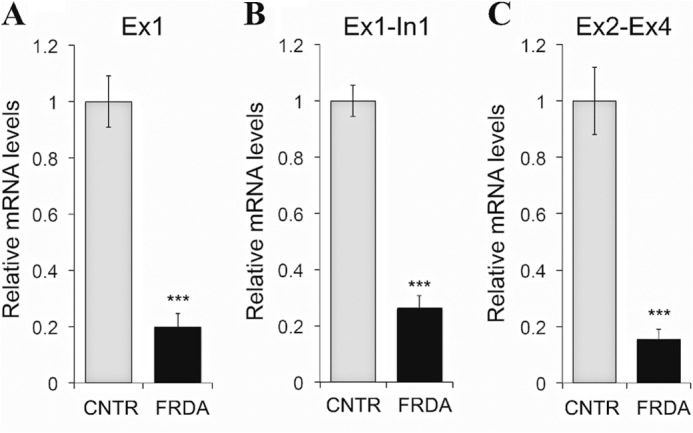
Reduced transcriptional activity upstream of the expanded GAA-TR in FRDA. A and B, quantitative RT-PCR showing significantly reduced amounts of FXN mRNA (Ex1 region (A) in Fig. 1A) and pre mRNA (Ex1-In1 region (B) in Fig. 1A) upstream of the GAA-TR in FRDA versus non-FRDA (CNTR) lymphoblast cells. C, quantitative RT-PCR showing a similar deficiency of FXN mRNA downstream of the GAA-TR in FRDA (Ex2-Ex4 region in Fig. 1A). Graphs represent the cumulative data from two complete experiments using three FRDA and three non-FRDA (CNTR) lymphoblast cell lines, each assayed in triplicate. Error bars represent ± S.E. ***, p < 0.001.
We next performed a complete transcriptome analysis (RNA-Seq) of two FRDA and two non-FRDA cell lines. Poly-A enrichment, and strand-specific library construction was followed by massively parallel, paired end sequencing using the Illumina HiSeq platform. Analysis of all hits spanning the FXN locus showed only 11.1% of normal (corrected for sequence depth; average per cell line = 2 × 26 million reads) in FRDA versus non-FRDA cell lines (Fig. 6A). No such difference was noted in the PIP5K1B gene, located immediately upstream of the FXN gene, or in RPS29, an unlinked housekeeping gene, indicating that transcriptional deficiency is limited to the FXN locus (Fig. 6A). Analysis of sequence reads specifically at the 5′ end of the FXN gene (i.e. including both FXN-TSS, the 5′-UTR, and exon 1) revealed several matches downstream of FXN-TSS2 (at −59) and none between FXN-TSS1 and FXN-TSS2 in the two control cell lines, confirming that FXN-TSS2 is the only active TSS in lymphoblasts (Fig. 6B; each box represents the 5′ end of a 100-bp sequence read). FRDA cell lines showed far fewer matches, amounting to 9.5% (25 versus 262) of normal (Fig. 6C), further substantiating the reduced amount of transcriptional activity upstream of the expanded GAA-TR, and indicating that the FXN promoter is much less active in FRDA.
FIGURE 6.
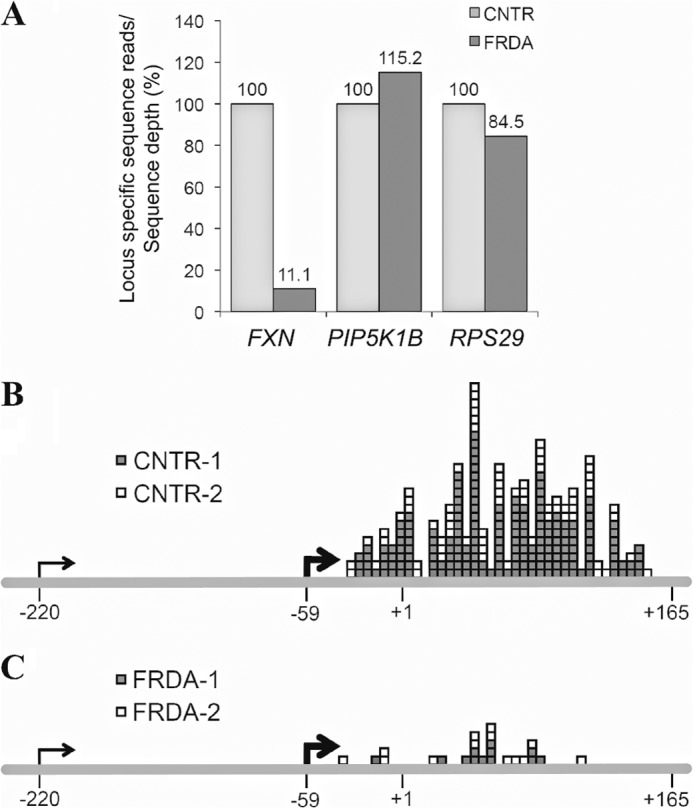
Transcriptome (RNA-Seq) analysis showing deficiency of transcriptional initiation in FRDA. A, RNA-Seq hits spanning the entire FXN gene showed 11.1% of normal in FRDA versus non-FRDA (CNTR) cell lines. No such difference was noted in the PIP5K1B gene, located immediately upstream of the FXN gene, or in RPS29, an unlinked housekeeping gene. Data are corrected for minor variations in sequence depth (average per cell line = 2 × 26 million reads) in each of two FRDA and two non-FRDA cell lines, with the non-FRDA cells depicted as 100%. B and C, RNA-Seq showing considerably less FXN mRNA sequence reads in the region upstream of the GAA-TR in two FRDA versus two non-FRDA (CNTR) lymphoblast cell lines (9.5% of normal; 25 versus 262). Each box indicates the 5′ end of a 100-bp sequence read (+165 denotes the 3′ end of exon 1). Note the lack of sequence reads between TSS1 (−220) and TSS2 (−59), indicating that TSS2 is the major FXN-TSS.
To directly test whether the FXN promoter is rendered less active in FRDA, FXN transcriptional initiation was measured via metabolic labeling of nascent transcripts in two FRDA and two non-FRDA cell lines. Nascent transcripts were labeled with ethynyl uridine, to which biotin was subsequently added via click chemistry, thus permitting a quantitative, dynamic, in vivo analysis of the newly synthesized FXN transcript. This is similar to a nuclear run-on assay, with the added advantage of measuring nascent transcripts in live cells. Quantitative RT-PCR was performed to measure FXN transcript levels both upstream (Ex1, which maps immediately downstream of FXN-TSS2 (Fig. 1A)) and downstream (Ex2-Ex4 (Fig. 1A)) of the expanded GAA-TR following 0.5, 1, 2, and 4 h of labeling. This revealed a severe (5–12-fold) deficiency of newly synthesized FXN transcript in FRDA at both locations (Fig. 7, A and B). The deficiency of dynamic accumulation of FXN transcript levels noted upstream of the expanded GAA-TR (but immediately downstream of FXN-TSS2) indicated a severe deficiency of transcriptional initiation in FRDA (note that the half-life of FXN transcript remains unchanged in FRDA (10)). Comparison of the levels of newly synthesized FXN transcript upstream versus downstream of the expanded GAA-TR revealed a consistent ∼2-fold decrease in the downstream location, indicating that transcriptional elongation further contributes to the transcriptional deficiency in FRDA. However, the 5–12-fold reduction in newly synthesized FXN mRNA upstream of the expanded GAA-TR indicates that deficient transcriptional initiation, and not elongation, is the predominant cause of FXN transcriptional deficiency in FRDA.
FIGURE 7.
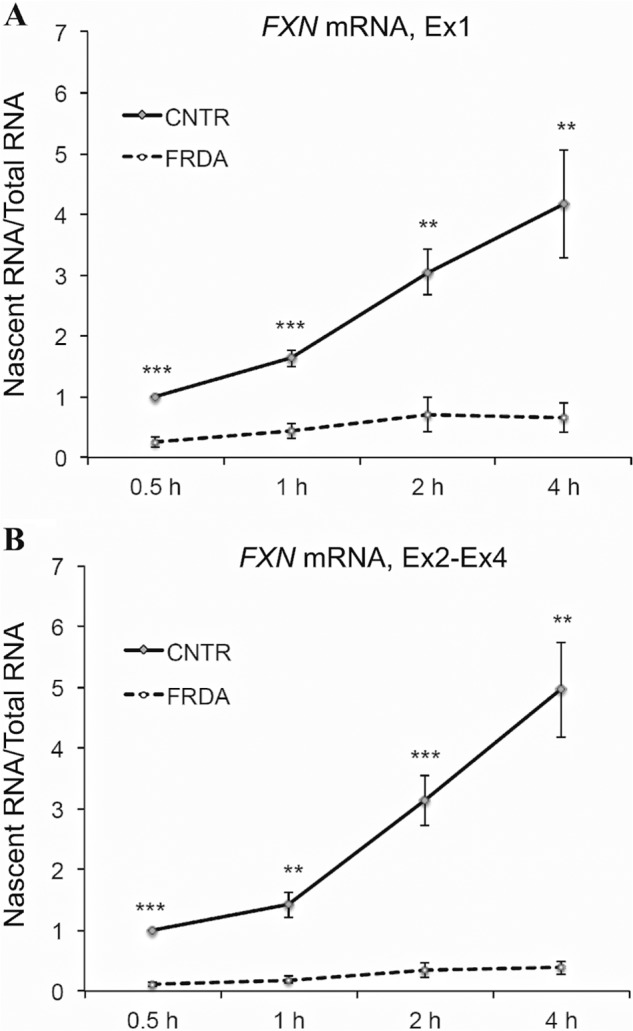
Metabolic labeling of nascent FXN transcript in living cells showing deficiency of transcriptional initiation in FRDA. A and B, quantitative RT-PCR of metabolically labeled nascent transcript for the indicated incubation times (0.5–4 h) is shown for FXN mRNA upstream (Ex1 region in Fig. 1A) and downstream (Ex2-Ex4 region in Fig. 1A) of the GAA-TR in intron 1. FRDA cells showed 5–12-fold less nascent FXN mRNA compared with non-FRDA cells (CNTR) at all time points assayed. Graphs represent the cumulative data from two complete experiments using two FRDA and two non-FRDA (CNTR) lymphoblast cell lines, each assayed in triplicate. Error bars represent ± S.E. **, p < 0.01; ***, p < 0.001.
DISCUSSION
We show that the expanded GAA-TR mutation in FRDA, located 1.6 kb away from the FXN-TSS results in disruption of its transcription-permissive chromatin structure and causes a deficiency of transcriptional initiation. Our data are consistent with the model that in FRDA, there is spread of repressive chromatin from the expanded GAA-TR in intron 1 to the upstream region of the FXN gene (Fig. 8). This spread results in alteration of nucleosome positioning, reduced accessibility of the FXN-TSS, which is normally in an open chromatin configuration, and severely deficient FXN transcriptional initiation (Fig. 8). As a consequence of reduced transcriptional initiation in FRDA, there is significantly diminished FXN transcriptional activity, detectable both upstream and downstream of the expanded GAA-TR. Notably, these findings are congruent with those of Saveliev et al. (5), who found that an expanded triplet repeat sequence (GAA or CTG) resulted in variegated silencing of a linked transgenic reporter gene. Indeed, silencing was mediated by an increase in nucleosomal density leading to a closed chromatin configuration of a nearby heterologous promoter, which is similar to what we have observed at the FXN promoter in FRDA.
FIGURE 8.

Model of deficient transcriptional initiation in FRDA. The expanded GAA-TR mutation in FRDA serves as a source of repressive chromatin (indicated here as trimethylation of H3K27 and hypoacetylation of H4K5), which spreads to the upstream regions of the FXN gene to involve the major FXN-TSS located 1.6 kb upstream. The major FXN-TSS, which is normally in a NDR, is rendered inaccessible because of increased nucleosome density. This results in a FXN promoter that is transcriptionally non-permissive (indicated here as obliteration of the NDR and depletion of CTCF, H3K4me3, and H2A.Z), leading to a severe deficiency of transcriptional initiation in FRDA.
A widely accepted model of the mechanism of transcriptional deficiency in FRDA is that there is deficient transcriptional elongation through the expanded GAA-TR in intron 1. This is predicated on the presence of repeat-proximal heterochromatin (6–9) and its reversal via a histone deacetylase inhibitor that reverses FXN transcriptional deficiency in FRDA (6, 25) and the ability of the expanded repeat to adopt secondary structures that can interfere with transcriptional elongation (3, 14–16, 26, 27). Indeed, some have even questioned the existence of any other transcriptional defect, claiming that the transcriptional defect in FRDA is due solely to deficient transcriptional elongation (10). However, our present data, along with those of Kumari et al. (11), confirm the existence of deficient transcriptional initiation in FRDA. In fact, the transcriptional initiation defect, which is related to the spread of repressive chromatin from the expanded GAA-TR mutation, seems to be the predominant contributor to the overall deficiency of FXN transcript in FRDA.
Several questions remain unanswered. For instance, what is the typical distance over which an expanded triplet repeat exerts its transcriptional silencing effect? Is the extent of spread or strength of repression related to repeat length? Answers to these questions would help uncover the mechanism of the known length-dependence of transcriptional deficiency (9, 13, 28) and severity of clinical phenotype (29, 30) in FRDA. There are numerous polymorphic GAA triplet repeat tracts in the genomes of mammals, many of which are located within genes, and some are of considerable length (31). It is plausible that these polymorphic repeats could serve as localized and allele-specific epigenetic regulators of transcription. Furthermore, it remains unclear if the repressive chromatin at the FXN-TSS in FRDA is variegated. Our NOMe-Seq data showed obliteration of the NDR in almost every chromatin fiber analyzed, which would tend to support the absence of variegation. However, a definitive answer would require sequencing of far more chromatin fibers than we have performed here, including chromatin associated with shorter than the typical expanded GAA alleles in FRDA. Another interesting unanswered question is what stops the spread of repressive chromatin emanating from an expanded triplet repeat? CTCF bound sequences are known to serve as chromatin insulators, some of which function as barriers to the spread of heterochromatin (32). However, CTCF does not seem to serve such a protective function in FRDA in the presence of the expanded GAA-TR. Not only is CTCF depleted from its binding site, which overlaps FXN-TSS2, but repressive chromatin in FRDA is established at FXN-TSS2 and also seems to spread further upstream. Among its many functions, CTCF is known to position nucleosomes (33), so it is also possible that CTCF depletion may have further contributed to the altered nucleosomal positioning in FRDA. An important caveat of our study is that it is based on findings from patient-derived lymphoblasts, which is not a cell type that is clinically affected in FRDA; clearly, additional confirmatory studies need to be carried out using more disease-appropriate cell types and/or tissues.
Finally, given the relative importance of deficient transcriptional initiation in FRDA, we propose that drugs that are designed to reactivate FXN transcriptional initiation would serve as a particularly efficient therapeutic strategy for FRDA.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS072418 (to S. I. B.).
- FRDA
- Friedreich ataxia
- TSS
- transcriptional start site(s)
- NDR
- nucleosome-depleted region
- TR
- triplet repeat
- CTCF
- CCCTC binding factor
- RNAP
- RNA polymerase.
REFERENCES
- 1. Parkinson M. H., Boesch S., Nachbauer W., Mariotti C., Giunti P. (2013) Clinical features of Friedreich's ataxia: classical and atypical phenotypes. J. Neurochem. 126, 103–117 [DOI] [PubMed] [Google Scholar]
- 2. Campuzano V., Montermini L., Moltò M. D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., Zara F., Cañizares J., Koutnikova H., Bidichandani S. I., Gellera C., Brice A., Trouillas P., De Michele G., Filla A., De Frutos R., Palau F., Patel P. I., Di Donato S., Mandel J. L., Cocozza S., Koenig M., Pandolfo M. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 [DOI] [PubMed] [Google Scholar]
- 3. Bidichandani S. I., Ashizawa T., Patel P. I. (1998) The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 62, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pastore A., Puccio H. (2013) Frataxin: a protein in search for a function. J. Neurochem. 126, 43–52 [DOI] [PubMed] [Google Scholar]
- 5. Saveliev A., Everett C., Sharpe T., Webster Z., Festenstein R. (2003) DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422, 909–913 [DOI] [PubMed] [Google Scholar]
- 6. Herman D., Jenssen K., Burnett R., Soragni E., Perlman S. L., Gottesfeld J. M. (2006) Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2, 551–558 [DOI] [PubMed] [Google Scholar]
- 7. Greene E., Mahishi L., Entezam A., Kumari D., Usdin K. (2007) Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 35, 3383–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al-Mahdawi S., Pinto R. M., Ismail O., Varshney D., Lymperi S., Sandi C., Trabzuni D., Pook M. (2008) The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 17, 735–746 [DOI] [PubMed] [Google Scholar]
- 9. Castaldo I., Pinelli M., Monticelli A., Acquaviva F., Giacchetti M., Filla A., Sacchetti S., Keller S., Avvedimento V. E., Chiariotti L., Cocozza S. (2008) DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 45, 808–812 [DOI] [PubMed] [Google Scholar]
- 10. Punga T., Bühler M. (2010) Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol. Med. 2, 120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumari D., Biacsi R. E., Usdin K. (2011) Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem. 286, 4209–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim E., Napierala M., Dent S. Y. (2011) Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res. 39, 8366–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans-Galea M. V., Carrodus N., Rowley S. M., Corben L. A., Tai G., Saffery R., Galati J. C., Wong N. C., Craig J. M., Lynch D. R., Regner S. R., Brocht A. F., Perlman S. L., Bushara K. O., Gomez C. M., Wilmot G. R., Li L., Varley E., Delatycki M. B., Sarsero J. P. (2012) FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 71, 487–497 [DOI] [PubMed] [Google Scholar]
- 14. Sakamoto N., Chastain P. D., Parniewski P., Ohshima K., Pandolfo M., Griffith J. D., Wells R. D. (1999) Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich's ataxia. Mol. Cell 3, 465–475 [DOI] [PubMed] [Google Scholar]
- 15. Grabczyk E., Mancuso M., Sammarco M. C. (2007) A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 35, 5351–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burnett R., Melander C., Puckett J. W., Son L. S., Wells R. D., Dervan P. B., Gottesfeld J. M. (2006) DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich's ataxia. Proc. Natl. Acad. Sci. U.S.A. 103, 11497–11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Biase I., Chutake Y. K., Rindler P. M., Bidichandani S. I. (2009) Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One 4, e7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cairns B. R. (2009) The logic of chromatin architecture and remodelling at promoters. Nature 461, 193–198 [DOI] [PubMed] [Google Scholar]
- 19. Kelly T. K., Liu Y., Lay F. D., Liang G., Berman B. P., Jones P. A. (2012) Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schones D. E., Cui K., Cuddapah S., Roh T. Y., Barski A., Wang Z., Wei G., Zhao K. (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vavouri T., Lehner B. (2012) Human genes with CpG island promoters have a distinct transcription-associated chromatin organization. Genome Biol 13, R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ozsolak F., Song J. S., Liu X. S., Fisher D. E. (2007) High-throughput mapping of the chromatin structure of human promoters. Nat. Biotechnol. 25, 244–248 [DOI] [PubMed] [Google Scholar]
- 23. Tolstorukov M. Y., Kharchenko P. V., Goldman J. A., Kingston R. E., Park P. J. (2009) Comparative analysis of H2A.Z nucleosome organization in the human and yeast genomes. Genome Res. 19, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Al-Mahdawi S., Sandi C., Mouro Pinto R., Pook M. A. (2013) Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS One 8, e74956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Plasterer H. L., Deutsch E. C., Belmonte M., Egan E., Lynch D. R., Rusche J. R. (2013) Development of frataxin gene expression measures for the evaluation of experimental treatments in Friedreich's ataxia. PLoS One 8, e63958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohshima K., Montermini L., Wells R. D., Pandolfo M. (1998) Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J. Biol. Chem. 273, 14588–14595 [DOI] [PubMed] [Google Scholar]
- 27. Sakamoto N., Ohshima K., Montermini L., Pandolfo M., Wells R. D. (2001) Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J. Biol. Chem. 276, 27171–27177 [DOI] [PubMed] [Google Scholar]
- 28. Pianese L., Turano M., Lo Casale M. S., De Biase I., Giacchetti M., Monticelli A., Criscuolo C., Filla A., Cocozza S. (2004) Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatry 75, 1061–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filla A., De Michele G., Cavalcanti F., Pianese L., Monticelli A., Campanella G., Cocozza S. (1996) The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 59, 554–560 [PMC free article] [PubMed] [Google Scholar]
- 30. Dürr A., Cossee M., Agid Y., Campuzano V., Mignard C., Penet C., Mandel J. L., Brice A., Koenig M. (1996) Clinical and genetic abnormalities in patients with Friedreich's ataxia. N. Engl. J. Med. 335, 1169–1175 [DOI] [PubMed] [Google Scholar]
- 31. Clark R. M., Bhaskar S. S., Miyahara M., Dalgliesh G. L., Bidichandani S. I. (2006) Expansion of GAA trinucleotide repeats in mammals. Genomics 87, 57–67 [DOI] [PubMed] [Google Scholar]
- 32. Holwerda S. J., de Laat W. (2013) CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R Soc. Lond. B Biol. Sci. 368, 20120369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fu Y., Sinha M., Peterson C. L., Weng Z. (2008) The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 4, e1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]