

Background: Nitroreductase reduces a broad range of nitroaromatics.
Results: Steady-state and pre-steady-state kinetics were combined with tests for aminoaromatic product formation.
Conclusion: Both half-reactions occur via a simple mechanism lacking detectable gating steps consistent with the broad substrate repertoire.
Significance: Nitroreductase does not generate p-aminobenzoic acid and, therefore, appears not to reduce nitro groups to amines.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Flavin, Flavoproteins, Reductase, Nitroreductase, Substrate Specificity
Abstract
The oxygen-insensitive nitroreductase from Enterobacter cloacae (NR) catalyzes two-electron reduction of nitroaromatics to the corresponding nitroso compounds and, subsequently, to hydroxylamine products. NR has an unusually broad substrate repertoire, which may be related to protein dynamics (flexibility) and/or a simple non-selective kinetic mechanism. To investigate the possible role of mechanism in the broad substrate repertoire of NR, the kinetics of oxidation of NR by para-nitrobenzoic acid (p-NBA) were investigated using stopped-flow techniques at 4 °C. The results revealed a hyperbolic dependence on the p-NBA concentration with a limiting rate of 1.90 ± 0.09 s−1, indicating one-step binding before the flavin oxidation step. There is no evidence for a distinct binding step in which specificity might be enforced. The reduction of p-NBA is rate-limiting in steady-state turnover (1.7 ± 0.3 s−1). The pre-steady-state reduction kinetics of NR by NADH indicate that NADH reduces the enzyme with a rate constant of 700 ± 20 s−1 and a dissociation constant of 0.51 ± 0.04 mm. Thus, we demonstrate simple transient kinetics in both the reductive and oxidative half-reactions that help to explain the broad substrate repertoire of NR. Finally, we tested the ability of NR to reduce para-hydroxylaminobenzoic acid, demonstrating that the corresponding amine does not accumulate to significant levels even under anaerobic conditions. Thus E. cloacae NR is not a good candidate for enzymatic production of aromatic amines.
Introduction
NAD(P)H:nitroaromatic reductases (nitroreductases) are flavoenzymes that catalyze the NAD(P)H-dependent reduction of the nitro groups of nitroaromatic and nitroheterocyclic compounds (1) (Fig. 1A).
FIGURE 1.

A, the reduction of nitroaromatic substrates catalyzed by nitroreductase for the example of nitrobenzene. B, ribbon structure of NR with lettering of the helices indicated. One monomer is in cyan, and the other is in magenta, the bound FMN is depicted with yellow sticks, and bound substrate analog benzoate is depicted with green sticks. Figure is based on 1KQB.pdb and generated using PyMOL (52).
Enterobacter cloacae nitroreductase (NR)2 was first isolated from bacteria collected at a weapon storage facility by Bryant et al. (2, 3) based on its ability to degrade explosive compounds such as 2,4,6-trinitrotoluene (TNT). The enzyme exists as a homodimer of 217-residue monomers with two identical active sites in the dimer interface, each containing a non-covalently bound FMN that derives H-bonds from one monomer and hydrophobic contacts from both (4) (Fig. 1B).
Nitroreductases are widely distributed among bacteria, but NR-like proteins are also found in archaea and eukaryotes including man (5). E. cloacae NR is able to reduce a variety of nitroaromatics ranging from nitrofurans, nitroarene pollutants such as 1-nitropyrene, nitro-containing drugs (metronidazole), herbicides (dinoseb), and most famously nitroaromatic and nitramine explosives such as TNT (2, 6, 7). The Escherichia coli homologue NfsB is being developed for use as a prodrug activator for treatment of cancer (8–10).
The NRs from E. cloacae and E. coli reduce nitroaromatics to the corresponding nitrosoaromatics and then to the corresponding hydroxylaminoaromatics via two successive two-electron reductions (6, 11) (Fig. 1A). It is difficult to isolate the nitroso intermediates because they react rapidly with NR, and the resulting hydroxylaminoaromatics react with their nitroso precursors to yield stable azoxy compounds (6, 12, 13). Nonetheless, there are reports of accumulation of a nitroso intermediate during biological reduction of certain nitroaromatics (14).
The hydroxylamine product is a basis for the toxicity of metronidazole and related antibiotics, which are activated in vivo by pathogen NRs (15, 16) (in anaerobes, other mechanisms reductively eliminate the nitro group producing nitro radicals that initiate oxidative stress (17, 18)). Thus chemistry of NR is being developed for clinical implementation against cancer via GDEPT (gene-directed enzyme prodrug therapy) and ADEPT (antibody-directed enzyme pro-drug therapy) wherein an NfsB-antibody fusion would be targeted to tumors where it would then reductively activate the prodrug CB1954 in ADEPT (8, 9, 19). Thus it is important to know what product NR produces.
Bioremediation and biocatalytic uses of NR have also been envisioned based on the possible production of aromatic amines by NR. Aromatic amines would be valuable synthons, and there are a few instances in which amine products have been detected (12, 13, 20, 21). In particular, the NR of Mycobacterium smegmatis (NR-Ms) converts the nitro group of benzothiazinone to an amine (20). The NR from Salmonella typhimurium (NR-Sal) displays both nitroreductase and enoate reductase activity, transforming nitrobenzene to nitrosobenzene, phenylhydroxylamine, and even the fully reduced product aniline (13). Similarly, the first NR of Klebsiella sp. C1 (NR-I) transforms 2,4,6-TNT to 2-amino-4,6-dinitrotoluene (12). However amine products can elude detection due to their reactivity with oxygen. Thus dissolved O2 can react with reduced products of NR including both the hydroxylamine and putative amine products in a futile cycle that will tend to diminish detected yields and mask formation of reduced products (7). This and the proclivity of the products for reactions among themselves has made it difficult to confirm accumulation of aminoaromatic products, especially given the common practice of conducting assays in air.
There are cases in which production of amines has been ruled out. For E. coli NfsB, it was demonstrated that only 2 eq. of NADH are oxidized per eq. of nitroaromatic reduced (11). Moreover Race et al. (11) showed that if reaction products were analyzed under aerobic atmosphere, hydroxylamines were converted to nitrosos. Thus full reduction of nitro groups to the corresponding amine is not a general property of the subfamily, and the identity of the substrate may be a major determinant of what final product can be achieved. Therefore, we have tested whether E. cloacae NR can produce para-aminobenzoic acid (p-ABA) from para-hydroxylaminobenzoic acid (p-HABA) under anaerobic conditions.
The broad substrate repertoire of nitroreductase makes it an attractive enzyme for bioremediation of nitroaromatics and for producing valuable pharmacological reagents because this one enzyme could be used to transform a variety of related compounds (6, 13). A number of enzymes are known that transform diverse substrates, including certain cytochrome P450s (22, 23), and pentaerythritol tetranitrate reductase (24, 25). Binding of diverse substrates appears to be associated with relatively weak and nonspecific substrate binding (for example, see Ref. 26). Indeed, NfsB substrates have been found to bind via water molecules and to have relatively large Km values (27). Thus one hypothesis is that the broad specificity of NR is related to its relatively high Kms and large solvent-accessible active site (4).
However, the broad substrate repertoire of NR could also have a mechanistic basis. In several enzymes that are highly discriminating regarding their substrates, the mechanisms have been found to incorporate multiple steps in which the prospective substrate is “tested” on the basis of its ability to elicit a conformation change in the enzyme or to enhance binding of additional substrates needed for the reaction to proceed. Thus a second hypothesis is that the mechanism of NR may lack gating steps that could constrain its use of diverse substrates.
Prior steady-state kinetics studies have shown that NR employs a ping-pong bi-bi mechanism in which the flavin is alternately reduced by a nicotinamide (preferably NADH or NADPH) and then reoxidized by a nitroaromatic (or quinone) substrate (6). Pre-steady-state studies of NfsB were used to identify variants of the enzyme that enhance its activity on CB1954 and tailor the site-specificity of the reduction with respect to the two nitro groups of CB1954 (28). In these, Jarrom et al. (28) determined the second order rate constant k1/Kd for the oxidative half-reaction (in which the flavin is reoxidized) but could not evaluate Kd due to the limited solubility of CB1954 as well as loss of a substantial portion of the kinetic trace in the instrument dead time for the higher CB1954 concentrations. The second-order rate constants were many times faster than the overall rate constant, indicating that the rate-limiting step is slow product release, possibly depending on conformational change (28). Thus the oxidizing substrates used in that study (CB1954 or nitrofurazone) were not able to address the possibility of a binding step preceding the chemical reaction. Nor did this study address the reductive half-reaction due to reaction times within the dead time of the instrument at 25 °C.
To slow down the NR reaction we have worked at lower temperatures and performed pre-steady-state as well as steady-state analyses of each of the two half-reactions of NR to identify elements of the reaction that could participate in defining the substrate specificity of NR (and lack thereof). We have chosen a poor substrate, para-nitrobenzoic acid (p-NBA) to allow chemical steps to contribute more to the observed rates, and we have simulated the reactions in terms of all the participating states of the enzyme to test for possible additional steps not explicit in the observed kinetics. The higher solubility of p-NBA in water also enabled us to probe binding events. Knowledge of the natures and lifetimes of all the participating states for this simple substrate provides a crucial baseline for understanding the different rates and product distributions displayed by different NR substrates as well as different variants of NR produced by mutagenesis.
EXPERIMENTAL PROCEDURES
Reagents
NADH (100% purity) was purchased from Roche Applied Science. p-NBA was from Acros Organics. p-Nitrosobenzoic acid (p-NOBA) was produced as per the procedure of Defoin (29) and validated by 1H NMR spectroscopy at 400 MHz. p-HABA was the generous gift of K. Ferguson and A. S. Bommarius (School of Chemical and Biomolecular Engineering, Georgia Institute of Technology) and was determined to be 99% pure based on NMR. Concentrations of reagents were determined using the known extinction coefficients at pH 7.0: ϵ340 = 6.22 × 103 m−1cm−1 or ϵ370 = 2.66 × 103 m−1cm−1 for NADH and ϵ273 = 10.1 × 103 m−1cm−1 for p-NBA. Wild-type NR was expressed and purified as previously described (1). The concentration of holo-NR was evaluated based on the extinction coefficient of the bound flavin, ϵ454 = 14.3 × 103 m−1cm−1, and rate constants reported refer to holo-enzyme (apoFMN enzyme represented <10% of the total protein based on the ratio of A454 to A280 (1).
Steady-state Kinetic Experiments
Initial velocities at various concentrations of NADH and p-NBA were measured using stopped-flow spectrophotometry at 4 °C (TgK Scientific). The reactions were monitored at 370 nm to observe conversion of NADH to NAD+. The assays contained 60 nm enzyme, various concentrations of NADH (0.1, 0.2, 0.3, and 0.4 mm) and various p-NBA concentrations (0.1, 0.2, and 0.4 mm) in 50 mm potassium phosphate, pH 7.50. Initial rates of the reaction were calculated using Program A (a generous gift of D. P. Ballou), and steady-state kinetic parameters were extracted using KaleidaGraph Version 3.6.2 to fit the data to the equation for a ping-pong mechanism (Equation 1) (30, 31) where KmNADH and KmPNBA are the Km values for NADH and p-NBA, respectively.
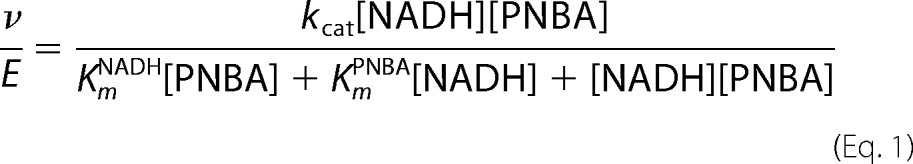 |
Rapid Reaction Experiments
Pre-steady-state reductive and oxidative half-reactions of NR were studied using a stopped-flow spectrophotometer (TgK Scientific). The flow system was made anaerobic by rinsing with an anaerobic buffer and incubating overnight with a solution of 50 mm sodium dithionite in 50 mm potassium phosphate, pH 7.50. The buffer to be used for the sodium dithionite solution was first sparged with N2 and then equilibrated overnight to remove oxygen in an anaerobic glove box (M-BRAUN UNIlab glove box with Siemens Corosop 15 controller). Sodium dithionite was added to the anaerobic buffer inside the glove box, and the resulting solution was transferred into tonometers. These were mounted on the stopped-flow apparatus to provide reservoirs of anaerobic solutions and used to flush the flow system, which was then allowed to stand overnight with dithionite solution throughout. Before experiments, the instrument was rinsed thoroughly with anaerobic buffer composed of 50 mm potassium phosphate, pH 7.50, equilibrated with N2 gas that had been passed through an oxygen removal column (Labclear).
In general NR was dissolved in 50 mm potassium phosphate, pH 7.50, and experiments were performed at 4 °C because the reaction rates at higher temperatures were too fast to permit detailed characterization. For single-mixing experiments a solution of ∼35 μm NR was loaded in one syringe, and a concentration of substrate twice the concentration desired for the reaction, in the same buffer, was loaded in the other syringe. Equal volumes of the two solutions were co-injected into the rapid mixer (75 μl each), and the dead-time was 1 ms.
For each substrate concentration, at least four replicate measurements were made once the instrument had stabilized, and these transients were averaged. Analysis was conducted by fitting the resulting kinetic trace to exponential equations for growth and decay using the Marquardt algorithm in Program A, developed by C. J. Chiu, R. Chung, J. Diverno, and D. P. Ballou at the University of Michigan (Ann Arbor, MI) or with the Kinesyst 3 software provided with the stopped-flow spectrophotometer (TgK Scientific). Plots of observed rate constants (kobs) versus the concentration of substrate used were fit with Equation 2 where k is the apparent first-order rate constant (for 17.5 μm ΝR), and Kd is the dissociation constant for substrate and enzyme.
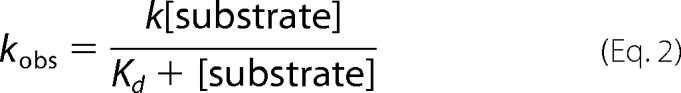 |
Rates of background reactions between substrate and dissolved oxygen were measured and constituted <1% of the rate observed in the presence of enzyme.
Kinetic traces were collected at a series of wavelengths from 340 to 700 nm at 5-nm intervals to look for the signatures of potential intermediates.
Reductive Half-reaction
A solution of 26 μm NR was made anaerobic by equilibration in the anaerobic glove box for 30 min then placed in a tonometer. All buffer and substrate solutions were made anaerobic by bubbling with N2 gas that had passed through an O2-removing cartridge. Substrate solutions were loaded in tonometers, which in turn were mounted on the stopped-flow spectrophotometer along with the NR solution. Upon rapid mixing of the enzyme with substrate, flavin reduction was monitored by measuring loss of absorbance at 454 nm. This wavelength is the peak of the oxidized enzyme absorption and provides the maximum difference between the oxidized and the reduced enzyme. After mixing, the final concentration of the enzyme was 13 μm, whereas the concentration of the substrate was kept at least 5-fold greater than that of the enzyme, so that the pseudo-first-order condition was maintained. Analysis was carried out as described above. Rate constants (kobs) were determined from fits of the kinetic traces obtained at 454 nm. Plots of kobs versus substrate concentration were analyzed using the non-linear least-square fitting algorithms in the KaleidaGraph package (Synergy Software).
To test the presumed kinetic mechanism, simulations of reaction transients predicted by various mechanisms, rate constants, and binding equilibria were performed using Berkeley Madonna, Version 8.3 (Macey and Oster, University of California-Berkeley) refined by the Runge-Kutta 4 integration method. In brief, in the reductive half-reaction, the known parameters, which are the concentration of oxidized NR, the extinction coefficients of oxidized and reduced NR, and the reduction rate constant, were provided to the simulation, then the other parameters, which were k1, k−1, and k3 and the extinction coefficients of Eox:NADH, and Ered:NAD+, were adjusted manually to produce the best agreement with experimental results based on the superposition of simulated transients with experimental transients (see Fig. 3). A whole set of transients was treated at once. We applied the same method to the oxidative half-reaction in which the reduced NR concentration, extinction coefficients of oxidized and reduced NR, and the oxidation rate constant from the experiments were provided to the simulation and allowed k4, k−4, and the extinction coefficients of other species to be optimized.
FIGURE 3.
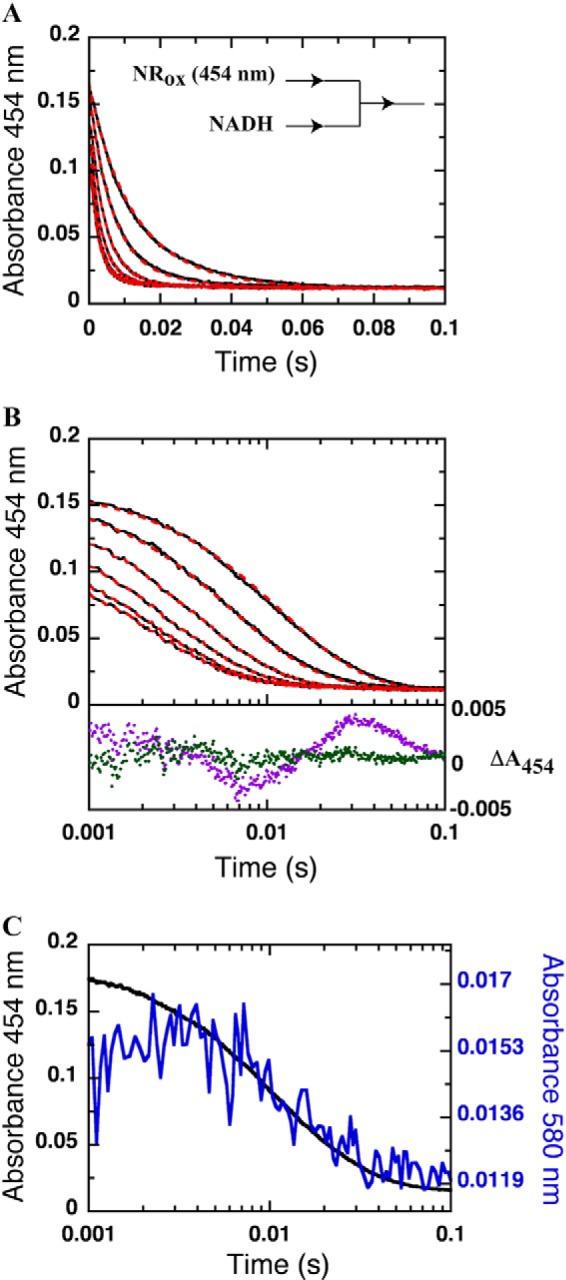
Reductive half-reaction of NR with NADH. Shown are the linear time scale (A) and log time scale (B) kinetic traces of the reaction of NR (13 μm) with various concentrations of NADH (0.08, 0.16, 0.32, 0.64, 1.28, and 2.56 mm) in 50 mm potassium phosphate pH 7.50 at 4 °C. Reactions were monitored in the stopped-flow spectrophotometer under anaerobic conditions. All concentrations are reported as final concentrations after mixing. Dashed red lines are simulated traces obtained using the kinetic constants listed in Table 1 and Scheme 1. Residuals of fitting the reduction of NR with 0.08 mm NADH using one exponential (magenta dots) and two exponentials (green dots) are shown below with a vertical ΔA454 axis on the right. C, kinetic traces of the reduction of NR at 454 nm (black line) and formation and decay of the charge-transfer complex observed at 580 nm (blue line) with a concentration of 0.08 mm NADH.
Oxidative Half-reaction
Reduced enzyme in 50 mm potassium phosphate at pH 7.50 (17.5 μm after mixing) was rapidly mixed with either p-NBA or p-NOBA in the same buffer. All substrate concentrations were at least 5-fold higher than the enzyme concentration used to maintain pseudo-first-order conditions. The reduced enzyme solution was prepared by equilibration with the inert atmosphere in the glove box followed by reduction with a slightly substoichiometric amount of dithionite (or NADH) in the anaerobic glove box (with equivalent results). The reduction process was monitored using a spectrophotometer at 4 °C. Oxidation of the enzyme by substrate was observed at 454 nm by the stopped-flow spectrometer, revealing formation of oxidized NR-bound flavin. Analysis was carried out as described above.
Testing for Formation of Aromatic Amine Product
The decomposition of p-HABA was monitored by UV-visible spectrophotometry using a CARY-300 spectrophotometer to observe the absorbance of the final product of p-HABA oxidation (see Fig. 6C). p-HABA was dissolved to a concentration of 1.3 mm in 50 mm potassium phosphate, pH 7.50, that had been made anaerobic, or equilibrated with air as a control. The anaerobic sample of p-HABA was produced by weighing p-HABA (15.3 mg) and transferring it into the anaerobic glove box. The p-HABA powder was equilibrated in the glove box for 10 min to ensure that there was no longer oxygen associated with it. A 5-ml stock solution of 20 mm was prepared in the glove box using anaerobic buffer. For analysis, 1.3 mm p-HABA was transferred to a gas-tight anaerobic cuvette before removing it from the glove box.
FIGURE 6.
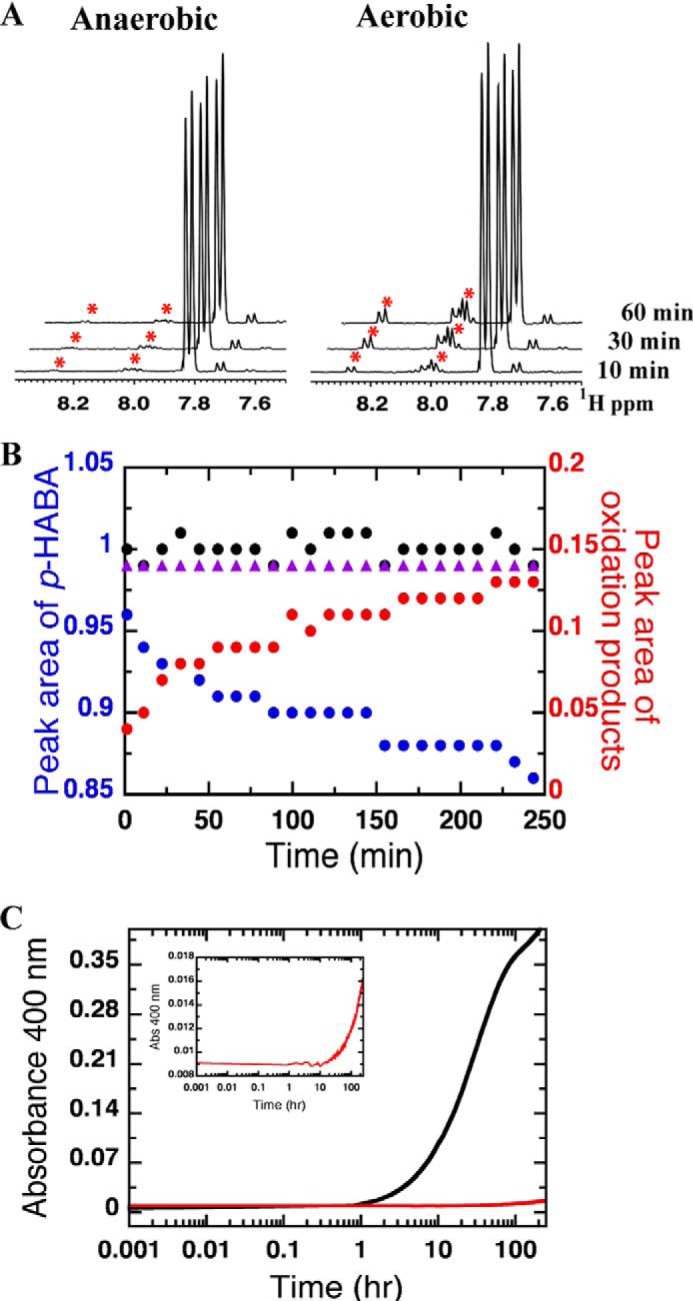
Decomposition of p-HABA under aerobic and anaerobic conditions. A, 1H NMR spectra of 13.3 mm p-HABA in 50 mm potassium phosphate, pH 7.50, 10% D2O, 0.1 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid under aerobic and anaerobic conditions after 10, 30, and 60 min. Asterisks indicate signals that increase with time. B, total integrated NMR peak area corresponding to p-HABA (at δ = 7.04, 7.06, 7.81, and 7.83, blue circles) or p-NOBA plus 4,4′-azoxydibenzoic acid (at δ = 7.99, 8.00, 8.01, 8.03, 8.26, and 8.28 ppm, red circles) as a function of time. Black circles represent the sum of the area corresponding to p-HABA and oxidation products. Purple triangles represent the total integrated NMR peak area corresponding to p-HABA under anaerobic conditions. C, kinetic traces of formation of decomposition product of p-HABA under aerobic (black trace) and anaerobic (red trace) conditions. The reactions were monitored via the absorbance at 400 nm at 25 °C. The inset shows a vertical expansion of the anaerobic reaction of p-HABA. The observed decomposition rates were calculated to be 3.0 × 10−2 h−1 and 5.4 × 10−4 h−1 for aerobic and anaerobic decomposition, respectively.
The stability and purity of p-HABA in anaerobic versus air-equilibrated buffer was also characterized by 1H NMR spectroscopy at 400 MHz. p-HABA (13.3 mm) was dissolved in 50 mm potassium phosphate, pH 7.50, containing 10% v/v 2H2O and 0.1 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as a chemical shift reference (δ = 0 ppm). The two samples were prepared as described above for optical samples but were transferred to NMR tubes instead of cuvettes. Wilmad NMR tubes, 5-mm diameter precision, were used to contain 600 μl of sample. For the anaerobic sample, Wilmad's gas-tight anaerobic NMR tube (535-TR-7) was used after prior degassing and incubation in the glove box overnight. The Wet1D sequence was used to achieve solvent suppression (32).
The possible formation of aromatic amines was assessed using 3-methyl-2-benzothiazolinone hydrazone (MBTH), which reacts with aromatic amines to produce a strongly absorbing chromophore (33, 34) (ϵ550 > 2 mm−1cm−1 in the presence of competing reactions with aromatic hydroxylamines). All manipulations were performed under inert atmosphere in a glove box ([O2] ≤ 10 ppm) at room temperature. With the exception of concentrated enzyme solution, all reagents and materials were equilibrated in the glove box overnight. Enzyme solutions were rendered anaerobic by 5–10 pump-purge cycles of ∼30 s followed by equilibration in the glove box for 1 h. Enzymatic reactions and controls were run for 0 min, 15 min, 30 min, or 10 h in 50 mm potassium phosphate, pH 7.50, containing 1.25 mm NADH, 17.5 μm NR, 0–1 mm p-ABA, and/or 0–1 mm p-HABA. At the end of the enzymatic reaction, protein was removed by ultrafiltration using a centrifugal filter (EMD-millipore, 10-kDa cutoff) for both experimental samples and controls. Removal of the protein resulted in lower background absorbance. The published protocol for quantification of substituted aromatic compounds using MBTH was optimized to accommodate the NADH. Under anaerobic conditions 1.5 mm MBTH and 6.5 mm Ce(SO4)2 in the presence of 0.5–1 mm p-ABA produced the largest increase in absorbance at 550 nm. The total time between termination of the reaction by protein removal and reading of the colorimetric assay results did not exceed 40 min.
RESULTS
Steady-state Kinetics of NR
Although steady-state kinetic analyses of NR have been reported before (6), they were repeated as part of this effort to obtain data sets with which to test the kinetic and equilibrium constants obtained by pre-steady-state methods and simulation (see below). The overall steady-state kinetics of NR were characterized using NADH and p-NBA as substrates at 4 °C. Either NADH or p-NBA was varied at each of several concentrations of the other substrate as described under “Experimental Procedures” and the previous study (6). The data (Fig. 2A) were fit with Equation 1 to give a KmNADH of 35 ± 8 μm, KmPNBA of 130 ± 5 μm, and a kcat value of 1.7 ± 0.3 s−1. The plot of initial velocity versus NADH concentration (0.02–0.33 mm) at high p-NBA concentration (0.4 mm) (Fig. 2B) and the plot of initial velocity versus p-NBA concentration (0.025–0.6 mm) at high NADH concentration (0.25 mm) (Fig. 2C) yielded similar parameters to those resulting from double-reciprocal plot (Fig. 2A). The complete steady-state study was repeated for two different preparations of NR. The results were within error of one another so only the parameters obtained from the larger study are reported.
FIGURE 2.
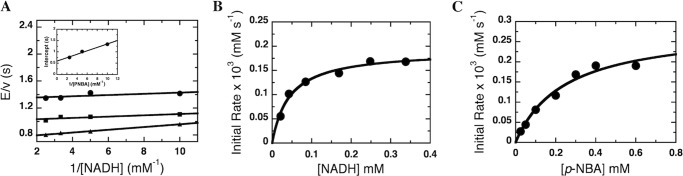
Steady-state kinetics of NR with NADH and p-NBA as substrates. A, double-reciprocal plots of the initial rates of reaction of NR with various concentrations of NADH (0.1, 0.2, 0.3, and 0.4 mm after mixing). Upper to lower lines of the primary plot are from the reactions of 0.1, 0.2, and 0.4 mm (after mixing) p-NBA, respectively. The assay reactions were performed using a stopped-flow spectrophotometer at 4 °C. The inset shows the plot of ordinate intercepts from the double-reciprocal plot versus the reciprocal of p-NBA concentrations. The parameters obtained from the fits are provided in Table 1. B and C, used an enzyme concentration of 118 nm versus 60 nm for panel A. B, plot of initial velocity versus NADH concentration (0.02, 0.042, 0.08, 0.17, 0.25, and 0.34 mm) at a fixed p-NBA concentration (0.4 mm). The results show hyperbolic dependence on NADH concentrations with KmNADH of 44 μm and kcat = 1.7 s−1. C, plot of initial velocity versus p-NBA concentration (0.025, 0.050, 0.1, 0.2, 0.3, 0.4, and 0.6 mm) at a fixed NADH concentration (0.25 mm). The results showed hyperbolic dependence on p-NBA concentrations with the KmPNBA of 0.25 mm and kcat = 1.7 s−1.
Reductive Half-reaction of oxidized NR with NADH
The reaction of NR with NADH was investigated by stopped-flow spectrophotometry under anaerobic conditions at 4 °C. An anaerobic solution of oxidized NR was mixed with various concentrations of NADH to yield final concentrations of 13 μm NR and 0.08, 0.16, 0.32, 0.64, 1.28, or 2.56 mm NADH (all concentrations are quoted after mixing here and below). Reduction of the enzyme-bound oxidized FMN was monitored via the loss of absorbance at 454 nm (Fig. 3, A and B), and formation and decay of a charge-transfer complex was monitored at 580 nm (Fig. 3C).
The results showed that reduction of NR by NADH is fast and the beginning of the kinetic trace (first phase) of all but the lowest concentration of NADH (0.08 mm) was within the dead time (0.001 s) of the stopped-flow instrument (Fig. 3B). Two rate constants were required to adequately describe the data; the use of only one resulted in systematic residuals that were eliminated by the use of a second rate constant (Fig. 3B). The first process (∼0.001–0.01 s for 0.08 mm NADH) was characterized by decreased absorbance at 454 nm and an increase in absorbance at 580 nm (Fig. 3C). Therefore, the first rate constant was attributed to hydride transfer and formation of a charge-transfer complex between reduced FMN and NAD+. The second phase (∼0.01–0.1 s for 0.08 mm NADH) was attributed to dissociation of NAD+. A plot of the observed rates of hydride transfer for reactions with 2.56 mm NADH and below yielded a hyperbolic dependence on NADH concentration and, therefore, was fit with Equation 2 (Fig. 4A) to yield the rate constant k2 = 700 ± 20 s−1 and the dissociation constant (Kd = k−1/k1) of 0.51 ± 0.04 mm depicted in Scheme 1.
FIGURE 4.
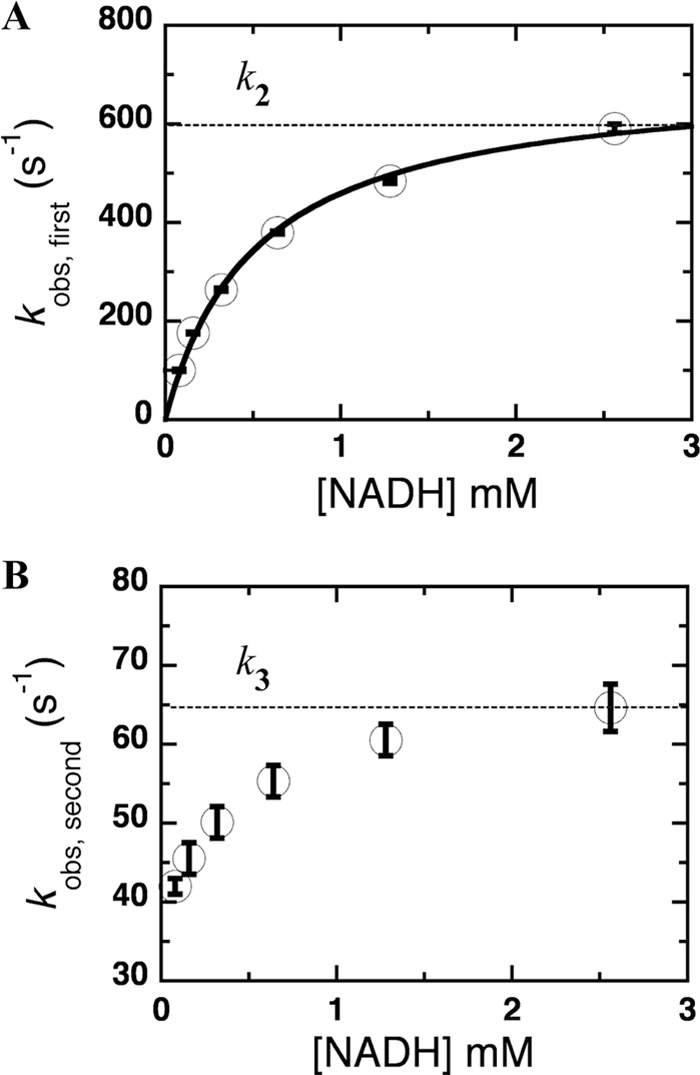
Plot of the observed rate constants of the reduction of NR. A, plot of the observed rate constants from the first phase; 100 ± 2, 176 ± 2, 264 ± 3, 380 ± 3, 484 ± 5, and 590 ± 9 s−1 (from low to high NADH concentrations; 0.08, 0.16, 0.32, 0.64, 1.28, and 2.56 mm) versus NADH concentration. The data including their error bars are well described by a rectangular hyperbola, which is the source of the values of k2 and Kd. B, plot of observed rate constants from the second phase; 42 ± 1, 46 ± 2, 50 ± 2, 55 ± 2, 61 ± 2, and 65 ± 3 s−1 (from low to high NADH concentrations as described above). The k3 value was estimated from the NADH asymptote as indicated by the dotted line.
SCHEME 1.
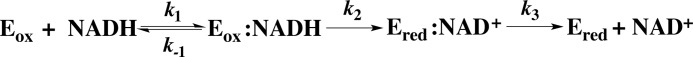
Reductive half-reaction of NR.
A rate constant for the second process was estimated from the high NADH concentration asymptote of the observed rate of the second phase (65 ± 3 s−1; Fig. 4B) and confirmed by simulations of the data using the obtained value of k3 = 60 s−1 (Table 1).
TABLE 1.
Kinetic parameters for NR at 4 °C: comparison of steady-state, pre-steady-state, and simulation results
The values were obtained from the experimental data performed in 50 mm potassium phosphate, pH 7.50, at 4 °C under anaerobic conditions using stopped-flow spectrophotometer or kinetic simulations. The kinetic parameters (steady-state kinetics) are as follows: kcat = 1.7 ± 0.3 s−1, KmNADH = 35 ± 8 μm; KmPNBA = 130 ± 5 μm.
| Kinetic parameters; pre-steady state |
Extinction coefficients |
|||
|---|---|---|---|---|
| From experiment | From simulation | Chemical species | From experiment | From simulation |
| m−1 cm−1 | ||||
| k1a = 1.3 × 107 m−1 s−1 | Eox | 14,300 | 14,340 | |
| k-1a = 6250 s−1 | Eox:NADH | 10,200 | ||
| k2 = 700 ± 20 s−1 | k2 = 700 s−1 | Ered:NAD+ | 1,940 | |
| k3 = 65 ± 3 s−1 | k3 = 60 s−1 | Ered | 931 | 920 |
| KdNADH = 0.51 ± 0.04 mm | KdNADH, k-1/k1 = 0.48 mm | Ered:PNBA | 2,850 | |
| k4a = 6.8 × 105 m−1 s−1 | Eox:Nitroso | 18,240 | ||
| k−4a = 181 s−1 | ||||
| k5 = 1.90 ± 0.09 s−1 | k5 = 2 s−1 | |||
| KdPNBA = 0.33 ± 0.04 mm | KdPNBA, k−4/k4 = 0.27 mm | |||
a The values provided are not necessarily unique but are able to fully explain the data observed in conjunction with the model we provide. The values of k1 and k−1 are inversely correlated, as are the values of k4 and k−4.
To determine whether the observed rate constants suffice to fully describe the observed behavior, simulations of the behavior predicted by the model in Scheme 1 with the parameters in Table 1 were performed, and the parameters were refined to obtain the best visual agreement with the experimental observations. The optimized simulation results (red dashed lines in Fig. 3, A and B) provide a good description of the observed kinetics (black lines in Fig. 3, A and B) and yield a “simulated” dissociation constant simKd of 0.48 mm (comparable to the Kd obtained from the experiments of 0.51 ± 0.04 mm). The simulated reduction rate constant (simk2) was 700 s−1 (versus 700 ± 20 s−1 from the experiment), and the rate of dissociation of NAD+ from NRred (simk3) was found to be 60 s−1 (Table 1). The study of NR reduction kinetics was repeated for two different preparations of NR. The results were again within error of one another so only the parameters obtained from the larger study are reported.
Thus we are able to report the first numerical values for the rate of reduction of the FMN of NR by NADH, which we find to occur very rapidly. Moreover these experiments yield a limiting value for the Km of NADH that is not complicated by concurrent operation of the other half-reaction.
Oxidative Half-reaction of Reduced NR with p-NBA
These reactions were investigated using stopped-flow experiments at 4 °C by rapidly mixing the reduced enzyme in 50 mm potassium phosphate, pH 7.50, with the same buffer containing different concentrations of p-NBA to yield a final NR concentration of 17.5 μm and p-NBA concentrations of 0.1, 0.2, 0.4, 0.8, and 1.6 mm (after mixing). The experiment was limited to a maximum p-NBA concentration of 1.6 mm by the modest solubility of p-NBA. The reduced enzyme was prepared by stoichiometric reduction with dithionite as described under “Experimental Procedures.” Kinetic traces at wavelengths from 340 to 700 nm at 5-nm intervals were collected. The kinetics at all wavelengths were the same, indicating that only formation of oxidized flavin contributes to optical spectrophotometric changes, and no transient intermediate was detected (data not shown).
The oxidation of NR the flavin of NR by p-NBA exhibited monophasic kinetics at 454 nm (Fig. 5). The observed rate constant (kobs) displayed saturation with respect to the p-NBA concentration as evinced by the hyperbolic curve (inset to Fig. 5). Therefore, the plot of kobs versus p-NBA concentrations was fit using Equation 2 yielding the limiting value of k5 = 1.90 ± 0.09 s−1 (Scheme 2). The intercept of the plot (k−5) approached a value of zero, indicating that the reverse reaction rate is negligible (inset to Fig. 5). The dissociation constant (Kd = k−4/k4) obtained from the inset in Fig. 5 and Equation 2 is 0.33 ± 0.04 mm. The hyperbolic dependence on substrate concentration indicates simple one-step binding before the flavin oxidation step (Scheme 2). The product of the oxidative half-reaction in this experiment was p-NOBA as no reductant was present to regenerate reduced enzyme and support a second cycle. The product was still bound to the active site at the end of the reaction (30 s) as indicated by a red shift of the final spectrum to 466 nm (from 454 nm for free oxidized enzyme) (data not show). The oxidative rate (1.90 ± 0.09 s−1) was close to the turnover value measured in steady-state kinetic experiments (kcat = 1.7 ± 0.3 s−1).
FIGURE 5.
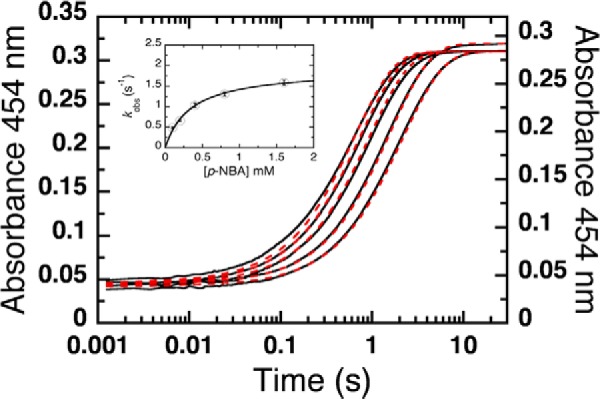
Oxidation of reduced NR by p-NBA. A solution of reduced NR (17.5 μm after mixing) was prepared by the stoichiometric addition of NADH, and this was then mixed with various concentrations of p-NBA to produce 0.1, 0.2, 0.4, 0.8, and 1.6 mm p-NBA in 50 mm potassium phosphate, pH 7.50. The reaction was monitored using the absorbance at 454 nm at 4 °C. The plot of kobs versus p-NBA concentration (inset) was fit with Equation 2 for a rectangular hyperbola. The calculated oxidation rate constant k5 and Kd are 1.90 ± 0.09 s−1 and 0.33 ± 0.04 mm, respectively. The kobs values from low to high p-NBA concentrations obtained from the transients are 0.49 ± 0.02, 0.66 ± 0.02, 1.03 ± 0.05, 1.31 ± 0.05, and 1.60 ± 0.06 s−1. The vertical line at each data point represents the S.D. of the fit from the experiments. The 0.2 mm and 1.6 mm p-NBA samples were slightly oxidized at the start of the transient, so a smaller fraction of the sample reacted with p-NBA. This is manifested by a slight deviation between the t = 0 absorbance of the sample (1.6 mm) and the need to employ a modified vertical axis for the sample containing 0.2 mm p-NBA. Dashed red lines are simulated traces obtained using the kinetic constants listed in Table 1 and defined in Scheme 2.
SCHEME 2.
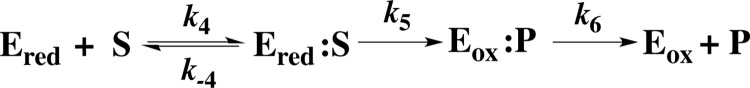
Oxidative half-reaction of NR.
Kinetic simulation of the model in scheme 2 and optimization of the kinetic parameters (Table 1) produced excellent agreement with the experimental data as shown in Fig. 5 (dashed lines versus solid, respectively). The simulations yielded simKd (=simk−4/simk4) of 0.27 mm and an oxidation rate constant (simk5) of 2 s−1 that are within error of the values determined directly from the data, indicating that this simple model and the parameters we report suffice to describe the optically perceptible events in the oxidative half-reaction of NR. All rate and equilibrium constants from the experiments and simulations are summarized in Table 1. These results reveal that oxidation of reduced flavin by p-NBA could be the rate-limiting step for overall turnover (k5).
The above results were confirmed using double-mixing stopped-flow spectrophotometry to first produce reduced NR by mixing oxidized NR with 20 μm NADH allowing an aging time of 0.5 s for complete reduction of NR and then combining the result with various concentrations of p-NBA (0.2, 0.4, 0.8, and 1.6 mm p-NBA and 17.5 μm NR were the final concentrations) in the second mix. The reactions were monitored at 454 nm to detect flavin reoxidation. The oxidation rate constant and dissociation constant were observed to be k5 = 2.00 ± 0.06 s−1 with an intercept close to zero and Kd = 0.20 ± 0.02 mm, respectively (not shown). The oxidative half-reaction of enzyme in the absence of NAD+ was studied using single-mixing stopped-flow spectrophotometry by reducing NR using stoichiometric sodium dithionite and rapidly mixing it with various concentrations of p-NBA as above. The observed rate constants were hyperbolically dependent on the p-NBA concentration. The limiting value was k5 = 1.90 ± 0.07 s−1, and the Kd value was 0.30 ± 0.04 mm (data not shown). These results confirm that dissociation of NAD+ from the active site of wild-type enzyme is complete within 0.5 s, and its presence in the reaction medium does not significantly affect the kinetics of the enzyme reaction with p-NBA. This supports a simple ping-pong bi-bi mechanism (6).
Oxidative Half-reaction with p-Nitrosobenzoic Acid
The dominant product resulting from reduction of nitroaromatics by NR is the corresponding hydroxylamino aromatic (6, 11). However, a few reports exist of the formation of a nitroso intermediate during biological reduction of certain nitroaromatics (14). It is difficult to isolate nitroso intermediates because they are reactive with hydroxylamines and rapidly reduced by NR (6). Thus reports of kinetic parameters are scarce.
To characterize the kinetics of nitroso aromatic reduction by NR, p-NOBA was generated by oxidation of p-ABA (see “Experimental Procedures”) and used as the substrate in a study of the kinetics of NR oxidation. An anaerobic solution of reduced NR was mixed with p-NOBA in 50 mm potassium phosphate, pH 7.50, to yield final concentrations of 17.5 μm NR and 0.1 mm p-NOBA. The kinetic trace was monitored at 454 nm and 4 °C using stopped-flow spectrophotometry. Most of the transients occurred during the dead time of the instrument (0.001 s) even with the lowest concentration of substrate (0.1 mm) at 4 °C, so the oxidation rate constant could not be determined directly. Based on the portion of the transient we could detect, the observed rate constant was roughly estimated to be ≈650 s−1 (at 0.1 mm p-NOBA) using Program A. This result is consistent with prior work.
A Third Possible Oxidative Half-reaction: Tests for Accumulation of Amino Aromatic Product
Several groups have shown that NR produces hydroxylamino products in multiple turnover reactions of nitroaromatic substrates (6, 11–13), but there are also a few reports of amine production (12, 13, 20, 21). Thus the capacity to produce amines may depend on the specific enzyme and substrate in use and require testing in each case. We have, therefore, optimized an assay for detecting p-ABA.
Low yields of anything are very difficult to ascertain; moreover use of multi-turnover assays beginning with a nitroaromatic substrate could produce the amino aromatic in the presence of the corresponding nitroso, which would react with the amine (35, 36). In addition, some of the prior studies were performed under air, whereas careful work has shown that hydroxylamino aromatic products can react with dissolved O2 to regenerate more oxidized precursors and establish a futile cycle (7) that would compete with further reduction and suppress formation of amine product. Finally, sample preparation and workup for detection can expose hydroxylamine and labile enzymatic products to O2. Again, hydroxylamino aromatics would be readily oxidized, and the resulting nitroso aromatics could then react with and mask amine products (37, 38). Therefore, to detect the possible formation of aromatic amines at low concentration, we reacted NR with p-HABA instead of p-NBA to focus directly on the reaction in question, and we have refined a sensitive direct chromogenic probe for aminoaromatic products that can be applied in the presence of hydroxylamines without any workup or exposure to air (33, 34).
To assess possible complications due to side-reactions and/or impurities, we first characterized the purity and stability of our p-HABA under the anaerobic conditions of our reaction by NMR and UV-visible spectrophotometry and compared the results with those obtained for p-HABA in conventional air-equilibrated solutions.
NMR spectra collected after incubation at room temperature for times ranging from 10 min to 24 h demonstrate that p-HABA is converted to another species in air-equilibrated buffer on a time scale of minutes to hours, but no reaction occurs under an inert atmosphere (Fig. 6). The p-HABA concentration was chosen to be 1.3 mm, 5 times the concentration of dissolved oxygen in air-saturated buffers (0.26 mm), to obtain pseudo-first order conditions. Within 5 h, the intensity of signals from 1.3 mm p-HABA had begun to visibly diminish, and new signals appeared and increased in successive 1H NMR spectra (Fig. 6A). The new signals at δ = 8.01, 8.03, 8.26, and 8.28 ppm match those of the aromatic protons of authentic p-NOBA, indicating that this is one of the oxidation products of p-HABA. Additional features in the spectrum are attributable to a small amount of contaminant (<5%) present in the p-HABA, and a compound that forms with the same time dependence as p-NOBA and that also forms when p-HABA is combined with p-NOBA under inert atmosphere, which we therefore tentatively assign to 4,4′-azoxydibenzoic acid (13). The decrease in the quantity of p-HABA present is fully accounted for by the sum of the increase in p-NOBA and putative azoxy product at all time points (Fig. 6B). Thus the optical signature of the proposed azoxy product could be used to monitor decay of p-HABA (Fig. 6C).
Azoxy compounds are intensely colored, whereas neither p-NOBA nor p-HABA absorbs in the visible range, so we monitored formation of the putative 4,4′-azoxydibenzoic acid at 400 nm. This was found to increase with a rate constant compatible with the growth of the new peaks in the NMR spectrum after accounting for concentration differences. Thus the initial oxidation rate constant was 3.0 × 10−2 h−1 for p-HABA. In contrast, for p-HABA in anaerobic solution the rate constant for appearance of the azoxy product was ≈3.2 × 10−5 h−1 (Fig. 6C). These results show that our anaerobic solutions of p-HABA do not spontaneously react to form any other products on a time scale extending to a day.
Amine Detection
We assayed directly for accumulation of p-ABA as a result of NR-catalyzed reduction of p-HABA in the presence of NADH. Reactions were incubated in the anaerobic glove box at ≈28 °C and terminated by filtration of the reaction mixture to remove NR (in the glove box). Controls lacking either NR or NADH were treated the same way. The MBTH assay for aromatic amines was performed in situ, avoiding any exposure to air or work-up.
MBTH reacts with aromatic amines forming an intensely colored adduct that can be detected with high sensitivity via its absorbance at 550 nm in a window between the optical signatures of NADH and FMN. The absorbance at 550 nm responded linearly to p-ABA concentration in the range of 0.05–0.3 mm p-ABA with a relatively low constant background attributable to products of reaction with NADH and/or p-HABA. To simulate the possible outcome of an enzymatic reaction beginning with 0.3 mm p-HABA and converting some amount to p-ABA, a second standard curve was constructed varying the mole fractions of p-ABA and p-HABA contributing to a total of 0.3 mm. The resulting response curve again reveals that the A550 increases with increasing amounts of p-ABA and decreasing amounts of p-HABA (Fig. 7). High concentrations of p-HABA appear to diminish the amount of dye available for reaction with p-ABA; therefore, controls were constructed to have the same initial concentration of p-HABA as experimental samples. Based on these data and their standard errors, we expect accumulation of >0.05 mm p-ABA to produce a significant A550 increase above background (Fig. 7).
FIGURE 7.
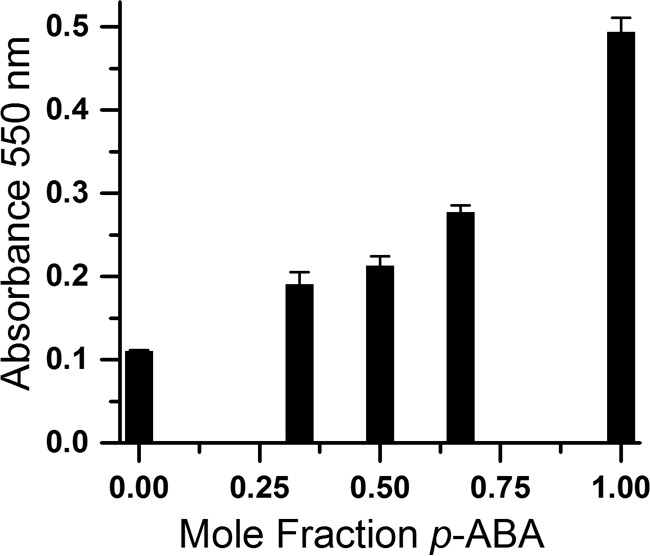
MBTH assay response to mole fraction of p-ABA in 0.3 mm combined concentration of p-ABA and p-HABA simulating increasing yields of p-ABA produced from p-HABA. Solutions totaling 0.3 mm p-ABA plus p-HABA but varying with respect to the mole fraction of p-ABA were augmented with 1.25 mm NADH and 17.5 μm NR and incubated for 15 min at 29 °C before being assayed for aromatic amine content using MBTH. The mole fraction is expressed as a function of the amine content: [p-ABA]/([p-ABA] + [p-HABA]).
We compared the outcome of MBTH assays on samples in which NR was allowed to react with p-HABA versus controls wherein NR was omitted to evaluate the extent to which any amine accumulation was enzyme-catalyzed. Table 2 shows slightly higher absorbance for the reactions initially containing 0.3 mm p-HABA compared with those initially containing 1 mm p-HABA, consistent with Fig. 7 and possible diversion of some MBTH by p-HABA. However, among reactions begun with equal p-HABA concentrations, no significant differences were found across treatments by one-way analysis of variance at 0.3 mm p-HABA (p = 0.097) or at 1 mm p-HABA (p = 0.097) (Table 2). The fact that omission of NR from the 10 h controls did not significantly affect absorbance at 550 nm indicates that there was little or no accumulation of aromatic amine due to enzymatic activity.
TABLE 2.
Analysis of reaction of p-HABA with NADH using MBTH
Samples were held at 29 °C in 50 mm potassium phosphate, pH 7.50, containing 1.25 mm NADH, 17.5 μm NR, and 0.3 or 1 mm p-HABA. Enzymatic reactions were terminated by protein removal using a centrifugal filter (EMD-millipore, 10-kDa cutoff). Aromatic amines were detected via the absorbance at 550 nm of the product of the reaction with 1.5 mm MBTH and 6.5 mm Ce(SO4)2.
| Reaction Time | p-HABA | Absorbance (550 nm) |
|---|---|---|
| mm | ||
| 0 min | 0.3 | 0.111(2)a |
| 10 hb | 0.3 | 0.120 (8) |
| 10 h control (no NR) | 0.3 | 0.120 (3) |
| 30 min | 1 | 0.065 (2) |
| 10 h | 1 | 0.080 (3) |
| 10 h control (no NR) | 1 | 0.074 (11) |
a S.D. based on three repetitions are provided in parentheses. No significant differences were found across treatments by one-way analysis of variance at 0.3 mm p-HABA (F(2,6) = 3.52, p = 0.097) or at 1 mm p-ABA (F(2,6) = 3.53, p = 0.097).
b Partial enzyme precipitation was observed after 6 h at 29 °C; however, aliquots of enzyme allowed to stand in buffer under the same conditions for 24 h retained 76% activity.
DISCUSSION
Nitroreductases are best known for their ability to catalyze reduction of the nitro groups of a wide range of aromatic substrates, but the biological function and the natural substrate of most nitroreductases are unknown. The prevalence of NR-family genes among bacteria nonetheless suggests that NRs confer a significant advantage on the organisms that bear them, and recent appreciation of the wide range of reactions conducted by members of the larger superfamily opens even wider the range of possibilities to be considered (5, 39). Recent data suggest a role as a dihydropteridine reductase for the NR homologue of Rhodobacter capsulatus (40).
Structural explanations for the broad substrate repertoires of NR homologues have been discussed (4, 41). The active site of NR is a large cavity between helices E and F, which extend out from the structural core of one monomer, and helix D and the preceding loop of the other monomer (Fig. 1). Diverse substrates bind by stacking on the flavin ring system (4); many exploit electrostatic interactions with the side chain of Lys-14 or employ water-mediated interactions with protein residues that are too far away for direct contact (41). The side chain of Phe-70 (helix D) was found to adopt diverse orientations and thereby accommodate different substrates (11, 41). Helix F also moves due to interactions between Phe-124 and substrate analogs (4, 27). The generally small number of interactions between substrate analogs and the enzyme is consistent with the generally large (>0.1 mm) Kd and Km values observed for oxidizing substrates (above and Refs. 6 and 11).
In other enzymes with broad substrate repertoires, the active site has been observed to alter its conformation to bind different substrates. The active site of the sulfotransferase SULT1A1 adopts different conformations to bind two molecules of nitrophenol at once versus estradiol (42), and isopropylmalate isomerase of Pyrococcus horikoshii was proposed to be able to bind either homocitrate or isopropylmalate due to the flexibility of a loop that replaces the helix that discriminates between similar substrates in the homologous aconitase (43). Similarly, a more flexible and accessible active site was credited with the expanded substrate repertoire of an evolved metallo-β-lactamase (44) and a glutathione S-transferase (GST) with a broader repertoire was found to be more flexible than a GST with greater substrate selectivity on the basis of H/D exchange as well as fluorescence lifetime analyses (45).
In contrast, mechanistic bases for specificity include conformational changes required as part of the catalytic cycle. For example, p-hydroxybenzoate hydroxylase (PHBH) exhibits substrate specificity in the formation of a reaction intermediate. For reducing equivalents to pass from NADPH to the tightly bound FAD flavin ring, the enzyme must undergo a conformational change. This only occurs in the WT enzyme when a substrate is bound and only when that substrate can be deprotonated (46, 47). α-Ketoglutarate-dependent dioxygenases also incorporate a similar “substrate trigger” (48): the substrate to be oxidized must be bound for a coordination site for O2 to become available on the catalytic Fe2+ ion (49). These and many other examples underscore the use of mechanistic intermediates in mediating substrate specificity. We have tested the corollary that an enzyme with a very broad substrate repertoire might be expected to employ a much simpler mechanism that does not include multiple events that could depend on different features of the substrate.
To test this we have attempted to elucidate all the rate-contributing steps of the NR mechanism of NR through analysis of the steady-state and pre-steady-state kinetics of the reductive and oxidative reactions of NR. We find that both the reductive and oxidative half-reactions of NR employ simple kinetic mechanisms with one-step binding and concluding with simple product release. There is no evidence for distinct binding steps in which specificity might be enforced. Release of product NAD+ does not appear to be rate-contributing for the simple substrate p-NBA, and the re-oxidation rates and dissociation constants of the reduced enzyme prepared by reduction with dithionite (1.90 ± 0.07 s−1 and 0.30 ± 0.04 mm, respectively) or NADH (2.00 ± 0.06 s−1 and 0.20 ± 0.02 mm, respectively) were comparable supporting a simple ping-pong bi-bi mechanism.
For NR, we find that oxidation of reduced flavin by p-NBA (1.90 ± 0.09 s−1) is likely an important contributor to the overall rate (1.7 ± 0.3 s−1) based on comparison of the steady-state turnover number kcat with the rate constants obtained by transient kinetics. For NfsB, product release (either NAD+ or nitroso aromatic) or steps other than the chemical reaction were found likely to be rate-limiting (28). The difference can be explained by the different substrates used, as the substrates CB1954 and nitrofurazone turn over at kcat ≈ 140 s−1 and 230 s−1, respectively (28), much faster than the kcat for p-NBA of 1.7 s−1. We chose the poor substrate, p-NBA, to be able to look for mechanistic steps that might define the substrate repertoire. Despite the very slow chemical conversion of this substrate, we were unable to observe any intermediates or evidence for additional steps. Thus, we have ascertained that the broad substrate repertoire of NR rests not only upon the structure of the enzyme but also on the very simple mechanism it employs.
The relatively high Kd of NR for NADH of 0.51 mm (±0.04) is in excellent agreement with the results of Koder and Miller (6). The Kd for p-NBA of 0.33 ± 0.04 mm is also consistent with the mm values obtained for other substrates in previous work (6). The large magnitudes of both these Kd values are compatible with binding via relatively few and water-mediated contacts as indicated by crystal structures of the enzyme complexed with other substrates (41).
The value of kcat = 1.7 ± 0.3 s−1 obtained from steady-state kinetics agrees well with k5 = 1.90 ± 0.09 s−1 obtained from pre-steady-state kinetics. Similarly, when a value is calculated for KmPNBA from the individual rate constants measured via pre-steady-state kinetics, the resulting KmPNBA = 0.27 mm agrees well with the experimental KmPNBA = 0.13 mm produced by the double-reciprocal plot (Fig. 2A) and the 0.25 mm describing the hyperbolic plot (Fig. 2C). However, the KmNADH = 1.5 μm we calculated from our individual rate constants is significantly different from our experimental values of 44 μm from the double-reciprocal plot and 35 μm from the hyperbolic plot. This may reflect the fact that genuinely saturating p-NBA concentrations could not be used due to their inhibitory effect (6). However, for kcat and KmPNBA our primary rate constants succeed well in accounting for observed overall behavior.
Using the substrates nitrobenzene and its two-electron reduced product nitrosobenzene, Koder et al. (6) found that the nitroso substrate was reduced some 720 times faster than was the nitro substrate. Race et al. (11) confirmed analogous behavior for NfsB and nitro/nitroso toluene or nitro/nitrosobenzene. For p-NOBA, we could only estimate the apparent rate of reduction at 0.1 mm to be 650 s−1, whereas the rate of p-NBA reduction is 0.50 s−1 at the same concentration so we find that reduction of the nitroso is some 1300 times faster than reduction of the nitro.
One reason for interest in NR is the hope that it might be useful for producing aromatic amines from the corresponding nitroaromatics (13). However, our current results confirm earlier findings that significant quantities of aromatic amines do not necessarily accumulate under the action of E. cloacae NR (6).
Further work is called for to understand the basis for the ability of M. smegmatis nitroreductase to reduce benzothiazinone 043 to the corresponding amine (20) and the success of Klebsiella nitroreductase in fully reducing one nitro group of TNT, (12) in contrast with the common reduction of nitroaromatics only as far as the hydroxylamine followed by use a hydroxylamino mutase to produce the corresponding aminophenol (50, 51). We find that the M. smegmatis nitroreductase has a flavin midpoint potential very similar to that of NR,3 arguing against greater reducing power on the part of the enzyme (53). Instead, the substrates in question may be more amenable to full reduction by virtue of additional functionalization of the aromatic ring by a trifluoromethyl group in the case of benzothiazinone 043 and two additional nitro groups that are not reduced in the case of TNT. Our refinement of a sensitive selective chromogenic assay to detect aromatic amines affirms that p-HABA and thus also p-NBA is not converted to p-ABA by E. cloacae NR. However, this assay provides a valuable tool for determining whether the same enzyme can produce aromatic amine products from different substrates.
Acknowledgments
We thank J. Park, K. Ferguson, and A. S. Bommarius for the gift of hydroxylamino aromatic substrates and discussions regarding assays for aromatic amines. We are grateful to E. Glazer for centrifuge and freezer use and A. Sebesta for repair of the anaerobic glove box. We also thank an anonymous reviewer for detailed helpful criticism.
This work was supported by the Vice President for Research of the University of Kentucky, the Center for Pharmaceutical Development (to A.-F. M.), the Department of Chemistry of the University of Kentucky, and a Research Challenge Trust Fund Fellowship (to W. P.).
W. Pitsawong and A. F. Miller, unpublished data.
- NR
- nitroreductase
- NRox
- oxidized NR
- NRred
- reduced NR
- MBTH
- 3-methyl-2-benzothiazolinone hydrazone
- NfsB
- E. coli homologue of E. cloacae nitroreductase
- p-ABA
- para-aminobenzoic acid
- p-HABA
- para-hydroxylaminobenzoic acid
- p-NBA
- para-nitrobenzoic acid
- p-NOBA
- para-nitrosobenzoic acid
- TNT
- trinitrotoluene.
REFERENCES
- 1. Koder R. L., Miller A. F. (1998) Overexpression, isotopic labeling, and spectral characterization of Enterobacter cloacae nitroreductase. Protein Expr. Purif. 13, 53–60 [DOI] [PubMed] [Google Scholar]
- 2. Bryant C., DeLuca M. (1991) Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J. Biol. Chem. 266, 4119–4125 [PubMed] [Google Scholar]
- 3. Bryant C., Hubbard L., McElroy W. D. (1991) Cloning, nucleotide sequence, and expression of the nitroreductase gene from Enterobacter cloacae. J. Biol. Chem. 266, 4126–4130 [PubMed] [Google Scholar]
- 4. Haynes C. A., Koder R. L., Jr., Miller A.-F., Rodgers D. W. (2002) Structures of nitroreductase in three states: effects of inhibitor binding and reduction. J. Biol. Chem. 277, 11513–11520 [DOI] [PubMed] [Google Scholar]
- 5. Friedman J. E., Watson J. A., Jr., Lam D. W., Rokita S. E. (2006) Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase superfamily. J. Biol. Chem. 281, 2812–2819 [DOI] [PubMed] [Google Scholar]
- 6. Koder R. L., Jr., Miller A.-F. (1998) Steady state kinetic mechanism, stereospecificity, substrate, and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim. Biophys. Acta 1387, 395–405 [DOI] [PubMed] [Google Scholar]
- 7. Nivinskas H., Koder R. L., Anusevicius Z., Sarlauskas J., Miller A. F., Cenas N. (2001) Quantitative structure-activity relationships in two-electron reduction of nitroaromatic compounds by Enterobacter cloacae NAD(P)H:nitroreductase. Arch. Biochem. Biophys 385, 170–178 [DOI] [PubMed] [Google Scholar]
- 8. Xu G., McLeod H. L. (2001) Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. 7, 3314–3324 [PubMed] [Google Scholar]
- 9. Prosser G. A., Copp J. N., Syddall S. P., Williams E. M., Smaill J. B., Wilson W. R., Patterson A. V., Ackerley D. F. (2010) Discovery and evaluation of Escherichia coli nitroreductases that activate the anti-cancer prodrug CB1954. Biochem. Pharmacol. 79, 678–687 [DOI] [PubMed] [Google Scholar]
- 10. Knox R. J., Friedlos F., Boland M. P. (1993) The bioactivation of CB 1954 and its use as a prodrug in antibody-directed enzyme prodrug therapy (ADEPT). Cancer Metastasis Rev. 12, 195–212 [DOI] [PubMed] [Google Scholar]
- 11. Race P. R., Lovering A. L., Green R. M., Ossor A., White S. A., Searle P. F., Wrighton C. J., Hyde E. I. (2005) Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone: reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 280, 13256–13264 [DOI] [PubMed] [Google Scholar]
- 12. Kim H. Y., Song H. G. (2005) Purification and characterization of NAD(P)H-dependent nitroreductase I from Klebsiella sp. C1 and enzymatic transformation of 2,4,6-trinitrotoluene. Appl. Microbiol. Biotechnol. 68, 766–773 [DOI] [PubMed] [Google Scholar]
- 13. Yanto Y., Hall M., Bommarius A. S. (2010) Nitroreductase from Salmonella typhimurium: characterization and catalytic activity. Org. Biomol. Chem. 8, 1826–1832 [DOI] [PubMed] [Google Scholar]
- 14. Liu D., Thomson K., Anderson A. C. (1984) Identification of nitroso compounds from biotransformation of 2,4-dinitrotoluene. Appl. Environ. Microbiol. 47, 1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goodwin A., Kersulyte D., Sisson G., Veldhuyzen van Zanten S. J., Berg D. E., Hoffman P. S. (1998) Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol. Microbiol. 28, 383–393 [DOI] [PubMed] [Google Scholar]
- 16. Upcroft P., Upcroft J. A. (2001) Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 14, 150–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitmore G. F., Varghese A. J. (1986) The biological properties of reduced nitroheterocyclics and possible underlying biochemical mechanisms. Biochem. Pharmacol. 35, 97–103 [DOI] [PubMed] [Google Scholar]
- 18. Goldman P., Koch R. L., Yeung T.-C., Chrystal E. J., Beaulieu B. B., Jr., McLafferty M. A., Sudlow G. (1986) Comparing the reduction of nitroimidazoles in bacteria and mammalian tissues and relating it to biological activity. Biochem. Pharmacol. 35, 43–51 [DOI] [PubMed] [Google Scholar]
- 19. Knox R. J., Friedlos F., Biggs P. J., Flitter W. D., Gaskell M., Goddard P., Davies L., Jarman M. (1993) Identification, synthesis and properties of 5-(aziridin-1-yl)-2-nitro-4-nitrosobenzamide, a novel DNA crosslinking agent derived from CB1954. Biochem. Pharmacol. 46, 797–803 [DOI] [PubMed] [Google Scholar]
- 20. Manina G., Bellinzoni M., Pasca M. R., Neres J., Milano A., Ribeiro A. L., Buroni S., Skovierová H., Dianišková P., Mikušová K., Marák J., Makarov V., Giganti D., Haouz A., Lucarelli A. P., Degiacomi G., Piazza A., Chiarelli L. R., De Rossi E., Salina E., Cole S. T., Alzari P. M., Riccardi G. (2010) Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiozinone resistance. Mol. Microbiol. 77, 1172–1185 [DOI] [PubMed] [Google Scholar]
- 21. LinWu S. W., Wu C. A., Peng F. C., Wang A. H. (2012) Structure-based development of bacterial nitroreductase against nitrobenzodiazepine-induced hypnosis. Biochem. Pharmacol. 83, 1690–1699 [DOI] [PubMed] [Google Scholar]
- 22. Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Bjorkhem I., Pikuleva I. A. (2003) Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry 42, 14284–14292 [DOI] [PubMed] [Google Scholar]
- 23. Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650 [DOI] [PubMed] [Google Scholar]
- 24. Khan H., Harris R. J., Barna T., Craig D. H., Bruce N. C., Munro A. W., Moody P. C., Scrutton N. S. (2002) Kinetic and structural basis of reactivity of pentaerythritol tetranitrate reductase with NADPH, 2-cyclohexenone, nitroesters, and nitroaromatic explosives. J. Biol. Chem. 277, 21906–21912 [DOI] [PubMed] [Google Scholar]
- 25. French C. E., Nicklin S., Bruce N. C. (1996) Sequence and properties of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2. J. Bacteriol. 178, 6623–6627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu Z., Dunaway-Mariano D., Allen K. N. (2011) The X-ray crystallographic structure and specificity profile of HAD superfamily phosphohydrolase BT1666: comparison of paralogous functions in B. thetaiotaomicron. Proteins Struct. Funct. Genet. 79, 3099–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lovering A. L., Hyde E. I., Searle P. F., White S. A. (2001) The structure of Escherichia coli nitroreductase complexed with nicotinic acid: three crystal forms at 1.7 Å, 108 Å, and 2.4 Å resolution. J. Mol. Biol. 309, 203–213 [DOI] [PubMed] [Google Scholar]
- 28. Jarrom D., Jaberipour M., Guise C. P., Daff S., White S. A., Searle P. F., Hyde E. I. (2009) Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 48, 7665–7672 [DOI] [PubMed] [Google Scholar]
- 29. Defoin A. (2004) Simple preparation of nitroso benzenes and nitro benzenes by oxidation of anilines with H2O2 catalysed with molybdenum salts. Synthesis 5, 706–710 [Google Scholar]
- 30. Cleland W. W. (1979) Substrate inhibition. Methods Enzymol. 63, 500–513 [PubMed] [Google Scholar]
- 31. Cornish-Bowden A. (1995) Fundamentals of Enzyme Kinetics, pp. 129–144, Portland Press, London [Google Scholar]
- 32. Smallcombe S. H., Patt S. L., Keifer P. A. (1995) WET solvent suppression and its applications to LC NMR and high-resolution NMR spectroscopy. J. Magn. Reson. A 117, 295–303 [Google Scholar]
- 33. Gasparic J., Svobodova D., Pospisilova M. (1977) Investigation of color-reaction of phenols with MBTH reagent - identification of organic-compounds. Mikrochim. Acta 1, 241–250 [Google Scholar]
- 34. el-Kommos M. E., Emara K. M. (1987) Spectrophotometric determination of certain local-anesthetics using 3-methylbenzothiazolin-2-one hydrazone. Analyst 112, 1253–1256 [DOI] [PubMed] [Google Scholar]
- 35. Ogata Y., Takagi Y. (1958) Kinetics of the condensation of anilines with nitrosobenzenes to form azobenzenes. J. Am. Chem. Soc. 80, 3591–3595 [Google Scholar]
- 36. Zhao R., Tan C. Y., Xie Y. H., Gao C. M., Liu H. X., Jiang Y. Y. (2011) One step synthesis of azo compounds from nitroaromatics and anilines. Tetrahedron Lett. 52, 3805–3809 [Google Scholar]
- 37. Sone T., Tokuda Y., Sakai T., Shinkai S., Manabe O. (1981) Kinetics and mechanisms of the Bamberger rearrangement. 3. Rearrangement of phenylhydroxylamines to p-aminophenols in aqueous sulfuric acid solutions. J. Chem. Soc. Perkin Trans. II, 298–302 [Google Scholar]
- 38. Yost Y. (1969) Oxidation of fluorenamines and preparation of 2,2′- and 4,4′-azofluorene. J. Med. Chem. 12, 961. [DOI] [PubMed] [Google Scholar]
- 39. Campbell G. R., Taga M. E., Mistry K., Lloret J., Anderson P. J., Roth J. R., Walker G. C. (2006) Sinorhizobium meliloti bluB is necessary for production of 5,6-dimethylbenzimidazole, the lower ligand of B12. Proc. Natl. Acad. Sci. U.S.A. 103, 4634–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pérez-Reinado E., Roldán M. D., Castillo F., Moreno-Vivián C. (2008) The NprA nitroreductase required for 2,4-dinitrophenol reduction in Rhodobacter capsulatus is a dihydropteridine reductase. Environ. Microbiol. 10, 3174–3183 [DOI] [PubMed] [Google Scholar]
- 41. Johansson E., Parkinson G. N., Denny W. A., Neidle S. (2003) Studies of the nitroreductase produg-activating system. Crystal structures of complexes with the inhibitor dicoumarol and dinitrobenzamide prodrugs and of the enzyme active form. J. Med. Chem. 46, 4009–4020 [DOI] [PubMed] [Google Scholar]
- 42. Gamage N. U., Tsvetanov S., Duggleby R. G., McManus M. E., Martin J. L. (2005) The structure of human SULT1A1 crystallized with estradiol: an insight into active site plasticity and substrate inhibition with multi-ring substrates. J. Biol. Chem. 280, 41482–41486 [DOI] [PubMed] [Google Scholar]
- 43. Yasutake Y., Yao M., Sakai N., Kirita T., Tanaka I. (2004) Crystal structure of the Pyrococcus horikoshii isopropylmalate isomerase small subunit provides insight into the dual substrate specificity of the enzyme. J. Mol. Biol. 344, 325–333 [DOI] [PubMed] [Google Scholar]
- 44. Tomatis P. E., Fabiane S. M., Simona F., Carloni P., Sutton B. J., Vila A. J. (2008) Adaptive protein evolution grants organismal fitness by improving catalysis and flexibility. Proc. Natl. Acad. Sci. U.S.A. 105, 20605–20610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hou L., Honaker M. T., Shireman L. M., Balogh L. M., Roberts A. G., Ng K. C., Nath A., Atkins W. M. (2007) Functional promiscuity correlates with conformational heterogeneity in A-class glutathione S-transferases. J. Biol. Chem. 282, 23264–23274 [DOI] [PubMed] [Google Scholar]
- 46. Frederick K. K., Palfey B. A. (2005) Kinetics of proton-linked flavin conformational changes in p-hydroxybenzoate hydroxylase. Biochemistry 44, 13304–13314 [DOI] [PubMed] [Google Scholar]
- 47. Palfey B. A., Moran G. R., Entsch B., Ballou D. P., Massey V. (1999) Substrate recognition by “password” in p-hydroxybenzoate hydroxylase. Biochemistry 38, 1153–1158 [DOI] [PubMed] [Google Scholar]
- 48. Bollinger J. M., Jr., Krebs C. (2006) Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 100, 586–605 [DOI] [PubMed] [Google Scholar]
- 49. Solomon E. I., Brunold T. C., Davis M. I., Kemsley J. N., Lee S.-K., Lehnert N., Neese F., Skulan A. J., Yang Y.-S., Zhou J. (2000) Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 100, 235–350 [DOI] [PubMed] [Google Scholar]
- 50. Schenzle A., Lenke H., Spain J. C., Knackmuss H.-J. (1999) 3-Hydroxylaminophenol mutase from Ralstonia eutropha JMP134 catalyzes a Bamberger rearrangement. J. Bacteriol. 181, 1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spain J. C. (1995) Biodegradation of nitroaromatics. Annu. Rev. Microbiol. 49, 523–555 [DOI] [PubMed] [Google Scholar]
- 52. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 53. Koder R. L., Haynes C. A., Rodgers M. E., Rodgers D. W., Miller A.-F. (2002) Flavin thermodynamics explain the oxygen sensitivity of Enteric nitroreductases. Biochemistry 41, 14197–14205 [DOI] [PubMed] [Google Scholar]