

Background: The E. coli pyruvate dehydrogenase complex catalyzes conversion of pyruvate to acetyl-CoA and comprises E1p, E2p, and E3 components.
Results: The structure of the E2 core domain was solved and shown to efficiently catalyze acetyl transfer between domains.
Conclusion: Mass spectrometry revealed hitherto unrecognized domain-induced interactions between E1 and E2 core domain.
Significance: A multifaceted approach is required to understand communication between intact multidomain components.
Keywords: Carbohydrate Metabolism, Enzyme Catalysis, Mass Spectrometry (MS), Nuclear Magnetic Resonance (NMR), Pyruvate Dehydrogenase Complex (PDC), X-ray Crystallography, HD Exchange
Abstract
The Escherichia coli pyruvate dehydrogenase complex (PDHc) catalyzing conversion of pyruvate to acetyl-CoA comprises three components: E1p, E2p, and E3. The E2p is the five-domain core component, consisting of three tandem lipoyl domains (LDs), a peripheral subunit binding domain (PSBD), and a catalytic domain (E2pCD). Herein are reported the following. 1) The x-ray structure of E2pCD revealed both intra- and intertrimer interactions, similar to those reported for other E2pCDs. 2) Reconstitution of recombinant LD and E2pCD with E1p and E3p into PDHc could maintain at least 6.4% activity (NADH production), confirming the functional competence of the E2pCD and active center coupling among E1p, LD, E2pCD, and E3 even in the absence of PSBD and of a covalent link between domains within E2p. 3) Direct acetyl transfer between LD and coenzyme A catalyzed by E2pCD was observed with a rate constant of 199 s−1, comparable with the rate of NADH production in the PDHc reaction. Hence, neither reductive acetylation of E2p nor acetyl transfer within E2p is rate-limiting. 4) An unprecedented finding is that although no interaction could be detected between E1p and E2pCD by itself, a domain-induced interaction was identified on E1p active centers upon assembly with E2p and C-terminally truncated E2p proteins by hydrogen/deuterium exchange mass spectrometry. The inclusion of each additional domain of E2p strengthened the interaction with E1p, and the interaction was strongest with intact E2p. E2p domain-induced changes at the E1p active site were also manifested by the appearance of a circular dichroism band characteristic of the canonical 4′-aminopyrimidine tautomer of bound thiamin diphosphate (AP).
Introduction
Along with a continuing fascination with the catalytic mechanism of the Escherichia coli pyruvate dehydrogenase complex (PDHc)4 at the entry to the citric acid cycle (1, 2), understanding complex assembly and active center coupling between components also presents multiple challenges (3–6). The PDHc is composed of three principal enzyme components. E1p (24 copies as 12 dimers) (5, 7) is a thiamin diphosphate (ThDP)-dependent decarboxylase, which proceeds via a series of ThDP-bound covalent intermediates, the reaction culminating with reductive acetylation of the covalently lipoylated acetyltransferase (3-lip E2p with three tandem lipoyl domains; 24 copies as 8 trimers) (Scheme 1). E2p is a multidomain protein, starting from the amino terminus with three tandem lipoyl domains (LD1, LD2, and LD3), followed by a peripheral subunit binding domain (PSBD; involved in binding of the E1 and E3 components) and terminating with the C-terminal catalytic domain (E2pCD), which forms the core of the complex and where acetyl-CoA is synthesized (8–10). All domains are connected by flexible linkers rich in Pro and Ala (11). Finally, E3 (12 copies as 6 dimers) is an FAD- and NAD+-dependent dehydrogenase charged with reoxidation of dihydrolipoyl moieties to lipoyl moieties (12). With the exception of cryoelectron microscopic imaging of the E2pCD from yeast (13, 14) and human E2p catalytic domain (15, 16) and a human E2-E3-binding protein core (16) reconstructed at low resolution, there is no high resolution structure of an intact E2 component from any source. Although the structure of E2CDs from different sources has been reported (15, 17–25), we here present the x-ray structure of the E2pCD from E. coli and its functions.
SCHEME 1.

Mechanism of E. coli PDHc. The individual intermediates and rate constants for their transformation starting from free pyruvate were detected by us (1) except for k−7, as determined in this paper. Left, tautomeric and ionization states of ThDP; right, consecutive reactions catalyzed by the three components.
Our principal contributions in this paper are as follows. 1) We present the x-ray structure of the E. coli E2pCD. When combined with our previous x-ray structures of E1p (26) and E3 (27), we now have a complete set of high resolution crystal structures for all three major components of the E. coli octahedral PDHc with a cubic core). 2) The functional competence of E2pCD was demonstrated upon reconstitution with C-terminally truncated E2p proteins in the presence of E1p and E3, showing NADH production even in the absence of the covalent bond between E2p domains. 3) We have demonstrated efficient catalysis by E2pCD of acetyl transfer between the lipoyl domain and coenzyme A (CoA), with a rate constant comparable with kcat, suggesting that acetyl transfer within E2p is not rate-limiting. 4) A domain-induced interaction was identified on E1p active centers upon assembly with E2p and C-terminally truncated E2p proteins (but not by recombinant LD or E2pCD) by hydrogen/deuterium exchange mass spectrometry (HDX-MS) and also supported by the appearance of a circular dichroism band characteristic of the canonical 4′-aminopyrimidine tautomer of bound thiamin diphosphate. A significant finding from HDX-MS is that the entire E2p chain with all domains is needed for optimal interaction between E1p and E2p, of importance to the superfamily comprising all such complexes.
EXPERIMENTAL PROCEDURES
Reagents
ThDP, NAD+, CoA, acetyl-CoA, DTT, and dl-α-lipoic acid were from U.S. Biochemical Corp. Sodium pyruvate and tris(2-carboxyethyl)phosphine (TCEP) were from Sigma-Aldrich.
Protein Expression and Purification
Expression and purification of E1p (28), 3-lip E2p (27), 1-lip E2p (28), and E3 (27) followed reported protocols.
Construction of Plasmid and, Expression and Purification of E2pCD
For expression of the E2pCD, DNA encoding residues 381–629 in wild-type 3-lip E2p was amplified by PCR using forward primer 5′-GGTGGTGGATCCATGGGTATCCCAGGTATGC-3′ and reverse primer 5′-GGTGGTCTCGAGCATCACCAGACGGCGAATG-3′ (IDT Technologies, Inc.) and was cloned into a modified pET-21a vector (Novagen) through the BamHI and XhoI restriction sites. The TEV cleavage site was introduced immediately after the XhoI site with an additional three histidines for a total of nine histidines (His9 tag), so the His9 tag could be removed from the purified E2pCD. The nucleotide sequence was confirmed by Genewiz Inc. DNA encoding E2pCD was introduced into Rosetta (DE3) cells (Novagen), and cells were grown in LB medium supplemented with 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol at 30 °C overnight. 40 ml of the overnight culture was inoculated into 2 liters of the LB medium supplemented with 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol, and cells were grown to an A600 of 0.8 at 37 °C, and then isopropyl 1-thio-β-d-galactopyranoside (1 mm) was added, and cells were grown for an additional 2 h at 37 °C. Cells were washed with 0.10 m sodium phosphate (pH 7.2) containing 0.15 m NaCl and stored at −80 °C. The harvested cells (10 g) were resuspended in 25 mm HEPES (pH 7.0), containing 75 mm KCl, 5 mm CaCl2, 2 mm β-mercaptoethanol, 20% glycerol, 640 μg/ml benzamidine·HCl, 0.6 μg/ml leupeptin, and 2.7 μg/ml pepstatin A (buffer A). Cells were treated with lysozyme (0.5 mg/ml) at 4 °C for 20 min and were disrupted using a sonic dismembrator. To remove nucleic acids, 1,000 units of DNase I (NEB) and 1,000 units of micrococcal nuclease (Pierce) were added, and the mixture was incubated for an additional 2 h at 4 °C. The clarified lysate was loaded onto a 5-ml low p.s.i. nickel affinity resin (Sigma) at 1 ml/min. The column was first washed with 20 mm HEPES (pH 7.0) containing 0.2 m KCl, 2 mm mercaptoethanol, 5 mm imidazole, and protease inhibitors: 640 μg/ml benzamidine·HCl, 0.6 μg/ml leupeptin, and 2.7 μg/ml pepstatin A as in buffer A above (buffer B). The next three washes were 30 ml of buffer B containing 25, 50, and 100 mm imidazole, respectively. The His9 tag E2pCD was eluted with 350 mm imidazole.
For crystallization, His9 tag E2pCD was treated by the addition of 250 μg of His tag TEV protease at 4 °C while dialyzing in buffer B overnight. To remove the cleaved His9 tag and TEV protease, the mixture was passed through 5 ml of the nickel affinity resin (Sigma). The E2pCD free of His9 tag was concentrated to 4 ml, and 100% glycerol (1 ml) was added before injecting into a Superdex G75 16/60 (GE Healthcare) column equilibrated with 25 mm HEPES (pH 7.5) containing 0.1 m KCl, 1 mm DTT, and 0.01% NaN3. Fractions with E2pCD were collected and were concentrated using a Vivaspin 20, 10,000 molecular weight cut-off concentrating unit (GE Healthcare) to 15 mg/ml and stored at −80 °C. For kinetic studies, His9 tag E2pCD was used.
Construction of Plasmids and Expression and Purification of C-terminally Truncated E2p Proteins
Construction of plasmids for expression of C-terminally truncated E2p proteins was as reported for E2p(1–190) didomain (28). Briefly, the pET-15b-E2p plasmid encoding His6 tag 1-lip E2p from the N-terminal end (containing a single hybrid lipoyl domain, LDh, in place of three LDs in the wild type 3-lip E2p) was used as a template, and the amplification primers and their complements were used for site-directed mutagenesis to introduce a TAA stop codon in place of Lys97, Lys165, or Lys191, leading to C-terminally truncated E2p proteins: E2p(1–96) (LDh, comprising LDh and part of the linker) and E2p(1–164) and E2p(1–190) didomains, both comprising LDh, PSBD, and linkers of different lengths (see Fig. 2A for amino acid sequence of 1-lip E2p and Fig. 5 (inset, left) for schematic representation of the C-terminally truncated proteins). The LDh is composed of residues 1–33 from LD1 and residues 238–289 from LD3 of the E. coli 3-lip E2p (29). The C-terminally truncated E2p proteins were expressed in BL21 (DE3) cells at 37 °C, similarly to that reported for the E2p(1–190) didomain (28). The culture was grown in LB medium supplemented with 50 μg/ml ampicillin and 0.30 mm lipoic acid, and protein expression was induced by 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 5–6 h at 37 °C. The C-terminally truncated proteins were purified using a nickel-Sepharose Fast Flow column (GE Healthcare). All proteins were additionally purified using G3000SW TSK size exclusion chromatography with a high performance liquid chromatography system (28).
FIGURE 2.

A, amino acid sequence of 1-lip E2p containing LDh. The C-terminally derived E2p proteins were created by introducing a TAA stop codon at certain position, leading to E2p(1–96) (LDh) comprising LDh (residues 1–85, in red) and partially linker (in black); E2p(1–164) and E2p(1–190) (both didomains), comprising LDh, PSBD (residues 108–164, in blue); linkers of a different length (in black); and E2pCD (residues 177–425, in violet). The additional 20 amino acid residues from vector including His6 tag at the N-terminal end are also presented (in red). B, amino acid sequence of 3-lip E2p with corrected residue 295 (Ala → Val) by LC-MS/MS and DNA sequencing. LD1, residues 1–78; LD2, residues 105–181; LD3, residues 282–313. The E2p(1–394) tetradomain comprised LD1, LD2, LD3, PSBD, and linkers (residues 1–394), and the E2pCD was residues 381–629.
FIGURE 5.

Comparative HDX-MS analysis of the interaction of E1p with 3-lip E2p- and 1-lip E2p-derived proteins. A difference plot shows decreased deuterium incorporation of selected peptic fragments of E1p in the presence of 3-lip E2p (black full circle) or C-terminally truncated 1-lip E2p domains: E2p(1–394) tetradomain (blue inverted triangle), E2p(1–190) didomain (green triangle), LDh (red square), and E2pCD (open cycle) upon 3 min of hydrogen/deuterium exchange at 25 °C (deuterons exchanged in free state minus deuterons exchanged in complexed state). Inset, right difference plot showing decreased deuterium incorporation of selected peptic fragments of E1p in the presence of 3-lip E2p compared with that in the presence of E2p(1–394) tetradomain (deuterons exchanged in the presence of E2p(1–394) tetradomain minus deuterons exchanged in the presence of 3-lip E2p). Vertical error bars, S.D. Inset, left domain structure of E2p proteins.
Construction, Expression, and Purification of E2p(1–394) Tetradomain
For expression of E2p(1–394) tetradomain, the pCA24N plasmid encoding 3-lip E2p (National Bio Resource Project, NIG, Japan) was used as a template, and the amplification primer 5′-GGTGGACTTCAGCTAATTTGGTGAAATCGAAGAAGTG-3′ and its complement were used for site-directed mutagenesis to introduce a TAA stop codon in place of Lys395, leading to the E2p(1–394) tetradomain, comprising LD1, LD2, LD3, PSBD, and linkers in the 3-lip E2p (see Fig. 2B for amino acid sequence of 3-lip E2p and Fig. 5 (inset, left) for schematic representation of E2p(1–394 tetradomain). The E2p(1–394) tetradomain was expressed in AG1 cells (Agilent Technologies). AG1 cells were grown in LB medium supplemented with 30 μg/ml chloramphenicol and 0.30 mm lipoic acid at 37 °C. The expression of E2p(1–394) tetradomain was induced by isopropyl 1-thio-β-d-galactopyranoside (0.5 mm) for 4 h at 37 °C. Purification of E2p(1–394) tetradomain used a nickel-Sepharose Fast Flow column, similar to that used for the E2p(1–190) didomain reported recently (27, 28).
Lipoylation of the C-terminally Truncated E2p Proteins in Vitro
To ensure full lipoylation of the C-terminally truncated E2p proteins and E2p(1–394) tetradomain, they were lipoylated in vitro by E. coli lipoyl protein ligase as reported earlier (1, 30). Lipoylation was confirmed by FT-MS.
Enzyme Activity Measurements; Overall Activity upon Reconstitution of PDHc from Individual Components
Overall PDHc activity was measured after reconstitution of E1p with 1-lip E2p and E3 components as reported earlier (31). The reaction medium contained the following in 1.0 ml: 0.1 m Tris-HCl (pH 8.0), 1 mm MgCl2, 0.20 mm ThDP, 2 mm sodium pyruvate, 2.5 mm NAD+, 0.1–0.20 mm CoA, 2.6 mm dithiothreitol at 30 °C. The reaction was initiated by the addition of PDHc and CoA. Steady-state velocities were taken from the linear portion of the progress curve. One unit of activity is defined as the amount of NADH produced (μmol/min/mg E1).
Overall PDHc Activity upon Reconstitution of E2p from E2pCD and C-terminally Truncated Proteins and E2p(1–394) Tetradomain
For reconstitution of PDHc using a 1:1:1:1 mass ratio of E1p/E2pCD/C-terminally truncated E2p/E3, C-terminally truncated E2p proteins (0.2 mg of each, 0.4 mg for E2p(1–394) tetradomain) with concentrations of E2p(1–96) (110 μm), E2p(1–164) didomain (69 μm), E2p(1–190) didomain (60 μm), and E2p(1–394) tetradomain (89 μm) were first mixed with 0.2 mg of E2pCD (33 μm) in 0.15 ml of 50 mm KH2PO4 (pH 7.0) and were incubated for 15 h at 4 °C. Next, 0.20 mg of E3 (26 μm) was added to this reaction mixture and was incubated for an additional 1 h at 25 °C. For PDHc assembly, E1p (0.02 mg, 1 μm concentration of subunits) was mixed with 0.015 ml of reaction mixture of reconstituted E2p-E3 subcomplex in 50 mm KH2PO4 (pH 7.0) in a total volume of 0.20 ml to yield mass ratios of individual components (μg/μg/μg/μg) of 1:1:1:1. A 50-μl aliquot was withdrawn to start the reaction, and the activity was measured in the overall PDHc assay as above. Additional information on the concentration of subunits is presented in Fig. 3 and Table 3.
FIGURE 3.
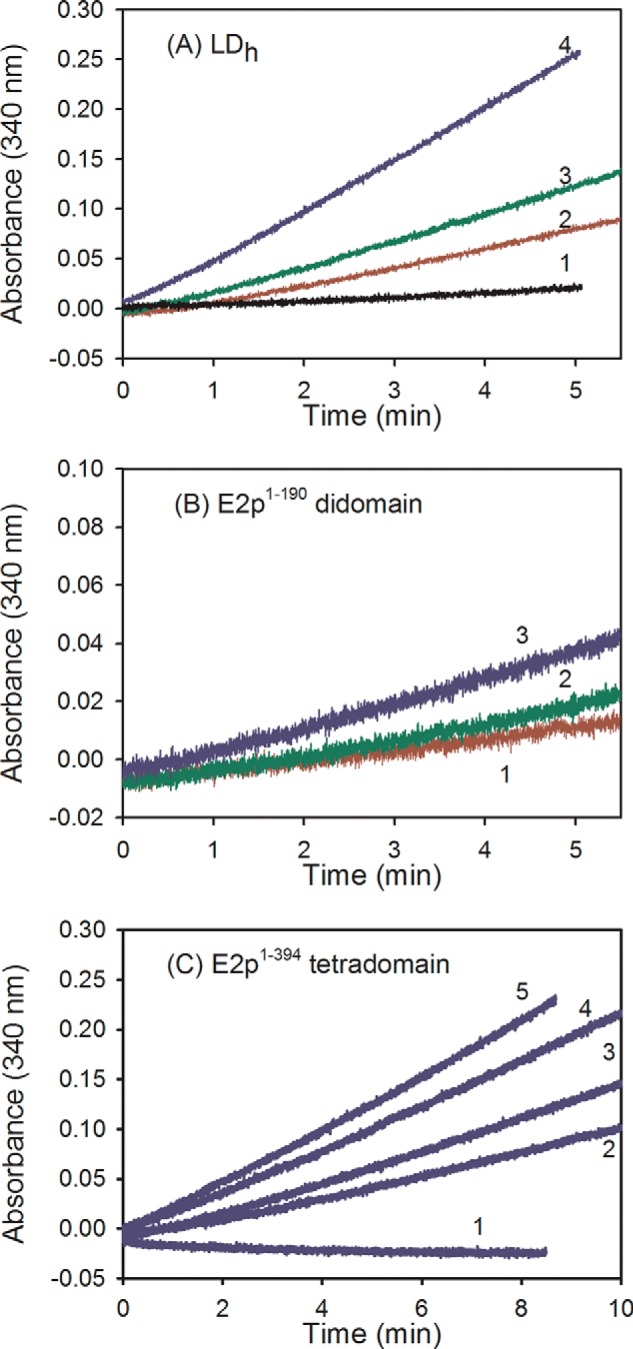
Progress curves for NADH production in the PDHc reaction. The E2p was assembled from C-terminally truncated E2p proteins and E2pCD. A, the E1p/LDh/E2pCD/E3 mass ratio (μg/μg/μg/μg) was as follows: 1:0.5:0.5:1 (2), 1:1:1:1 (3), 1:2:2:2 (4), and control in the absence of E1p (1). For the 1:1:1:1 mass ratio, the concentrations of individual subunits were 1 μm (E1p)/8.2 μm (LDh)/3.3 μm (E2pCD)/1.9 μm (E3). B, the E1p/E2p(1–190) didomain/ E2pCD/E3 ratio was as follows: 1:1:1:1 (1); 1:2:2:2 (2); 1:3:3:3 (3). C, 1, control in the absence of E1p. For the 1:1:1:1 mass ratio, the concentrations of individual subunits were as follows: 1 μm (E1p), 4.5 μm (E2p(1–190) didomain), 3.3 μm (E2pCD), and 1.9 μm (E3). The E1p/E2p(1–394) tetradomain/E2pCD/E3 ratio was as follows: 1:1.5:1.5:1 (2); 1:2:2:2 (3); 1:3:3:3 (4); 1:4.5:4.5:3 (5); control in the absence of E1p (1). For the 1:1:1:1 mass ratio, the concentrations of individual subunits were as follows: 1 μm (E1p), 4.6 μm (E2p(1–394) tetradomain), 3.3 μm (E2pCD), and 1.9 μm (E3). For assembly of E2p from C-terminally truncated E2p proteins and E2pCD and E2p reconstitution with E1 and E3 components, see “Experimental Procedures.”
TABLE 3.
The overall PDHc activity on reconstitution of PDHc from E1p, E3, and E2p, where E2p was assembled from C-terminally truncated E2p proteins and E2pCD
| E2p-derived proteins | Overall PDHc activitya |
|||
|---|---|---|---|---|
| 1:0.5:0.5:1 mass ratiob | 1:1:1:1 mass ratiob | 1:2:2:2 mass ratiob | Other mass ratiosb | |
| μmol·min−1·mg E1p−1 | ||||
| 1-lip E2p | 38 ± 1.6 (100%) | NAc | NA | NA |
| E2p(1–96) LDh | 0.62 ± 0.01 (1.6%) | 0.87 ± 0.01d (2.3%) | 2.46 ± 0.08 (6.4%) | NA |
| E2p(1–164) didomain | NA | 0.19 ± 0.01 (0.50%) | 0.31 ± 0.01 (0.8%) | 0.34 ± 0.01 (0.9%) (1:2.5:2.5:2) |
| E2p(1–190) didomain | NA | 0.16 ± 0.01 (0.43%) | 0.27 ± 0.01 (0.70%) (1:1.5:1.5:1.5) | 0.42 ± 0.02 (1.1%) (1:3:3:3); 0.45 ± 0.01 (1.2%) (1:3.5:3.5:4) |
| E2p(1–394) tetradomain | NA | 0.42 ± 0.04 (1.1%) (1:1.5:1.5:1) | 0.55 ± 0.02 (1.4%) | 0.72 ± 0.02 (1.9%) (1:3:3:3); 0.89 ± 0.01 (2.3%)e,f (1:4.5:4.5:3) |
a All experiments were done in triplicate. No background formation of NADH was detected for C-terminally truncated E2p proteins reconstituted with E2pCD and E3p in the absence of E1p.
b Mass ratio for E1p/E2p-derived proteins/E2pCD/E3 (μg/μg/μg/μg).
c NA, data not available.
d The mass ratio of 1:1:1:1 corresponds to the following concentrations of the individual subunits: E1p (1 μm)/E2p(1–96) (8.2 μm)/E2pCD (3.3 μm)/E3 (1.9 μm). No background formation of NADH was observed for E2pCD reconstituted with E2p(1–96) in the absence of E1p and E3.
e No background formation of NADH was detected for E2p(1–394) tetradomain by itself or E2p(1–394) tetradomain mixed with E1p and E3 but in the absence of E2pCD.
f The mass ratio of 1:4.5:4.5:3 corresponds to the following concentrations of subunits: E1p (1.0 μm)/E2p(1–394) tetradomain (6.3 μm)/E2pCD (0.3 μm)/E3 (3.9 μm). See Fig. 3 for progress curves of NADH production and “Experimental Procedures” for assembly of E2p from C-terminally truncated E2p proteins and E2pCD and reconstitution of E2p with E1p and E3 components.
Acetyltransferase Activity of the E2pCD in the Reverse Direction
The following protocol outlines simultaneous detection of acetylated and unacetylated LDs from 3-lip E2p after peptic digestion of the reaction mixture. When using 3-lip E2p as catalyst, the 3-lip E2p (80 μm) was first reduced by TCEP (1 mm) at 25 °C for 5 min, followed by the addition of 1 mm acetyl-CoA to initiate the acetyltransferase reaction lasting for 1 min. Then 2-μl aliquots were withdrawn and were quenched into 78 μl of ice-cold quench solution of 0.2 m KH2PO4 (pH 2.6). As controls, 2-μl aliquots were withdrawn without and with TCEP, but in the absence of acetyl-CoA, and then quenched as above. The samples were immediately frozen in liquid nitrogen and stored at −80 °C before analysis. The frozen sample was thawed and loaded into a 20-μl sample loop inside the refrigeration system (27). The protein sample (∼40 pmol) was carried by a 0.3 ml/min digestion flow (0.1% formic acid) into an immobilized pepsin column (Poroszyme Immobilized Pepsin Cartridge, 2.1 × 30 mm, Applied Biosystems) and digested at 15 °C for 20 s. The resultant peptides were immediately cooled down to 0 °C through a heat exchanger and were concentrated and desalted on a peptide trap (Michrom Peptide MacroTrap, 3 × 8 mm). The peptides were eluted and separated in 15 min through a reverse-phase C18 HPLC column (Agilent Poroshell 300SB-C18, 2.1 × 75 mm) at a flow rate of 0.2 ml/min with a 0 °C 2–40% acetonitrile gradient containing 0.1% formic acid. Bruker Daltonics DataAnalysis version 4.0 was used for spectrum analysis and data treatment as in Ref. 27 and below.
When using E2pCD as catalyst, 15 μl of the reaction mixture containing dihydro-LDh (40 μm), 5 mm TCEP, and E2pCD (0.2 μm) in 35 mm NH4HCO3 (pH 7.4) in one syringe was mixed in the reaction loop with 15 μl of 0.7 mm acetyl-CoA in the same buffer from the second syringe on a KinTek Chemical-Quench-Flow instrument, and the reaction was quenched at 0.1–10 s by the addition of 50% methanol and 1% formic acid from the third syringe. Samples were diluted into 50% methanol and 0.1% formic acid and were analyzed by FT-MS for the relative amounts of acetyldihydro-LDh and dihydro-LDh. The fraction of acetyldihydro-LDh at different times was determined by taking a ratio of the relative intensity of the acetyldihydro-LDh to the total relative intensity (sum of dihydro-LDh and acetyldihydro-LDh) and was plotted versus time. The rate constant was calculated from the linear fit to initial rate conditions.
Acetyltransferase Activity of the E2pCD in the Physiological Direction
The LDh (40–70 μm) was first reductively acetylated by 0.2 μm E1p and 1.5 mm pyruvate in 35 mm NH4HCO3 (pH 7.4) containing 1 mm MgCl2 and 0.050 mm ThDP at 25 °C (confirmed by FT-MS). After the addition of 2 mm TCEP, a 15-μl aliquot of this reaction mixture was mixed with 15 μl of a solution containing 0.4 μm E2pCD and 0.7 mm CoA. The reaction was quenched manually at different times by the addition of 50% methanol and 1% formic acid. For FT-MS, samples were further diluted 10-fold into 50% methanol with 0.1% formic acid. The intensities of the masses of the dihydro-LDh and acetyldihydro-LDh were recorded and plotted as a ratio of dihydro-LDh versus total LDh (a sum of dihydro-LDh and acetyldihydro-LDh) versus time. In a control experiment, the LDh (70 μm) was incubated with 0.2 μm E1p and 1.5 mm pyruvate in the absence of E2pCD in the reaction mixture containing 1 mm MgCl2, 0.050 mm ThDP, and 2 mm TCEP. Aliquots were withdrawn at different times, and the reaction was quenched into 1 ml of 50% methanol and 0.1% formic acid.
Reductive Acetylation in Vitro
In a steady state experiment, the LDh (30–60 μm) and E1p (0.2–0.4 μm) in 35 mm NH4HCO3 (pH 7.4) containing MgCl2 (4 mm) and ThDP (0.40 mm) in syringe A was mixed with pyruvate (2 mm) in syringe B, and the reaction was stopped at times in the range of 0.2–16 s by the addition of 84 μl of quench solution (50% methanol and 1% formic acid) from syringe C on a KinTek Chemical-Quench-Flow instrument. Samples were diluted in 50% methanol and 0.1% formic acid to result in a concentration of LDh of 1–2 μm and were analyzed for the relative amounts of acetyldihydro-LDh and dihydro-LDh by FT-MS. For longer time scales, the reaction was conducted as above and was quenched manually. Single-turnover experiments were carried out as reported earlier (1).
Circular Dichroism Spectrometry
CD spectra were recorded at 25 °C on a Chirascan spectrometer (Applied Photophysics, Leatherhead, UK) in the near-UV region (290–500 nm) using a 1-cm path cell. CD spectra of apo-E1p (free of ThDP; 30 μm subunits) by itself or in the presence of 3-lip E2p (2.1 mg/ml), E2p(1–394) tetradomain (1.3 mg/ml), E2p(1–190) didomain (0.69 mg/ml), or LDh (0.36 mg/ml). In other words, each E2p-derived protein was present at a 30 μm concentration. CD spectra were recorded 5 min after ThDP (0.30 mm) and MgCl2 (3 mm) were added in 50 mm KH2PO4 (pH 7.0) in a total volume of 2.4 ml at 25 °C. Three to five spectra were averaged with baseline subtraction.
Hydrogen/Deuterium Exchange Experiment Monitored by FT-MS
The hydrogen/deuterium exchange was performed as described (27). Prior to hydrogen/deuterium exchange, E1p, 3-lip E2p, LDh, E2p(1–190) didomain, and E2p(1–394) tetradomain were exchanged into 10 mm KH2PO4 (pH 7.0) with 50 mm KCl, 0.2 mm ThDP, and 1 mm MgCl2. Five samples were prepared as follows: 80 μm E1p, a mixture of 80 μm E1p and 80 μm 3-lip E2p, or C-terminally truncated E2p proteins (LDh, E2p(1–190) didomain, and E2p(1–394) tetradomain). All samples were equilibrated for at least 1 h at 25 °C. The deuterium labeling reaction was initiated by diluting 2 μl of protein sample into 38 μl of labeling buffer (10 mm KH2PO4 (pH 7.0), 50 mm KCl, 0.2 mm ThDP, 1 mm MgCl2, 99.9% D2O) and lasted for 3 min at 25 °C. Next, a 30-μl aliquot from the labeling reaction was rapidly quenched by the addition of an equal volume of ice-cold quench solution (0.2 m KH2PO4, pH 2.6). The samples were immediately frozen in liquid nitrogen and stored at −80 °C before analysis. Non-deuterated samples (E1p, 3-lip E2p, and E2pCD) were generated following the same procedure except that protein samples were diluted into aqueous buffer and incubated for 1 min, followed by the quench process (see supplemental Table S1 for E1p and supplemental Table S2 for E2pCD). All experiments were run in triplicates.
Peptic Digestion and FT-MS Analysis
Peptic digestion and FT-MS analysis of the resultant peptides were carried out as described under “Acetyltransferase Activity of the E2pCD in the Reverse Direction”) with minor modifications (27). Digestion time was 20 s. The FT-MS settings were as follows: ESI+ mode; capillary, 4,500 V; spray shield, 4,000 V; drying gas temperature, 190 °C, mass acquisition range, 400–2,000 m/z; scan rate, 0.5 scans/s. Bruker Daltonics DataAnalysis version 4.0 was used for spectrum analysis and data treatment. Peptides were identified from undeuterated samples by a customized program, DXgest, which matches experimental peptide mass with theoretically generated peptic peptide mass by using statistical data for the pepsin cleavage pattern under hydrogen/deuterium exchange conditions (32). To determine deuterium uptake, mass tolerance was set at <2.0 ppm. hydrogen/deuterium exchange data for each individual peptide were processed using the program HX-Express (33) to determine deuterium uptake and peak width. No correction was needed for back-exchange. The number of backbone amides and peptide coverage were generated by using MSTools (34). Difference plots were produced by Origin (OriginLab, Northampton, MA) and Microsoft Excel (35).
X-ray Structure Determination of the E2pCD
Single crystals of the E2pCD (His9 tag removed) were obtained by vapor diffusion (15 mg/ml protein) against a precipitant solution of 0.2 m potassium thiocyanate and 20% (w/v) polyethylene glycol 3350 at 23 °C and with pH 7.0 ± 0.2. The crystals were orthorhombic, with a = 77.56, b = 92.83, c = 110.54 Å in space group P212121. The crystals diffracted to 2.25 Å resolution, and the x-ray structure was determined by molecular replacement using a polyalanine search model obtained from the known crystal structure of the related, E2 catalytic domain (Protein Data Bank code 1DPC) fragment from Azotobacter vinelandii PDHc (23). The results clearly indicated three monomers of E2pCD per crystallographic asymmetric unit, and these are related by a non-crystallographic triad axis. Sequence adjustment and refinement by PHENIX (36) included the addition of 318 water molecules and yielded final values of R = 0.207, RFree = 0.246. Relevant crystallographic data and refinement statistics are shown in Table 1.
TABLE 1.
Crystallographic data and refinement statistics for the E2pCD
Values in parenthesis are for the last shell.
| Data statistics | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 77.56, b = 92.83, c = 110.54 |
| Diffraction limit (Å) | 2.25 |
| Completenes (%) | 100.0 (99.9) |
| Total no. of reflections | 233,033 |
| No. of unique reflections | 38,560 |
| Rmerge (on I) | 0.093 (0.572) |
| I/ δ (unaveraged) | 3.7 (1.8) |
| Average redundancy | 6.04 (5.77) |
| X-ray source | Synchrotron (APS, SERCAT, sector 22BM) |
| Wavelength (Å) | 1.00 |
| Refinement | |
| Resolution range (Å) | 21.40–2.25 |
| No. of reflections | 37,762 |
| No. of reflections for test set | 1,885 |
| R factor | 0.207 (0.362) |
| Rfree | 0.246 (0.365) |
| No. of residues | 741 |
| No. of protein atoms | 5,725 |
| No. of solvent atoms | 318 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (degrees) | 0.70 |
NMR Spectroscopy
All NMR data for backbone sequence-specific resonance assignments were recorded at 25 °C on a Varian INOVA 600-MHz spectrometer equipped with 5-mm HCN room temperature probes. Chemical shifts for E2p(1–190) didomain were assigned using conventional triple resonance NMR methods, which were selected from the VARIAN standard Biopack pulse sequence library. Spectra were processed with NMR DRAW version 5.96 (37) and transferred to CARA version 1.9.0b3 for further analysis (38). The backbone 1H, 15N, 13Cα, and 13C′ resonances were assigned using two-dimensional and three-dimensional experiments, including [15N,1H]HSQC TROSY, three-dimensional HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HNCO, and HNCACO experiments (39). Chemical shifts were calibrated to the waterline at 4.7 ppm, and nitrogen shifts were referenced indirectly to liquid NH3 (40). The interaction of E2p(1–190) didomain with E1p was followed by acquiring two-dimensional 15N,1H HSQC TROSY spectra. All samples for NMR spectroscopy were made up in 20 mm KH2PO4 (pH 7.0), containing 150 mm NaCl, 10% D2O. For measurement of interactions by chemical shift mapping methodology, 15N-labeled E2p(1–190) didomain was used at a concentration of 0.5 mm, and unlabeled E1p (0.12 mm) was present at an E1p/E2p(1–190) didomain molar ratio of 1:4. ThDP, Mg2+, and pyruvate were added to final concentrations of twice the E1p concentration. 15N,1H HSQC TROSY spectra were recorded using 32 scans for each t1 point and 256 increments at 25 °C, with spectral widths of 7,629 Hz (1H) and 2,200 Hz (15N). Combined chemical shift differences (Δδ) of peaks were extracted from the 15N,1H HSQC TROSY spectra and computed according to the equation (60), Δδ = sqrt(Δ(δ1H)2 + 0.2 × Δ(δ15N)2), with a scaling factor (αN) of 0.2, where Δδ is the change in chemical shift, Δ(δ1H) is the observed change in chemical shift of the 1H nucleus, and Δ(δ15N) is the observed chemical shift change in the chemical shift of the 15N nucleus on complexation. Completed sequence-specific chemical shift assignments for the same E2p(1–190) didomain have been presented previously (27), where its interaction with E3 was reported, whereas in this paper, its interaction with E1p is being studied.
RESULTS AND DISCUSSION
Overall Description of the E2pCD X-ray Structure
Three E2pCDs form a trimer (Fig. 1a), with the internal interactions between monomers being essentially the same as in other reported E2 CD trimers (15, 17–25). Each E2pCD subunit consists of a V-shaped N-terminal arm (residues 381–407) and a main globular portion (residues 408–629). Approximately 40% of the residues in each E2pCD subunit assume helical conformations, whereas an additional 33% are in the form of β-sheets. Of the globular portion, 89 residues are composed in five α-helices, 10 residues in two short 310 helical segments, and 83 residues in 10 β strands. The N-terminal arm does not interact with the rest of the monomer but does participate in hydrophobic as well as hydrogen bond contacts with the body of another E2pCD within the trimer. No water-filled channel is present along the triad axis, and only two solvent molecules lie within 0.4 Å of the axis. The trimer interface is stabilized by an intricate network of hydrogen bonds centered around a water molecule 0.19 Å from the non-crystallographic triad axis and involves His424, Thr426, and Ala561 from each monomer. The packing interactions within trimers are similar to those in the Protein Data Bank having the codes 1DPC (23), 1E2O (18), and 1EAE (19), all of which gave rise to 24-subunit cubic cores. In those structures, however, the location and orientation of a basic trimer unit coupled with the presence of 3-fold axes along the body diagonals of their cubic unit cells ensured that only one subunit is crystallographically unique, and all three monomers forming each trimer participate in exactly the same interactions with symmetry-related molecules. Obviously, the absence of any crystallographic 3-fold axis precludes this strict equivalence of the three subunits in the E. coli E2pCD trimer. Note that in the E2pCD structure reported here, the long, unstructured, N-terminal ends of each E2pCD subunit wrap around the perimeter and interact with another subunit in the trimer, as seen in Fig. 1. In the intact PDHc, this may not be the case because the N-terminal ends are probably extended outward from the core to allow the PSBD and LD domains (not present in the E2pCD used for crystallization) to reach the peripheral E1p and E3 components.
FIGURE 1.

a, ribbon drawing showing the trimer of E2pCDs in the crystallographic asymmetric unit, viewed roughly down the non-crystallographic 3-fold axis and with each subunit shown in a different color. b, active site region at a dimeric subunit interface within a trimer showing positions of Ser550, His602, and Asp606. The substrates (dihydrolipoamide and CoA) are shown at their respective locations as observed in the corresponding A. vinelandii E2pCD ternary complex (19).
The E2pCD Active Site Channel
The catalytic activity of the E. coli E2pCD is presumed to depend on the proximity of the highly conserved residues His602 and Ser550 located at the catalytic center formed at the interface between two subunits (Fig. 1b). These residues were predicted from comparison of a number of E2 sequences with that of chloramphenicol acetyltransferase (41) and were confirmed by site-directed substitutions of E2p. For the E. coli PDHc with H602C E2p substitution, no overall PDHc activity or E2p acetyltransferase activity were detected, and with the S550A substitution, the PDHc activity was drastically reduced; however, the ability to bind the E1p and E3 components and the reductive acetylation of the lipoyl domain by E1p were not affected (42, 43). Based on kinetic studies above and x-ray structures of A. vinelandii E2pCD in binary and ternary complexes with CoA and dihydrolipoamide, it was suggested that the histidine-serine pair present in the catalytic center of E2CDs plays an essential role in the acetyltransferase reaction mechanism, with histidine acting as a general base in this reaction (15, 19, 21, 23).
In E. coli E2pCD, the residues Asp601 and Arg603, flanking the highly conserved His602, form a salt bridge involving both NE and NH2 atoms of Arg603, one bonding to each of the carboxylate oxygens of conserved Asp601, perhaps thereby restricting the conformation assumed by the His602. The same phenomenon was observed in Protein Data Bank entries 2II4 (21), 1DPC (23), 1C4T (22), 1EAE (19), and 1EAC (19). The conformations observed allow a hydrogen bond to form between ND1 and the carbonyl oxygen of the same amino acid, perhaps thereby further stabilizing the side chain conformation of the histidine. This histidine conformation may facilitate hydrogen bonding between the S8 atom of dihydrolipoamide with the histidine NE2 atom, supporting the specificity of E2p for the acetylation on S8 (44). In E. coli E2pCD, the reactive sulfur atom of the substrate dihydrolipoamide is probably binding in the proximity of His602 (Fig. 1b). It has been suggested that a highly conserved serine (Ser550 in E. coli E2pCD) is involved in CoA binding and stabilizing the negatively charged tetrahedral intermediate of the acetyltransferase reaction in the active site channel (19, 23). Approximate coordinates of two substrates, dihydrolipoamide and CoA (Fig. 1b), present at the interface between the two subunits were obtained by superposition of the E. coli E2pCD structure with the related, ternary complex structure (Protein Data Bank code 1EAC) of A. vinelandii E2pCD that contained the CoA and dihydrolipoamide substrates (19). Furthermore, it is presumed that binding of dihydrolipoamide substrate to the E. coli E2p catalytic center promotes reorientation of the Asp606 side chain by participating in electrostatic interactions with the active site His602 (histidine residue) similar to that reported for the A. vinelandii E2pCD (19, 23). The x-ray structure of E2pCD here reported, together with the E1p and E3 structures reported by us (26, 27) culminates the determination of the high resolution structures for all three major components of an octahedral PDHc from E. coli. We note that the E3 components are common to all such 2-oxo acid dehydrogenase complexes in E. coli.
Functional Competence of E2pCD Is Revealed by PDHc Activity upon Its Reconstitution with C-terminally Truncated E2p Proteins
Observation of overall PDHc activity upon non-covalent reconstitution of E2p from E2pCD and C-terminally truncated E2p proteins would constitute an advance in our understanding regarding active center coupling of the E2pCD with the peripheral E1p and E3 components (5, 6). We tested the following C-terminally truncated E2p proteins upon assembly with E2pCD: E2p(1–96) (LDh), E2p(1–164) and E2p(1–190) didomains (they comprise LDh and PSBD with linkers of different lengths), and E2p(1–394) tetradomain comprising LD1, LD2, LD3, and PSBD (Fig. 2 and Table 2). It was first confirmed by FT-MS that all C-terminally truncated E2p proteins were fully lipoylated and were capable of being reductively acetylated by E1p and pyruvate (Table 2). As seen in Table 3 and in Fig. 3, all of the assembled PDH complexes confirmed NADH production. Perhaps unexpectedly, at a similar mass ratio of assembled components, the LDh displayed the highest activity (6.4% in Table 3) as compared with E2p(1–164) and E2p(1–190) didomains (0.7–0.8%), contrary to expectation because they provide the binding site (PSBD) for both the E1p and E3 components. In comparison, 2.3% of activity was detected for E2p(1–394) tetradomain reconstituted with E2pCD, E1p, and E3, suggesting that the amount of LDs supplied for the reaction is an important determinant. Increasing the time of incubation of the E2p(1–394) tetradomain with E2pCD for up to 5 h failed to improve the overall PDHc activity. The following controls were performed to assure that NADH is produced in the overall PDHc reaction upon assembly of all necessary components. 1) For all C-terminally truncated E2p proteins that were assembled with E2pCD and E3 but in the absence of E1p, no NADH production was detected. 2) No background formation of NADH was observed for E2pCD reconstituted with E2p(1–96) but in the absence of E1p and E3. 3) For E2p(1–394) tetradomain by itself or for E2p(1–394) tetradomain assembled with E1p and E3 but in the absence of E2pCD, no NADH production was detected. These controls clearly confirm and demonstrate that E2pCD is a catalytic component that is essential for the overall PDHc reaction.
TABLE 2.
FT-MS evidence for post-translational modifications of C-terminally truncated E2p proteins
| E2p-derived proteinsa | Theoretical mass | FT-MS massb |
|---|---|---|
| Da | Da | |
| E2p(1–96) (LDh)c | 12,144.6 (lipoylated) | 12,145.29 |
| 12,188.6 (acetylated) | 12,189.19 | |
| E2p(1–164) didomain | 19,447.00 (lipoylated) | 19,448.36 |
| 19,491.00 (acetylated) | ||
| E2p(1–190) didomain | 22,054.02 (lipoylated) | 22,054.48 |
| 22,098.02 (acetylated) | 22,098.53 | |
| E2p(1–394) tetradomain | 42,626.09 (lipoylated) | 42,626.4c |
| 42,758.17 (acetylated) | 42,756.0 | |
| E2p CD | 30,028.7 Da | 30,030.23 |
This study provides the first evidence that reaction intermediates are transferred between active centers of E1p, E2p, and E3 upon reconstitution of PDHc, where the E2p is assembled from C-terminally truncated E2p proteins and independently expressed E2pCD, with no covalent bond linking E2pCD to the remaining E2p domains. The results have direct bearing on future enzyme engineering of these important complexes because it will be sufficient to use independently expressed domains to optimize for desired altered substrate specificity.
Direct Demonstration of Acetyl Transfer between E2p Domains
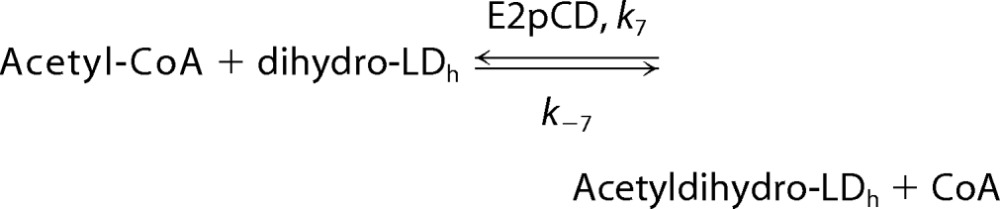
It had been reported that E2p and independently expressed E2pCD catalyze the model reaction using acetyl-CoA as acetyl donor and a dl-dihydrolipoamide as acetyl acceptor (dl-dihydrolipoamide replacing the dihydrolipoyl moieties covalently bound to the lipoyl domain) in the reaction reversed from the physiological reaction (see Reaction 1) (23, 24, 45–47). The acetyltransferase activity had been measured in a coupled assay with phosphotransacetylase, where the formation of a thioester bond (S-acetyldihydrolipoamide) was monitored at 240 nm (233 nm) (23, 24, 46), or by detection of the radioactive S-acetyldihydrolipoamide formed from [1-14C]acetyl-CoA (45, 47). The current study is the first to demonstrate direct transfer of the acetyl moiety from acetyl-CoA to the reduced hybrid lipoyl domains (dihydro-LDh) (29), leading to determination of catalytically competent rate constants for acetyl transfer.
First, using 3-lip E2p with all three lipoyl domains lipoylated and reduced by TCEP (95% of lipoyl moieties are in reduced form according to FT-MS), we demonstrated that about 83% of reduced lipoyl moieties were acetylated by acetyl-CoA within 1 min of reaction, and the percentage remained the same even after 10 min. Our evidence for reduction and acetylation of the lipoyl domains in 3-lip E2p using acetyl-CoA as acetyl donor relied on detection of the lipoylated ITVEGDKASM peptide by FT-MS pursuant to pepsin digest of 3-lip E2p. The mass of this peptide was 1,238.5443 Da before and 1,240.5594 Da after reduction of lipoic moieties by TCEP, and the peptide mass was increased to 1,282.5712 Da after acetylation of 3-lip E2p by acetyl-CoA. The intensity of the 1,282.5712 Da peak represents 83% of the total intensity summing oxidized, reduced, and reductively acetylated peptides. These data provide direct evidence of reverse acetyl transfer within the entire 3-lip E2p, from the E2p core active center to the dihydrolipoamide covalently bound to the lipoyl domains. Earlier, reversibility of the overall PDHc reaction was demonstrated based on the observation of hydrolysis of acetyl-CoA by E. coli PDHc and detection of acetyl-ThDP formation on the E1p component (48).
Second, to evaluate the rate constant k−7 in Scheme 1, a mixture of a dihydro-LDh (LDh reduced by TCEP) and E2pCD, both expressed independently, was reacted with acetyl-CoA according to Reaction 1. Differences between our results and those in the literature are the following. 1) Reduced lipoyl domain (with dihydrolipoyl moieties bound to lipoyl domain (dihydro-LDh)), rather than dihydrolipoamide, a poor substrate for E2pCD, was used as substrate. The reported values of Km for dihydrolipoamide are in the range of 0.13–0.40 mm (45), 1.2 mm (46), 4 mm (47), and 5.4 mm (23). 2) Direct mass measurements of the masses of dihydro-LDh and acetyldihydro-LDh by FT-MS were used to quantify the extent of acetyl transfer.
 |
For steady-state detection of the rate constants of acetyldihydro-LDh formation in Reaction 1, the Km values for dihydro-LDh and acetyl-CoA were determined by FT-MS. At concentrations of dihydro-LDh (50 μm) and acetyl-CoA in the range of 0.025–0.65 mm and in the presence of E2pCD (0.1 μm), the dihydro-LDh is a substrate for E2pCD, and the acetyldihydro-LDh is accumulated as a product of the reaction. The reaction was stopped at different times (20–180 s); however, a maximum degree of acetylation was already reached after 20 s of reaction at all analyzed concentrations of acetyl-CoA. A plot of the ratio of acetyldihydro-LDh/total LDh versus concentrations of acetyl-CoA resulted in a hyperbolic curve yielding a Km of acetyl-CoA of 0.07 ± 0.001 mm, which correlates well with the Km of acetyl-CoA reported elsewhere (0.03–0.10 mm (45): 0.013 mm (46) and 0.022 mm (23). Under similar conditions but with saturating acetyl-CoA (0.35 mm) and concentrations of dihydro-LDh in the range of 10–54 μm, a Km for dihydro-LDh of <10 μm was estimated, clearly demonstrating that dihydro-LDh, but not dihydrolipoamide (24, 45–47) is a valid substrate for the reverse model reaction. As a control, when dihydro-LDh was tested by itself or in a reaction mixture with 0.35 mm acetyl-CoA (but in the absence of E2pCD), a species with a molecular mass of 12,147.7 Da was detected by FT-MS corresponding to dihydro-LDh only. No species corresponding to acetyldihydro-LDh (molecular mass of 12,189.7 Da) was detected. The presence of all three components of the reaction (acetyl-CoA, dihydro-LDh, and E2pCD) is required for the formation of acetyldihydro-LDh as detected by FT-MS.
At saturating concentrations of dihydro-LDh and acetyl-CoA, ∼80% of dihydro-LDh was converted to acetyldihydro-LDh by E2pCD within 1.6 s, leading to a k−7 of 199 s−1 (Fig. 4A) calculated from the initial rate of acetyldihydro-LDh formation. The estimated k−7 of 199 s−1 was not very different from the kapp of 134 ± 24 s−1 for reductive acetylation of the E2p(1–190) didomain by E1p and pyruvate and from the value of kcat of 95 s−1 for conversion of pyruvate to NADH in the overall PDHc reaction (1). From a comparison of the microscopic rate constants presented above, it is evident that acetyl transfer between acetyl-CoA and dihydro-LDh is not rate-limiting in the overall PDHc reaction starting with free pyruvate and culminating in acetyl-CoA formation (see Scheme 1). The determined k−7 of 199 s−1 was similar to that of 144 s−1 reported for E2pCD from A. vinelandii (23) and was much higher than that reported for E2CD from Thermoplasma acidophilum (47 min−1) (24) or for E2pCD from Saccharomyces cerevisiae (7.8 s−1) (47), indicating that E. coli E2pCD is an efficient catalyst in acetyl transfer between E2p domains as demonstrated by direct observation of acetyldihydro-LDh formation.
FIGURE 4.
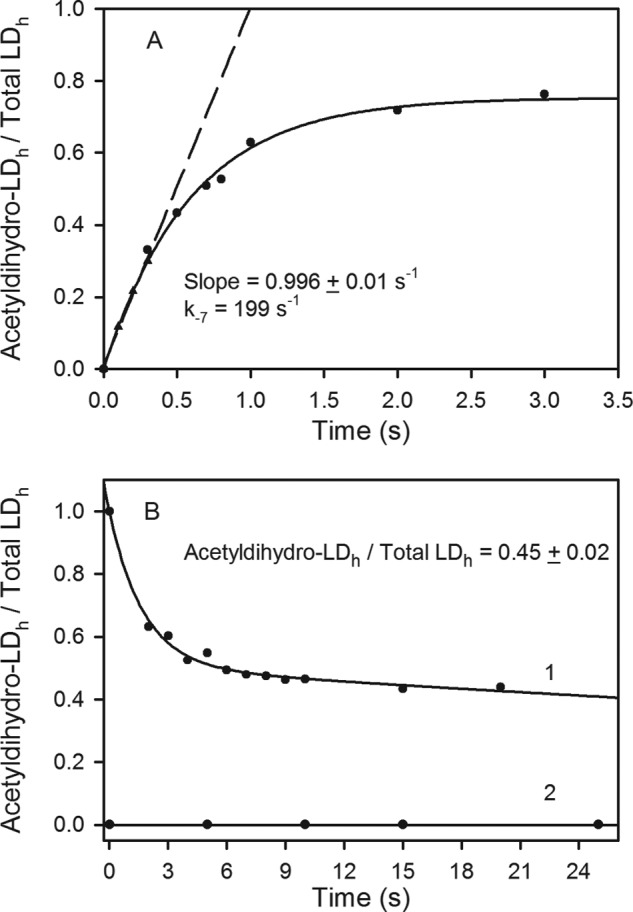
Kinetic fate of the acetyldihydro-LDh in the acetyltransferase reactions catalyzed by E2pCD. A, progress curve for acetyldihydro-LDh formation from acetyl-CoA and dihydro-LDh catalyzed by E2pCD in the reverse acetyltransferase reaction. The dihydro-LDh (20 μm), 2.5 mm TCEP, and E2pCD (0.1 μm) in 50 mm NH4HCO3 (pH 7.4) in the reaction loop were mixed with acetyl-CoA (0.3 mm) on a KinTek Chemical-Quench-Flow instrument (see “Experimental Procedures” for details); the reaction was stopped at 0.1–5 s by the addition of quench solution; and samples were analyzed by FT-MS. The intensity of the MS signals for acetyldihydro-LDh and dihydro-LDh was detected. The relative intensity of acetyldihydro-LDh versus total intensity (sum of acetyldihydro- and dihydro-LDh) was plotted versus time. The trace is a nonlinear regression fit to a single exponential rise to maximum, and the dashed line represents a linear fit to initial rate conditions. The three points drawn as triangles were calculated from a non-linear regression fit of the experimental data to demonstrate the good fit of the initial slope to the curve. B, time dependence for the approach to equilibrium between consumption of the acetyldihydro-LDh in the direct reaction and its formation in the reverse reaction. 1, the LDh (70 μm) was first reductively acetylated by E1p (0.2 μm) and 1.5 mm pyruvate. Then 15 μl of acetyldihydro-LDh was mixed with a 15-μl solution containing 0.4 μm E2pCD, 0.7 mm CoA, and 1 mm TCEP, and the reaction was quenched at different times. The presence of the acetyldihydro-LDh and dihydro-LDh was analyzed by FT-MS. 2, a control experiment demonstrated that no acetyldihydro-LDh was produced by E1p and pyruvate on LDh reduced by TCEP. See “Experimental Procedures” for details.
Evidence for Reactivity of the E2pCD in the Physiological Acetyltransferase Reaction
To study the acetyl transfer in the physiologically relevant direction (k7 in Reaction 1 and in Scheme 1), the LDh, reductively acetylated by E1p and pyruvate to form acetyldihydro-LDh was reacted with CoA in the presence of E2pCD and TCEP (to maintain LDh in the reduced form). Two forms of LDh were detected when the reaction was quenched after 2 s or even after 120 min, dihydro-LDh (55%) and acetyldihydro-LDh (45%; Fig. 4B), indicating an approach to equilibrium between consumption of acetyldihydro-LDh in the direct, physiological reaction and its formation in the reverse direction. The detected acetyldihydro-LDh is a product of the reverse acetyltransferase reaction rather than of reductive acetylation of dihydro-LDh by E1p and pyruvate. This was confirmed by a control experiment that demonstrated that no acetyldihydro-LDh was produced by E1p and pyruvate on dihydro-LDh (Fig. 4B). These results allow us to conclude that the forward k7 and reverse k−7 transacetylase reactions in Reaction 1 are both catalyzed by the E2pCD, and kinetics display a rapid approach to equilibrium. The reaction interconverts two thiol esters and is expected to have an equilibrium constant near unity. For this single-step reaction, the kinetic barrier should be similar in both directions according to the principle of microscopic reversibility. Recently, the kcat of 122 s−1 for [C2-13C]acetyl-CoA formation from [C3-13C]pyruvate was reported by us using gCHSQC NMR (1), which is similar to other rate constants on the pathway in Scheme 1 and to the value of k−7 of 199 s−1 detected in this paper, indicating that transfer of the acetyl group to the CoA is not the rate-limiting step (1).
Induced Changes in the Active Center of E1p upon Interaction with 3-lip E2p as Evidenced by FT-MS Spectrometry
Next, the interaction of 3-lip E2p with E1p was evaluated using HDX-MS by comparing the deuterium uptake of the peptic peptides originating from E1p in 1) E1p by itself, 2) E1p subcomplex with 3-lip E2p, 3) E1p subcomplexes with the C-terminally truncated E2p proteins, and 4) E1p in the presence of E2pCD. The peptic digest of E1p resulted in 98% sequence coverage with 62 peptides identified. Of the 62 peptides, 17 were selected for comparative analysis, including peptides (1–17, 18–23, 24–39, and 40–58) from the N-terminal region of E1p, covering residues 1–58, which, as reported by us earlier, are involved in interaction with E2p (28, 50). The peptide 388–414 encompasses the inner active center loop (residues 401–413); peptide 532–555 encompasses the outer active center loop (residues 541–557); and finally, there are two regions surrounding the ThDP active site (residues 262–277 and residues 317–347) (supplemental Table S1). The comparative HDX-MS analysis revealed only a modest effect of the independently expressed LDh on deuterium uptake by E1p with a difference of no more than 0.5 Da, mainly at the inner and outer active center loops of E1p (Fig. 5); nor were significant changes in deuterium uptake of E1p observed in the presence of E2pCD, with the exception of the outer active center loop region (peptide 532–555), indicating limited direct interaction between E1p and E2pCD, in agreement with data reported earlier (51).
The presence of the E2p(1–190) didomain and E2p(1–394) tetradomain comprising the PSBD for E1p/E3 binding in addition to lipoyl domains displayed significant effects on deuterium uptake by E1p. Major changes were observed in peptides 1–17, 24–39, and 40–58, all from the N-terminal region of E1p, confirming our previous findings that the N-terminal region of E1p is involved in interaction with the PSBD of E2p (Fig. 5) (28, 50). Upon increasing the number of N-terminal E2p domains from LDh to E2p(1–190) didomain and then to E2p(1–394) tetradomain, the deuterium uptake by E1p decreases at peptides corresponding to residues 388–414 and 532–555 from the inner (residues 401–413) and outer (residues 541–557) active center loops of E1p. In the presence of E2p(1–394) tetradomain, there is also a significant change in deuterium uptake by peptides corresponding to residues 262–277 and 317–347 from the ThDP binding site. These results also affirm that all C-terminally truncated E2 proteins are correctly folded.
As shown in Fig. 5 and the right inset, deuterium uptake by E1p is reduced significantly more by complexation with 3-lip E2p than with E2p(1–394) tetradomain or with E2p(1–190) didomain, as is most evident in peptides encompassing the outer loop (residues 532–555), inner loop (residues 388–414), and two peptides related to the ThDP-Mg2+ binding site (residues 262–277 and 317–347) and even at the N-terminal region of E1p (residues 40–58). These results affirm once more that E2p(1–394) tetradomain is correctly folded and indeed interacts with E1p. The inset bar graph shows a difference in interaction of E1p with E2p(1–394) tetradomain and 3-lip E2p according to deuterium uptake. The difference in deuterium uptake is as much as 1.2 Da on peptide residues 262–277 in the ThDP-binding region, demonstrating induced changes in the active centers of E1p upon assembly with 3-lip E2p. These results also signal for the first time that interaction maps with truncated PDHc components do not give an accurate picture of the entire interaction map and that the HDX-MS method used here is indeed capable of elucidating interactions in PDHc between intact components.
LDh of the E2p(1–190) Didomain Senses the Interaction with E1p According to NMR Spectroscopy
In a parallel experiment, the interaction of E1p with E2p(1–190) didomain was probed by NMR. Recently, we reported partial backbone assignments for the E2p(1–190) didomain, mainly for the LDh; many backbone chemical shifts of assigned residues are available (27). Two-dimensional 15N TROSY HSQC NMR spectra of E2p(1–190) didomain in the presence of E3 revealed negligible changes in chemical shift of the E2p(1–190) didomain, indicating little or no interaction between E3 and LDh (27). In contrast to its interaction with E3, the interaction of E2p(1–190) didomain with E1p resulted in reduction of the intensity of multiple peaks in the two-dimensional 15N HSQC TROSY spectra of E2p(1–190) didomain, mainly in the LDh, with some peak intensities below the detection level: Thr17, Leu20, Leu33, Thr35, Ser42, Met43, Val45, Ala47, Lys54. Some of the chemical shifts could not be determined, implying an intermediate exchange regime on the chemical shift time scale upon interaction with E1p. Chemical shift changes of E2p(1–190) didomain observed upon interaction with E1p are shown in Fig. 6. When the chemical shifts corresponding to the LDh residues above were overlaid onto the chemical shifts corresponding to residues from the inner lipoyl domain (L3 in 3-lip E2p; Protein Data Bank code 1QJO) (52), all of these residues except for Lys54 mapped onto one face of the LDh, mainly in the β strands 4 and 5, which is close in space to the lipoyl-lysine β-turn. Additional changes were observed in chemical shift (changes greater than 0.01 ppm) of the resonances for residues 6–20 and 70–80, also located around the lipoyl lysine β-turn region. Residues 6–20 are mainly from the surface loop region of the LDh linking β-strands 1 and 2, close in space to the lipoyl-lysine β-turn, whereas residues 70–80 are mainly from β-strand 8 from the first sheet. Globally, the intensity of resonances of LDh diminished overall (Fig. 6), and the most likely explanation for this phenomenon is that the overall tumbling has been slowed down by formation of the complex (mass of E2p(1–190) didomain + (E1p)2 totals 220 kDa). However, the peak intensity is drastically reduced for the residues around the lipoyl lysine region. Regarding the NMR results, we point out that more extensive changes are observed (i.e. more interactions are evident) between E2p(1–190) didomain and E1p than between LDh by itself and E1p (53).
FIGURE 6.

Representative NMR spectra of E2p(1–190) didomain in the absence and presence of E1p. Top, two-dimensional 1H-15N HSQC TROSY spectrum of E2p(1–190) didomain (0.5 mm, in red) overlaid with that of the E2p(1–190) didomain (0.5 mm)-E1p (0.12 mm with 0.24 mm ThDP/pyruvate) subcomplex (in blue) recorded at 25 °C in 20 mm KH2PO4 (pH 7.0) with 0.15 m NaCl. Assigned peaks are indicated with one-letter amino acid code and residue number. Bottom, chemical shift deviation in LDh upon complexation of E2p(1–190) didomain with E1p.
Evidence for Induced Changes in the Active Centers of E1p upon Interaction with C-terminally Truncated Proteins and 3-lip E2p from Circular Dichroism Studies
Further evidence of the induced effect of the E2p(1–190) didomain, E2p(1–394) tetradomain and 3-lip E2p on the ThDP at the E1p active centers was obtained from CD spectroscopy.
It was first demonstrated that the lipoyl domain in LDh, E2p(1–190) didomain, E2p(1–394) tetradomain, and 3-lip E2p all give evidence for a strong negative CD band at 330 nm in their oxidized forms only, which could be eliminated when reduced by TCEP (Fig. 7). This provides a new CD signature for oxidized lipoyl domain not reported before. The reason for the smaller amplitude in the spectrum of the 3-lip E2p could be attributed to its oligomerization.
FIGURE 7.
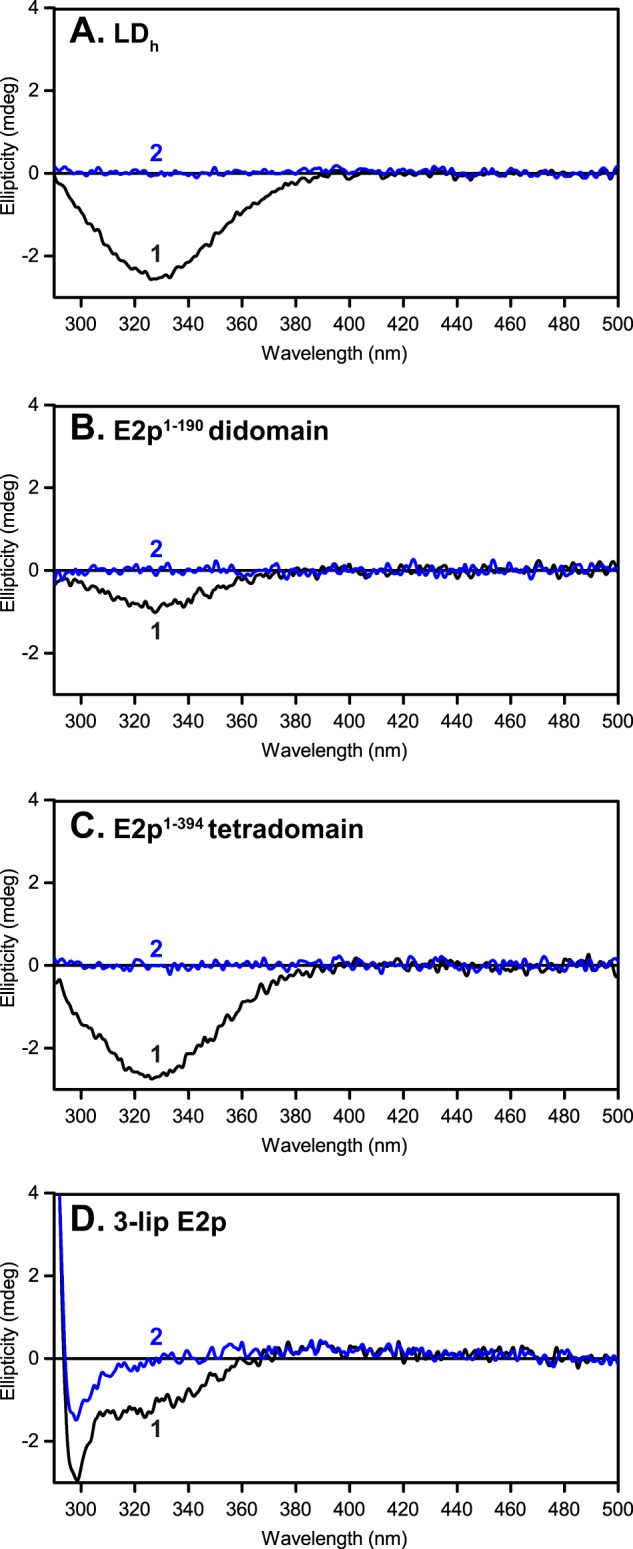
Lipoyl domain has a characteristic negative CD band at 330 nm. Shown are CD spectra of C-terminally truncated E2p proteins and of 3-lipoyl E2p with lipoyl domains in their oxidized state (spectrum 1) and TCEP-reduced form (spectrum 2). A, LDh (0.36 mg/ml). B, E2p(1–190) didomain (0.69 mg/ml). C, E2p(1–394) tetradomain (1.3 mg/ml). D, 3-lip E2p (2.1 mg/ml). The concentration of each protein is ∼30 μm in 50 mm KH2PO4 (pH 7.0) in a total volume of 2.4 ml at 25 °C.
Next, to an E1p-E2p subcomplex (where 1) ThDP was removed and 2) TCEP was used to reduce the lipoyl domains in all of the C-terminally truncated proteins) was added ThDP (0.3 mm), producing a new negative CD band near 320 nm (Fig. 8), distinct from that observed for oxidized lipoyl domains in Fig. 7. We assign this negative CD band to the aminopyrimidine tautomeric form of ThDP, based on numerous observations on 11 ThDP enzymes (54–58). The amplitude of the induced change at 320 nm is in the order 3-lip E2p > E2p(1–394) tetradomain ∼ E2p(1–190) didomain (Fig. 8). In a similar experiment with oxidized lipoyl domain-containing E2p proteins, the addition of ThDP only produced modest effects on the E1p CD spectra because the two bands overlap (330 nm for lipoyl domain and 320 nm for the aminopyrimidine form of ThDP; data not shown).
FIGURE 8.
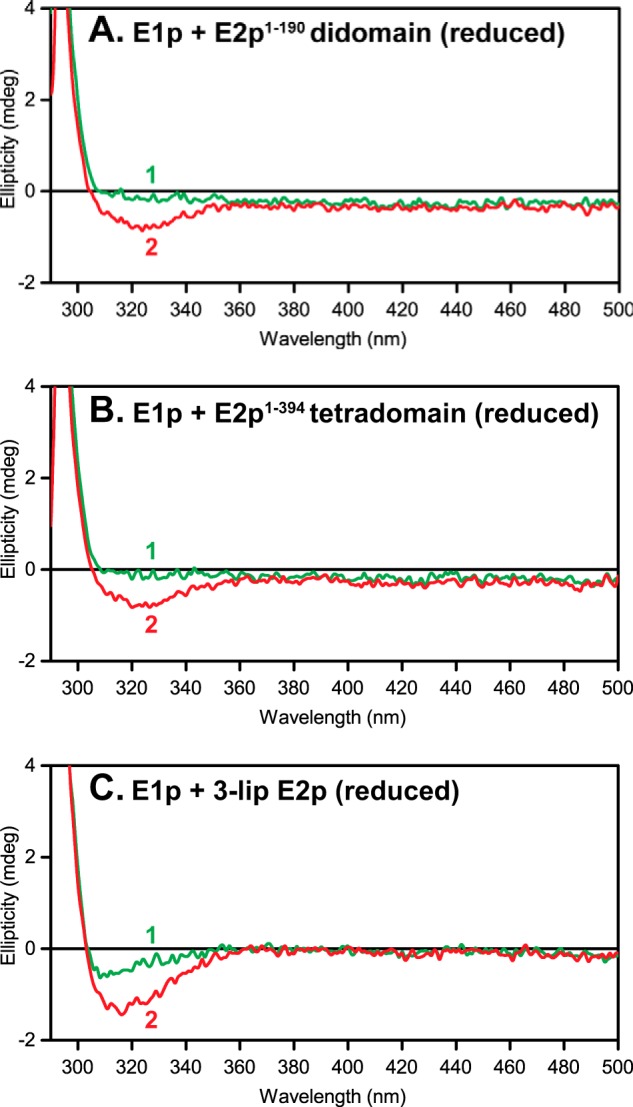
CD spectra of apo-E1p in the presence of C-terminally truncated E2p proteins and 3-lip E2p. A–C, spectrum 1, apo-E1p (3 mg/ml or 30 μm active centers) + E2p proteins + TCEP (60 μm) but no ThDP added. Spectrum 2, same as spectrum 1 upon the addition of 0.3 mm ThDP. Other conditions are as described in the legend to Fig. 7.
CONCLUSION
In E. coli PDHc, three E2pCD subunits form a trimer, displaying interactions between monomers and packing interactions within trimers that are similar to those reported for other E2CDs in the Protein Data Bank. Based on the x-ray structures of A. vinelandii E2pCD in complexes with CoA and dihydrolipoamide, the following residue interactions were identified as important in E. coli E2p by superposition as well as by biochemical studies: 1) the His602-Ser550 pair plays an essential role in the acetyltransferase reaction mechanism; 2) Asp601 and Arg603 stabilize the His602 conformation; and 3) Ser550 is involved in CoA binding and stabilization of the tetrahedral intermediate of the acetyltransferase reaction. Assignment of specific roles to these residues is still premature; more details regarding their functional roles are needed. Most importantly, as reported by Guest and colleagues (42, 43) and confirmed at Rutgers (49), substitution of the “key” His602 with Ala leads to 5.6% residual activity, only modestly affecting acetyl-CoA production, not consistent with a general base role.
Functional competence of the E2pCD by the demonstration that independently expressed E2pCD and LDh could produce 6.4% NADH activity upon reconstitution with E1p and E3 suggests that the domains LDh and E2pCD by themselves can maintain partial active center coupling within the assembled PDHc. The results also allow us to conclude that the forward k7 and reverse k−7 transacetylase reactions are both catalyzed by the E2pCD, and kinetics display a rapid approach to equilibrium. Recently, a kcat of 122 s−1 for acetyl-CoA formation from pyruvate was reported by us for E. coli PDHc (1), similar within experimental error to other rate constants on the pathway in Scheme 1 and to the value of k−7 of 199 s−1 here reported, indicating that transfer of an acetyl group to the CoA is not the rate-limiting step (1).
To place the magnitude of the rate constant k−7 for interdomain acetyl transfer in E2p in a relevant context, we can consider the following. The Keq for Reaction 1 should be very near unity because it is the conversion of one alkyl thiol ester to another; what is the size of the barrier? There is excellent model chemistry for baseline from Hupe and Jencks (59), where the rate of reaction for β-mercaptoethanol with unhindered thiol ester was quoted as 12 m−1 s−1, compared with k−7 of 199 s−1 for E2pCD and a second-order rate constant of kcat/Km = 2.84 × 106 m−1 s−1. We can estimate the rate acceleration compared with the Hupe-Jencks model of 105-fold (7 kcal/mol barrier reduction) by the E2pCD. In other words, the active center residues identified from the x-ray structure each may contribute modestly to achieve this rate acceleration.
The comparative HDX-MS analysis revealed only a modest effect of the independently expressed LDh on deuterium uptake by E1p or E2pCD. The deuterium uptake by E1p is reduced significantly more by complexation with 3-lip E2p than with E2p(1–394) tetradomain or with E2p(1–190) didomain. These results also signal for the first time that interaction maps with truncated PDHc components do not give an accurate overall picture and that the HDX-MS method here used is indeed capable of elucidating interactions in PDHc between intact components. In contrast to the HDX-MS analysis, NMR studies could detect interaction of the lipoyl domain in E2p(1–190) didomain with E1p, showing the important complementarity of and need for multiple structural methods to obtain a more complete interaction picture among the components in these important complexes.
The results presented in this paper also allowed us to complete the determination of individual rate constants for the interconversion of intermediates in the E. coli PDHc (a molecular machine with a mass of 4,500 kDa) catalytic cycle through acetyl-CoA formation, clearly signaling that the rate-limiting step is formation of the predecarboxylation intermediate LThDP on E1p, which appears to be controlled by the dynamics of E1p active center mobile loops (Scheme 1) (1). Considering the variety of approaches used to obtain these rate constants, they are all very similar within experimental error, suggesting that the kinetic barriers of different steps are also rather similar, including, most importantly, the one describing intercomponent communication (acetyl transfer between the E1 and E2 components). It is of obvious importance to demonstrate if these findings are general for other such complexes.
An important conclusion from the study never detected or discussed before is the E2p domain-induced changes in the active center of E1p (for a review of domain-induced allostery, see Ref. 61). This is very pronounced according to HDX-MS (Fig. 5), where the E2pCD covalently attached in 3-lip E2p has a significantly greater impact on the interactions than the E2p(1–394) tetradomain (lacking only E2pCD). The evidence is especially compelling because all E2p proteins show identical interactions along the N-terminal region of E1p, also suggesting that they are correctly folded, and the comparison carries its own control experiments. The domain-induced changes at the E1p active site are also supported by the CD results in Fig. 8, by the appearance of the aminopyrimidine tautomer of ThDP induced by the E2p proteins; this signal was never observed on E. coli E1p with any substitutions (58, 62).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM061791 (to W. F.) and GM050380 (to F. J.). This work was also supported by a Veterans Affairs Merit Review (to W. F.).

This article contains supplemental Tables S1 and S2.
The atomic coordinates and structure factors (code 4N72) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PDHc
- pyruvate dehydrogenase complex
- E1p
- the first pyruvate dehydrogenase component
- 1-lip E2p
- dihydrolipoyl acetyltransferase component with a single lipoyl domain
- 3-lip E2p
- wild type dihydrolipoyl acetyltransferase component with three lipoyl domains
- E3
- dihydrolipoyl dehydrogenase component
- E2pCD
- E2p core domain
- LD
- lipoyl domain
- LDh
- a single hybrid lipoyl domain
- ThDP
- thiamin diphosphate
- PSBD
- peripheral subunit binding domain
- HDX-MS
- hydrogen/deuterium exchange mass spectrometry
- FT-MS
- Fourier transform mass spectrometry
- TCEP
- tris(2-carboxyethyl)phosphine
- TEV
- tobaccoetch virus.
REFERENCES
- 1. Balakrishnan A., Nemeria N. S., Chakraborty S., Kakalis L., Jordan F. (2012) Determination of pre-steady-state constants on the Escherichia coli pyruvate dehydrogenase complex reveals that loop movement controls the rate-limiting step. J. Am. Chem. Soc. 134, 18644–18655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jordan F., Arjunan P., Kale S., Nemeria N. S., Furey W. (2009) Multiple roles of mobile active center loops in the E1 component of the Escherichia coli pyruvate dehydrogenase complex. Linkage of protein dynamics to catalysis. J. Mol. Catal. B Enzym. 61, 14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reed L. J., Hackert M. L. (1990) Structure-function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem. 265, 8971–8974 [PubMed] [Google Scholar]
- 4. Perham R. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 [DOI] [PubMed] [Google Scholar]
- 5. Perham R. (2000) Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu. Rev. Biochem. 69, 961–1004 [DOI] [PubMed] [Google Scholar]
- 6. Perham R. N., Jones D. D., Chauhan H. J., Howard M. J. (2002) Substrate channeling in 2-oxo acid dehydrogenase multienzyme complexes. Biochem. Soc. Trans. 30, 47–51 [DOI] [PubMed] [Google Scholar]
- 7. Stephens P. E., Darlison M. G., Lewis H. M., Guest J. R. (1983) The pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence encoding the pyruvate dehydrogenase component. Eur. J. Biochem. 133, 155–162 [DOI] [PubMed] [Google Scholar]
- 8. Stephens P. E., Darlison M. G., Lewis H. M., Guest J. R. (1983) The pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence encoding the dihydrolipoamide acetyltransferase component. Eur. J. Biochem. 133, 481–489 [DOI] [PubMed] [Google Scholar]
- 9. Bleile D. M., Munk P., Oliver R. M., Reed L. J. (1979) Subunit structure of dihydrolipoyl transacetylase component of pyruvate dehydrogenase complex from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 4385–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reed L. J., Pettit F. H., Eley M. H., Hamilton L., Collins J. H., Oliver R. M. (1975) Reconstitution of the Escherichia coli pyruvate dehydrogenase complex. Proc. Natl. Acad. Sci. U.S.A. 72, 3068–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Green J. D., Perham R. N., Ullrich S. J., Appella E. (1992) Conformational studies of the interdomain linker peptides in the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase multienzyme complex of Escherichia coli. J. Biol. Chem. 267, 23484–23488 [PubMed] [Google Scholar]
- 12. Stephens P. E., Lewis H. M., Darlison M. G., Guest J. R. (1983) Nucleotide sequence of the lipoamide dehydrogenase gene of Escherichia coli K12. Eur. J. Biochem. 135, 519–527 [DOI] [PubMed] [Google Scholar]
- 13. Stoops J. K., Cheng R. H., Yazdi M. A., Maeng C.-Y., Schroeter J. P., Klueppelberg U., Kolodziej S. J., Baker T. S., Reed L. J. (1997) On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J. Biol. Chem. 272, 5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou Z. H., Liao W., Cheng R. H., Lawson J. E., McCarthy D. B., Reed L. J., Stoops J. K. (2001) Direct evidence for the size and conformational variability of the pyruvate dehydrogenase complex revealed by three-dimensional electron microscopy. J. Biol. Chem. 276, 21704–21713 [DOI] [PubMed] [Google Scholar]
- 15. Yu X., Hiromasa Y., Tsen H., Stoops J. K., Roche T. E., Zhou Z. H. (2008) Structures of the human pyruvate dehydrogenase complex cores: a highly conserved catalytic center with flexible N-terminal domains. Structure 16, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vijayakrishnan S., Kelly S. M., Gilbert R. J., Callow P., Bhella D., Forsyth T., Lindsay J. G., Byron O. (2010) Solution structure and characterisation of the human pyruvate dehydrogenase complex core assembly. J. Mol. Biol. 399, 71–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mattevi A., Obmolova G., Kalk K. H., Westphal A. H., de Kok A., Hol W. G. (1993) Refined crystal structure of the catalytic domain of dihydrolipoyl transacetylase (E2p) from Azotobacter vinelandii at 2.6 Å resolution. J. Mol. Biol. 230, 1183–1199 [DOI] [PubMed] [Google Scholar]
- 18. Knapp J. E., Mitchell D. T., Yazdi M. A., Ernst S. R., Reed L. J., Hackert M. L. (1998) Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J. Mol. Biol. 280, 655–668 [DOI] [PubMed] [Google Scholar]
- 19. Mattevi A., Obmolova G., Kalk K. H., Teplyakov A., Hol W. G. (1993) Crystallographic analysis of substrate binding and catalysis in dihydrolipoyl transacetylase (E2p). Biochemistry 32, 3887–3901 [DOI] [PubMed] [Google Scholar]
- 20. Leslie A. G. W., Moody P. C. E., Shaw W. V. (1988) Structure of chloramphenicol acetyltransferase at 1.75 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 85, 4133–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kato M., Wynn R. M., Chuang J. L., Brautigam C. A., Custorio M., Chuang D. T. (2006) A synchronized substrate-gating mechanism revealed by cubic core structure of the bovine branched chain α-ketoacid dehydrogenase complex. EMBO J. 25, 5983–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knapp J. E., Carroll D., Lawson J. E., Ernst S. R., Reed L. J., Hackert M. L. (2000) Expression, purification, and structural analysis of the trimeric form of the catalytic domain of the Escherichia coli dihydrolipoamide succinyltransferase. Protein Sci. 9, 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hendle J., Mattevi A., Westphal A. H., Spee J., de Kok A., Teplyakov A., Hol W. G. (1995) Crystallographic and enzymatic investigations on the role of Ser558, His610, and Asn614 in the catalytic mechanism of Azotobacter vinelandii dihydrolipoamide acetyltransferase (E2p). Biochemistry 34, 4287–4298 [DOI] [PubMed] [Google Scholar]
- 24. Marrott N. L., Marshall J. J., Svergun D. I., Crennell S. J., Hough D. W., Danson M. J., van den Elsen J. M. (2012) The catalytic core of an archaeal 2-oxoacid dehydrogenase multienzyme complex is a 42-mer protein assembly. FEBS J. 279, 713–723 [DOI] [PubMed] [Google Scholar]
- 25. Izard T., Aevarsson A., Allen M. D., Westphal A. H., Perham R. N., de Kok A., Hol W. G. (1999) Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U.S.A. 96, 1240–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arjunan P., Nemeria N., Brunskill A., Chandrasekhar K., Sax M., Yan Y., Jordan F., Guest J. R., Furey W. (2002) Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 Å resolution. Biochemistry 41, 5213–5221 [DOI] [PubMed] [Google Scholar]
- 27. Chandrasekhar K., Wang J., Arjunan P., Sax M., Park Y.-H., Nemeria N. S., Kumaran S., Song J., Jordan F., Furey W. (2013) Insight to the interaction of the dihydrolipoamide acetyltransferase (E2) core with the peripheral components in the Escherichia coli pyruvate dehydrogenase complex via multifaceted structural approaches. J. Biol. Chem. 288, 15402–15417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song J., Park Y. H., Nemeria N. S., Kale S., Kakalis L., Jordan F. (2010) Nuclear magnetic resonance evidence for the role of the flexible regions of the E1 component of the pyruvate dehydrogenase complex from gram-negative bacteria. J. Biol. Chem. 285, 4680–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ali S. T., Guest J. R. (1990) Isolation and characterization of lipoylated and unlipoylated domains of the E2p subunit of the pyruvate dehydrogenase complex of Escherichia coli. Biochem. J. 271, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones D. D., Horne H. J., Reche P. A., Perham R. N. (2000) Structural determinants of post-translational modification and catalytic specificity for the lipoyl domains of the pyruvate dehydrogenase multienzyme complex of Escherichia coli. J. Mol. Biol. 295, 289–306 [DOI] [PubMed] [Google Scholar]
- 31. Nemeria N., Volkov A., Brown A., Yi J., Zipper L., Guest J. R., Jordan F. (1998) Systematic study of the six cysteines of the E1 subunit of the pyruvate dehydrogenase multienzyme complex from Escherichia coli: none is essential for activity. Biochemistry 37, 911–922 [DOI] [PubMed] [Google Scholar]
- 32. Hamuro Y., Coales S. J., Molnar K. S., Tuske S. J., Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046 [DOI] [PubMed] [Google Scholar]
- 33. Weis D. D., Engen J. R., Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 [DOI] [PubMed] [Google Scholar]
- 34. Kavan D., Man P. (2011) MSTools: Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58 [Google Scholar]
- 35. Nambi S., Badireddy S., Visweswariah S. S., Anand G. S. (2012) Cyclic AMP-induced conformational changes in micobacterial protein acetyltransferases. J. Biol. Chem. 287, 18115–18129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMR Pipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 38. Keller R. L. (2004) The Computer Aided Resonance Assignment, CANTINA Verlag, Goldau, Switzerland [Google Scholar]
- 39. Yamazaki T., Lee W., Arrowsmith C. H., Muhandiram D. R., Kay L. E. (1994) A suite of triple resonance NMR experiments for the backbone assignment of 15N-, 13C-,2H-labeled proteins with high sensitivity. J. Am. Chem. Soc. 116, 11655–11666 [Google Scholar]
- 40. Live D. H., Davis D. G., Agosta W. C., Cowburn D. (1984) Observation of 1000-fold enhancement of 15N NMR via proton-detected multiquantum coherences: studies of large peptides. J. Am. Chem. Soc. 106, 6104–6105 [Google Scholar]
- 41. Russell G. C., Guest J. R. (1991) Sequence similarities within the family of dihydrolipoamide acetyltransferases and discovery of a previously unidentified fungal enzyme. Biochim. Biophys. Acta 1076, 225–232 [DOI] [PubMed] [Google Scholar]
- 42. Russell G. C., Guest J. R. (1990) Overexpression of restructured pyruvate dehydrogenase complexes and site-directed mutagenesis of a potential active-site histidine residue. Biochem. J. 269, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Russell G. C., Guest J. R. (1991) Site-directed mutagenesis of the lipoate acetyltransferase of Escherichia coli. Proc. Biol. Sci. 243, 155–160 [DOI] [PubMed] [Google Scholar]
- 44. Yang Y. S., Frey P. A. (1986) Dihydrolipoyl tranaacetylase of Escherichia coli: formation of 8-S-acetyldihydrolipoamide. Biochemistry 25, 8173–8178 [DOI] [PubMed] [Google Scholar]
- 45. Butterworth P. J., Tsai C. S., Eley M. H., Roche T. E., Reed L. J. (1975) A kinetic study of dihydrolipoyl transacetylase from bovine kidney. J. Biol. Chem. 250, 1921–1925 [PubMed] [Google Scholar]
- 46. Allen M. D., Perham R. N. (1997) The catalytic domain of dihydrolipoyl acetyltransferase from the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus: expresssion, purification and reversible denaturation. FEBS Lett. 413, 339–343 [DOI] [PubMed] [Google Scholar]
- 47. Niu X.-D., Stoops J. K., Reed L. J. (1990) Overexpression and mutagenesis of the catalytic domain of dihydrolipoamide acetyltransferase from Saccharomyces cerevisiae. Biochemistry 29, 8614–8619 [DOI] [PubMed] [Google Scholar]
- 48. CaJacob C. A., Gavino G. R., Frey P. A. (1985) Pyruvate dehydrogenase complex of Escherichia coli: thiamin pyrophosphate and NADH-dependent hydrolysis of acetyl-CoA. J. Biol. Chem. 260, 14610–14615 [PubMed] [Google Scholar]
- 49. Song J., Jordan F. (2012) Interchain acetyl transfer in the E2 component of bacterial pyruvate dehydrogenase suggests a model with different roles for each chain in a trimer of the homooligomeric component. Biochemistry 51, 2795–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park Y. H., Wei W., Zhou L., Nemeria N., Jordan F. (2004) Amino-terminal residues 1–45 of the Escherichia coli pyruvate dehydrogenase complex E1 subunit interact with the E2 subunit and are required for activity of the complex but not for reductive acetylation of the E2 subunit. Biochemistry 43, 14037–14046 [DOI] [PubMed] [Google Scholar]
- 51. Schulze E., Westphal A. H., Obmolova G., Mattevi A., Hol W. G., de Kok A. (1991) The catalytic domain of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii and Escherichia coli: expression, purification, properties and preliminary x-ray analysis. Eur. J. Biochem. 201, 561–568 [DOI] [PubMed] [Google Scholar]
- 52. Jones D. D., Stott K. M., Howard M. J., Perham R. N. (2000) Resticted motion of the lipoyl-lysine swinging arm in the pyruvate dehydrogenase complex of Escherichia coli. Biochemistry 39, 8448–8459 [DOI] [PubMed] [Google Scholar]
- 53. Jones D. D., Stott K. M., Reche P. A., Perham R. N. (2001) Recognition of the lipoyl domain is the ultimate determinant of substrate channeling in the pyruvate dehydrogenase multienzyme complex. J. Mol. Biol. 305, 49–60 [DOI] [PubMed] [Google Scholar]
- 54. Jordan F., Nemeria N. S., Zhang S., Yan Y., Arjunan P., Furey W. (2003) Dual catalytic apparatus of the thiamin diphosphate coenzyme:acid-base via the 1′,4′-iminopyrimidine tautomer along with its electrophilic role. J. Am. Chem. Soc. 125, 12732–12738 [DOI] [PubMed] [Google Scholar]
- 55. Nemeria N., Baykal A., Joseph E., Zhang S., Yan Y., Furey W., Jordan F. (2004) Tetrahedral intermediates in thiamin diphosphate-dependent decarboxylations exist as a 1′,4′-imino tautomeric form of the coenzyme, unlike the Michaelis complex or the free coenzyme. Biochemistry 43, 6565–6575 [DOI] [PubMed] [Google Scholar]
- 56. Nemeria N., Chakraborty S., Baykal A., Korotchkina L. G., Patel M. S., Jordan F. (2007) The 1′,4′-iminopyrimidine tautomer of thiamin diphosphate is poised for catalysis in asymmetric active centers on enzymes. Proc. Natl. Acad. Sci. U.S.A. 104, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nemeria N., Korotchkina L., McLeish M. J., Kenyon G. L., Patel M. S., Jordan F. (2007) Elucidation of the chemistry of enzyme-bound thiamin diphosphate prior to substrate binding: defining internal equilibria among tautomeric and ionization states. Biochemistry 46, 10739–10744 [DOI] [PubMed] [Google Scholar]
- 58. Nemeria N. S., Chakraborty S., Balakrishnan A., Jordan F. (2009) Defining states of ionization and tautomerization of the coenzyme at individual steps on thiamin diphosphate dependent enzymatic pathways, FEBS J. 276, 2432–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hupe D. J., Jencks W. P. (1977) Nonlinear structure-reactivity correlations: acyl transfer between sulfur and oxygen nucleophiles. J. Am. Chem. Soc. 99, 451–464 [Google Scholar]
- 60. Shuker S. B., Hajduk P. J., Meadows R. P., Fesik S. W. (1996) Discovering high affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 61. Tompa P. (2014) Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chem. Rev., in press [DOI] [PubMed] [Google Scholar]
- 62. Jordan F., Nemeria N. (2005) Experimental observation of thiamin diphosphate-bound intermediates on enzymes and the mechanistic information derived from these observations. Bioorg. Chem. 33, 190–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.