

Background: Seprase is an integral membrane serine protease implicated in the cellular invasiveness of tumor, endothelial, and fibroblast cells of various human tumors.
Results: Seprase is transcriptionally targeted in invasive melanoma cells via TGF-β signaling.
Conclusion: Seprase-expressing melanoma cells produce high levels of autocrine TGF-β and display invasive phenotypes in vitro.
Significance: This study provides the first insight into TGF-β-driven seprase transcription in cancer.
Keywords: Gene Regulation, Gene Transcription, Invasion, Melanoma, Serine Protease, Transcription Promoter, Transcription Regulation
Abstract
The tumor invasive phenotype driven by seprase expression/activity has been widely examined in an array of malignant tumor cell types; however, very little is known about the transcriptional regulation of this critical protease. Seprase (also named fibroblast activation protein-α, antiplasmin-cleaving enzyme, and dipeptidyl prolyl peptidase 5) is expressed at high levels by stromal fibroblast, endothelial, and tumor cells in a variety of invasive tumors but is undetectable in the majority of normal adult tissues. To examine the transcriptional regulation of the gene, we cloned the human seprase promoter and demonstrated that endogenous seprase expression and exogenous seprase promoter activity are high in invasive melanoma cells but not in non-invasive melanoma cells/primary melanocytes. In addition, we identified a crucial TGF-β-responsive cis-regulatory element in the proximal seprase promoter region that enabled robust transcriptional activation of the gene. Treatment of metastatic but not normal/non-invasive cells with TGF-β1 caused a rapid and profound up-regulation of endogenous seprase mRNA, which coincided with an abolishment of the negative regulator c-Ski, and an increase in binding of Smad3/4 to the seprase promoter in vivo. Blocking TGF-β signaling in invasive melanoma cells through overexpression of c-Ski, chemically using SB-431542, or with a neutralizing antibody against TGF-β significantly reduced seprase mRNA levels. Strikingly, RNAi of seprase in invasive cells greatly diminished their invasive potential in vitro as did blocking TGF-β signaling using SB-431542. Altogether, we found that seprase is transcriptionally up-regulated in invasive melanoma cells via the canonical TGF-β signaling pathway, supporting the roles of both TGF-β and seprase in tumor invasion and metastasis.
Introduction
Tumor cell invasion involves induction of the cell surface protease seprase in a variety of cancer tissues, but the regulatory mechanisms governing the gene in invasive/metastatic tumor cells remain to be determined (1). The human gene encoding FAPα2 was originally cloned in 1994 (2) and was mapped to chromosome band 2q23 by fluorescence in situ hybridization (3). Independently, seprase was identified and cloned from the LOX human metastatic melanoma cell line and in fact deduced to be the same gene as FAPα (4, 5).
Seprase was initially described in the LOX melanoma cell line and found to be a 760-amino acid type II transmembrane glycoprotein whose 97-kDa monomers can dimerize to form a 170-kDa enzymatically active gelatinase·dipeptidyl prolyl peptidase complex (4–6). FAPα protein expression and proteolytic activity were also independently identified in reactive tumor stromal fibroblasts but not in tumor or endothelial cell types tested (7–9). Seprase functions as a serine integral membrane protease and has been implicated in the cellular invasiveness of tumor cells, endothelial cells, and fibroblasts of various human tumors (1, 4, 6, 10–18). Specifically, seprase is up-regulated in infiltrating ductal carcinomas of the breast and in resulting tumor metastases (19) as well as in peritoneal metastases in ovarian cancer (16, 20). Increased seprase expression has also been associated with a more aggressive disease state in colon cancer (21, 22), in osteosarcoma (23), and with lymph node metastases in human colorectal (14), pancreatic (24), and gastric cancers (25).
Recently, the mouse FAPα promoter was cloned and shown to have some conserved regions as compared with the human seprase promoter, and basal transcription was found to be regulated by EGR1 in a panel of human cancer cell lines (26). In addition, an in silico electronic Northern blot study showed that normal tissues generally lack FAPα RNA signal apart from the endometrium, whereas the majority of tumor tissues express FAPα RNA (27). FAPα gene expression was found to be up-regulated by a combination of interleukin-1 and oncostatin M in both chondrocytes and cartilage explant cultures (28). FAPα protein levels were found to be induced in FB20 leptomeningeal fibroblasts upon addition of TGF-β1, 12-O-tetradecanoylphorbol-13-acetate, retinoids, or a combination of TGF-β1 and 12-O-tetradecanoylphorbol-13-acetate (29). TGF-β1 was also able to induce FAPα protein expression in immortalized human dermal fibroblasts (30) as well as in the NIH3T3 fibroblast cell line (31). Taken together, these results suggested to us the possibility that the TGF-β pathway may regulate seprase in some direct capacity.
TGF-β signaling controls a wide range of cellular processes, including differentiation, proliferation, embryonic development, tissue regeneration, apoptosis, cellular homeostasis, and regulation of the immune system (32). In the canonical pathway, the TGF-β ligand signals through a protein complex consisting of two type II receptors and two type I receptors, both of which are serine/threonine kinases. Upon binding of the ligand to this complex, the type II receptors phosphorylate and ultimately activate the type I receptors. Once activated, the type I receptors can phosphorylate and activate the so-called receptor-regulated Smads (R-Smads), namely Smad2 and Smad3. The now activated R-Smads can form complexes and bind to the co-Smad, Smad4, forming heteromeric complexes, which can accumulate in the nucleus. These Smad complexes act as transcription factors in the regulation of target gene expression and can do so in both positive and negative manners (33, 34).
Because the TGF-β pathway normally functions in an antiproliferative, tumor-suppressive fashion, alterations in components of the pathway such as deletions, mutations, down-regulation of positive regulators, and overexpression or amplification of negative regulators are observed in many types of human cancer (35, 36). Contradictory to this, TGF-β can control processes such as tumor cell invasion, modification of the tumor microenvironment, and modulation of immune system surveillance, all of which promote tumor progression (37, 38). In some studies, TGF-β overexpression in tumor cells was shown to correlate with tumor vascularization, metastasis, and poor patient prognosis (39). However, precise elements involved in the induction of tumor cell invasiveness remain to be investigated.
To examine the transcriptional regulation of the seprase gene in metastatic tumor cells, we cloned a significant portion of the human seprase promoter and discovered that the gene is a bona fide transcriptional target of the canonical TGF-β/Smad pathway in a pair of metastatic melanoma cell lines. Seprase promoter targeting by TGF-β is absent/impaired in both a non-invasive melanoma cell line and non-transformed primary human melanocytes. Furthermore, the level of TGF-β signaling and ultimately seprase expression determined the invasive capability of these melanoma cells in vitro.
EXPERIMENTAL PROCEDURES
Cell Culture and Retroviral Infection
The human malignant melanoma cell line A735, human melanotic melanoma cell line SK-MEL-28, and human fibroblast cell line CCD-28SK were obtained from American Type Culture Collection (Manassas, VA). Human epidermal melanocytes isolated from lightly pigmented skin (HEMa-LP) were obtained from Cascade Biologics (Portland, OR). The HaCat keratinocyte cell line was originally described in Boukamp et al. (40), and the LOX human amelanotic melanoma cell line was supplied by Fodstad et al. (41). Cell lines were grown in cancer cell culture (CCC) media, a 1:1 mixture of DMEM (Invitrogen) and RPMI 1640 medium (Invitrogen) supplemented with 10% fetal calf serum (Invitrogen), 5% Nu-Serum (BD Biosciences), 1% l-glutamine (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 0.2% Fungizone (Invitrogen). HEMa-LP cells were grown in Medium 254 (Cascade Biologics) supplemented with human melanocyte growth supplement (Cascade Biologics). The retroviral packaging cell line GPG29, supplied by Dr. M. Sadelain, was cultured and used as described previously (42). Briefly, transfections of retroviral plasmids were performed using Lipofectamine 2000 (Invitrogen). Post-transfection, GPG29 cells were cultured in CCC media. Virus-containing supernatants were harvested 24 and 48 h later, added to 70% confluent cultures of target cells in the presence of 8 μg/ml Polybrene (Sigma), and incubated overnight. Cells were allowed to recover for 24 h in CCC media. Stable cell lines were generated by selecting with 2 μg/ml puromycin. HEK-293T cells were used to generate lentivirus in a similar manner for induction of target A375 cells. Stable A375 cells were generated by selecting with 2 μg/ml puromycin. LOX pGUS-NT and LOX pGUS-Sep KD (pGUS-SEP1384) are cell lines described previously (10, 16). A375 pGUS-NT and A375 pGUS-Sep KD cells were generated in a similar fashion. All cell lines were maintained in a humidified 37 °C incubator with 5% CO2.
Plasmids
Three wild-type (WT) human seprase promoter fragments of differing lengths: 2.637 kbp (−2432 to +205), 1357 bp (−1152 to +205), and 674 bp (−469 to +205) were generated by PCR using the HotStar HiFidelity Polymerase kit (Qiagen, Valencia, CA), and C.H.O.R.I. bacterial artificial chromosome clone RP11-576I16 (Children's Hospital Oakland Research Institute, Oakland, CA) as template. Each promoter fragment was individually cloned into the KpnI and HindIII sites of the promoterless pGL4.15[luc2P/Hygro] (pGL4) luciferase reporter vector (Promega, Madison, WI). Site-directed mutagenesis to inactivate nine putative Smad binding sites, a TATA box, and a non-binding site identified in the WT 674-bp promoter was carried out according to the DpnI-based QuikChange site-directed mutagenesis kit protocol (Stratagene, La Jolla, CA). Sequences of primers are given in Table 1. All resulting plasmids were sequence-verified. pGUS-NT and pGUS-Sep KD (pGUS-SEP1384) are previously described (10, 16) RNAi vectors allowing stable expression of hairpins against either a scrambled non-target (NT) or seprase, respectively. pGUS vectors concomitantly express GFP in addition to hairpins of interest (10, 16). pLKO.1 eGFP shRNA (LKO1-GFP-KD; used as negative control) and pLKO.1 SKI shRNA Clone TRCN0000010437 (LKO1-SKI-KD) plasmids (Open Biosystems, Waltham, MA) and packaging plasmids pMDLg/pRRE, pRSV-Rev, pMD2.G (43) were used to generate lentivirus for induction of A375 cells in c-Ski loss-of-function experiments. pBabe-Puro and pBabe-Puro/c-Ski retroviral constructs (44) were provided by Dr. K. Luo. 4x-SBE-Luc (45) and (CAGA)9-MLP-Luc (46) are previously described TGF-β-responsive reporters. The pRL-TK control plasmid (Promega) contains the herpes simplex virus thymidine kinase promoter to provide low to moderate levels of Renilla luciferase expression in co-transfected mammalian cells.
TABLE 1.
| Mutant | Sequence of site-directed mutagenesis primers |
|---|---|
| SBE site 1 mutant | |
| Forward | 5′-GCAGCCGTGGGTTTAAATAGTTGAATTTTTAAAC-3′ |
| Reverse | 5′-GTTTAAAAATTCAACTATTTAAACCCACGGCTGC-3′ |
| SBE site 2 mutant | |
| Forward | 5′-GAAAAACATTTCTTTTTGCAATACCTC-3′ |
| Reverse | 5′-GAGGTATTGCAAAAAGAAATGTTTTTC-3′ |
| SBE site 3 mutant | |
| Forward | 5′-GCAATACCTCATAATCTTCTTTAGGAAAAAAAAGTGCAGTTA-3′ |
| Reverse | 5′-TAACTGCACTTTTTTTTCCTAAAGAAGATTATGAGGTATTGC-3′ |
| SBE site 4 mutant | |
| Forward | 5′-AAAAAGTAGATATATGTTTAATGTAAAAACCTGCAAGTTTCATTATTTTA-3′ |
| Reverse | 5′-TAAAATAATGAAACTTGCAGGTTTTTACATTAAACATATATCTACTTTTT-3′ |
| SBE site 5 mutant | |
| Forward | 5′-CCTCTGTATGTCAACGTAAGTTTTTGTTGGTGTAGTTACAAGG-3′ |
| Reverse | 5′-CCTTGTAACTACACCAACAAAAACTTACGTTGACATACAGAGG-3′ |
| SBE site 6 mutant | |
| Forward | 5′-CTGTTTCTAATTTTAAAAAAATCTTTTGAAACTTGGC-3′ |
| Reverse | 5′-GCCAAGTTTCAAAAGATTTTTTTAAAATTAGAAACAG-3′ |
| SBE site 7B mutant | |
| Forward | 5′-CCAACTACAAAGACTTTTTTGGTCCTTTTCAACG-3′ |
| Reverse | 5′-CGTTGAAAAGGACCAAAAAAGTCTTTGTAGTTGG-3′ |
| SBE site 7A mutant | |
| Forward | 5′-CCAACTACAATTTTAGACTTGGTCC-3′ |
| Reverse | 5′-GGACCAAGTCTAAAATTGTAGTTGG-3′ |
| SBE site 7AB mutant | |
| Forward | 5′-GCTTCAGCTTCCAACTACAATTTTTTTTTTGGTCCTTTTCAACGG-3′ |
| Reverse | 5′-CCGTTGAAAAGGACCAAAAAAAAAATTGTAGTTGGAAGCTGAAGC-3′ |
| SBE site 8 mutant | |
| Forward | 5′-CCTTTTCAACGGTTTTCATTTTTCCAGTGACCCACGC-3′ |
| Reverse | 5′-GCGTGGGTCACTGGAAAAATGAAAACCGTTGAAAAGG-3′ |
| SBE site 9 mutant | |
| Forward | 5′-CCAGTGACCCACGCTCTGATTTTAGAATTAGCTAACTTTCAAA-3′ |
| Reverse | 5′-TTTGAAAGTTAGCTAATTCTAAAATCAGAGCGTGGGTCACTGG-3′ |
| TATA mutant | |
| Forward | 5′-GTTACAAGGATGAGAAGGCTCCGAAACTTCCCTTGAGTCACTC-3′ |
| Reverse | 5′-GAGTGACTCAAGGGAAGTTTCGGAGCCTTCTCATCCTTGTAAC-3′ |
| Non-site mutant | |
| Forward | 5′-GAAACTTGGCACGGTATTTGGGAGTCCGTGGAAAGAAAAAAA-3′ |
| Reverse | 5′-TTTTTTTCTTTCCACGGACTCCCAAATACCGTGCCAAGTTTC-3′ |
Quantitative Real Time RT-PCR (qRT-PCR)
Total RNAs were purified from cell lines using the RNeasy Mini kit (Qiagen). The following primer pairs were used for qRT-PCR: seprase: forward, 5′-TTGCCATCTAAGGAAAGAAAGG-3′; reverse, 5′-TTTTCTGACAGCTGTAATCTGG-3′; c-Ski: forward, 5′-GAGGTGGAAGTTGAAAGCAGG-3′; reverse, 5′-TCATGCAGGAACTTCTCTTTGG-3′; and endogenous control β-actin: forward, 5′-AGATGACCCAGATCATGTTTGA-3′; reverse, 5′-GCACAGCTTCTCCTTAATGTCA-3′. The QuantiTect SYBR Green RT-PCR kit (Qiagen) was used to prepare PCRs according to the manufacturer's specifications. Serial dilutions of LOX cell total RNA were used to generate standard curves for every experiment on seprase expression. Inter-experimental data for seprase mRNA levels were comparable because LOX cell RNA standard was purified from a 500-cm2 cell culture dish, pooled, quantified, and frozen as −80 °C aliquots. qRT-PCR conditions were: 50 °C for 30 min; 95 °C for 15 min; 40 cycles of 94 °C for 15 s, 59 °C for 30 s, 72 °C for 30 s, and 76 °C for 10 s; and 10 min at 72 °C. qRT-PCR was conducted on the Opticon II (MJ Research, Watertown, MA) and analyzed using OpticonMONITORTM analysis software Version 3.0 (MJ Research). All samples were run in triplicate, and non-template controls were included in each run. The RNA levels of seprase and c-Ski were normalized against β-actin in every case, and experiments were repeated at least three times.
Luciferase Assays
Assays were performed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions and a SPECTRAmax M5 plate reader (Molecular Devices, Sunnyvale, CA) equipped with SoftMax Pro software (Molecular Devices). Briefly, cells cultured in 12-well plates were transfected as indicated with 0.5 μg of seprase promoter plasmid (WT or mutant), 0.3 μg of the 4x-SBE-Luc plasmid, or 0.3 μg of the (CAGA)9-MLP-Luc construct, respectively, and 50 ng of pRL-TK vector as an internal control. All transfections were carried out under standard protocols using Lipofectamine (Invitrogen) except in experiments comparing all five cell lines in which case Lipofectamine 2000 was used. SK-MEL-28 cells were transfected with FuGENE® HD transfection reagent (Roche Applied Science). Twenty-four hours post-transfection, cells were harvested and assayed for luciferase activity, normalizing reporter activity to Renilla luciferase control. In experiments with TGF-β1 addition, 2 ng/ml was added for the final 6 or 12 h of incubation depending on the experiment. All experiments were performed in triplicate and independently repeated at least three times.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were conducted using the Chromatin Immunoprecipitation Assay kit (Upstate, Lake Placid, NY) following the provided standard protocol. Cells were sonicated on ice five times (30 s each time) using a Kontes microultrasonic cell disruptor with AS1 probe and an output of 55. This was optimized to consistently generate 200–500-bp fragments of cross-linked DNA. The following polyclonal antibodies were used for this assay: 4 μg of anti-Smad2/3 (E-20, Santa Cruz Biotechnology, Santa Cruz, CA), 4 μg of anti-Smad 4 (C-20, Santa Cruz Biotechnology), 4 μg of anti-Ski (H-329, Santa Cruz Biotechnology), and for use as a negative control 4 μg of normal rabbit IgG (Cell Signaling Technology, Beverly, MA). The following primer pairs (flanking binding sites of interest) were used for real time PCR: Smad site 5: forward, 5′-GCAACATAAACCTGAACTGG-3′; reverse, 5′-GCCCTCAAATGAACTGTGAG-3′; 134-bp amplicon; Smad site 7: forward, 5′-ATTCAAAAGTCCGTGGAAAG; reverse, 5′-GCTAATTCTGTCTTCAGAGCG-3′; 125-bp amplicon; and control primers flanking a distal promoter region with no putative binding sites (non-site): forward, 5′-TCACTCTCATCATTTCCCAC-3′; reverse, 5′-GGATTCTATGACTGTGTGTGG-3′; 125-bp amplicon. The QuantiTect SYBR Green PCR kit (Qiagen) was used to prepare PCRs according to the manufacturer's specifications. Dilutions of experimental input DNA were used for comparative analysis. Real time PCR conditions were: 95 °C for 15 min and 40 cycles of 94 °C for 15 s, 50 °C for 30 s, and 72 °C for 30 s. (See “Quantitative Real Time RT-PCR (qRT-PCR)” for PCR run and analysis.) All samples were run in triplicate, non-template controls were included in each run, and experiments were independently repeated at least three times.
Invasion Assays
BD BioCoatTM growth factor-reduced MatrigelTM invasion chambers (BD Biosciences) were used to conduct invasion assays according to the manufacturer's instructions. Briefly, cells were serum-starved for 12 h in either CCC media or Medium 254 containing 0.1% BSA. 100,000 cells were added to the top well of a 24-well growth factor-reduced Matrigel insert. Media added to the bottom well contained 10% fetal bovine serum. After 12 h (a 6–24-h time course was conducted to determine optimal time), cells that remained in the top chamber were removed with a cotton swab, the invaded cells were fixed in 1% paraformaldehyde, and DNA was labeled with Hoechst (1:500). The number of invading cells per field of view was counted on a Nikon ECLIPSE TE300 inverted microscope with an epifluorescence attachment under a 20× magnification. For seprase RNAi invasion experiments, cells were counted at 10× magnification based on their GFP fluorescence. All four cell lines had similar GFP expression/intensities. Experiments were independently repeated at least three times.
ELISA and Western Blotting
To quantitatively determine the amount of TGF-β1 produced by melanoma cell lines, the Quantikine Human TGF-β1 ELISA kit (R&D Systems) was used according to the kit instructions. Only activated (bioactive) TGF-β1 is recognized by the kit. Samples were acid-activated, and the readout indicated total TGF-β1 produced. To determine any bioactive TGF-β1 secreted, acid activation was skipped before conducting ELISAs. Experiments were performed at least three times. Western blotting was carried out as described previously (47) with minor modification. Whole-cell extracts were lysed in radioimmune precipitation assay buffer (25 mm Tris-HCl (pH 7.6), 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with PhosSTOP phosphatase inhibitor mixture tablets (Roche Applied Science), and Complete protease inhibitor mixture tablets (Roche Applied Science). The following antibodies were used: antibodies against actin (AC-40, Sigma), TGF-β RII (H-567), TGF-β RI (R-20), Smad4 (C-20), Smad2/3 (E-20), and Ski (H-329), all from Santa Cruz Biotechnology; phospho-Smad2 (138D4) from Cell Signaling Technology; and seprase (D8) generated in our laboratory and described previously (6).
Cell Treatments
Cells were treated for the indicated amount of time with either 2 or 5 ng/ml recombinant human TGF-β1 (R&D Systems), 20 ng/ml recombinant human latent TGF-β1 (R&D Systems), various concentrations of anti-TGF-β neutralizing antibody (R&D Systems), 10 μm MG132 (Sigma), or 10 μm SB-431542 (Sigma).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA). Two-way analysis of variance and unpaired t tests were used to determine statistical significance. Values shown are the means ± S.D. Statistical significance was defined as p < 0.05.
RESULTS
Identification of the Human Seprase Gene Promoter
To identify and clone the human seprase promoter, the genomic sequence 5′ of the initiation codon was determined using GenBankTM (NCBI), and putative promoter fragments were PCR-amplified starting from within the 5′-UTR to upward of about 2.6 kbp relative to ATG. Three fragments were generated: 2.637 kbp, 1357 bp, and 674 bp, all of which were identical in sequence to a region of chromosome 2 located 5′ of the seprase coding sequence (GenBankTM accession number NT_005403.17). An Incyte (Open Biosystems) complete full-length human seprase cDNA sequence was available for analysis, although it had no accession number. The sequence was an exact match to human FAPα mRNA (GenBankTM accession number NM_004460.2) with the exception of being 1 bp longer at the 5′-end and what we designate as the transcriptional start site (+1) (Fig. 1A). In comparison, this sequence is 168 bp longer on the 5′-end than the Mammalian Gene Collection seprase cDNA (GenBankTM accession number BC026250.1) and 55 bp longer on the 5′-end than the most abundant transcript found in the HeLa cell line identified by 5′ rapid amplification of cDNA ends (26). The full 5′-UTR of the human seprase gene identified spans 209 bp in length.
FIGURE 1.

Characterization of the human seprase gene 5′-regulatory region (promoter) in cell lines with differential seprase expression and invasive potential. A, DNA sequence of the 674-bp human seprase proximal promoter region spanning −469 to +205 and cloned upstream of the luciferase reporter gene. Black arrows designate the PCR amplified region. Essential promoter elements identified were the transcriptional start site (+1) based on the complete full-length human seprase cDNA sequence available, a TATA box at position −41 to −36, and a 209-bp 5′-UTR. Putative cis-regulatory elements are in bold and labeled according to which transcription factors putatively bind them. Putative Smad-binding elements are labeled 1–9 starting from 5′-end. B, relative seprase mRNA levels were determined by qRT-PCR in the panel of six human cell lines indicated. Seprase mRNA was normalized against β-actin levels for all samples. Results are the mean ± S.D. of three qRT-PCR results conducted in triplicate (n = 9). Error bars represent S.D. C, parallel transient transfections comparing reporter activity of seprase promoter regions pGL4-Sep2.637 (−2432/+205), pGL4-Sep1357 (−1152/+205), pGL4-Sep674 (−469/+205), and the pGL4 vector in the indicated cell lines. All samples were normalized for transfection efficiency by co-transfection with pRL-TK vector. Reporter activity is expressed as relative luciferase units (RLUs). Results are the mean ± S.D. of triplicate samples from two of three experiments (n = 6). Error bars represent S.D. D, representative images of the Matrigel invasion assay after 12 h. Magnification, 20×. E, analysis of the average (Avg) number of invading cells of three independent Matrigel invasion assays run in triplicate ±S.E. (***, p < 0.0001; n = 9). Error bars represent S.E.
Putative seprase promoter fragments were cloned upstream of the luciferase gene into pGL4 and labeled as pGL4-Sep2.637 (−2432 to +205), pGL4-Sep1357 (−1152 to +205), and pGL4-Sep674 (−469 to +205) and used in subsequent reporter assays. A canonical TATA box (−41 to −36) was readily identified 36 bp 5′ upstream of +1 (Fig. 1A) and found to be conserved between rat and human sequences (Fig. 2). MatInspectorTM software (Genomatix) was used to identify an array of putative cis-acting promoter elements. We systematically analyzed only the 674-bp putative seprase promoter because the transcriptional activity of this minimal 674-bp reporter was essentially equal to that of the larger versions (Fig. 1C). Putative canonical cis-elements found were an E-box (−331 to −326), a CCAAT/enhancer-binding protein site (−180 to −176), two Ets family sites (−53 to −50 and +75 to +78), an AP1 site (−27 to −21), and a lymphoid enhancer factor/T cell factor site (+113 to +120) (Fig. 1A).
FIGURE 2.

ClustalW2 alignment of the seprase gene promoter. ClustalW2 was used to align the 674-bp human seprase promoter with rat and mouse promoter sequences. Putative Smad cis-binding elements are outlined and labeled 1–9. Putative TATA boxes are also outlined.
We placed the MatInspector threshold low to identify sites resembling known Smad3/4 binding sequences, in essence screening the entire 674-bp putative promoter for possible TGF-β-responsive elements. As a result, nine putative Smad sites were identified: site 1 (−458 to −455), site 2 (−368 to −365), site 3 (−345 to −342), site 4 (−245 to −241), site 5 (−73 to −70), site 6 (+34 to +37), site 7A/B (+118 to +121/+122 to +125), site 8 (+149 to +152), and site 9 (+173 to +179) (Fig. 1A). The identification of putative Smad3/4 binding sites would be consistent with the induction of seprase expression in fibroblasts by TGF-β1 (29, 30) and further suggested that TGF-β may regulate the seprase gene at the transcriptional level.
Determination of Seprase as a Transcriptionally Regulated Gene
To test the hypothesis that the seprase gene is transcriptionally regulated, we conducted qRT-PCR for seprase gene expression using a panel of five cell lines and one type of primary cell. We found that both LOX and CCD-28SK cell lines had high levels of seprase mRNA relative to the A375 cell line, which had a moderate level, and to HEMa-LP, SK-MEL-28, and HaCat cells, which were devoid of seprase mRNA (Fig. 1B). Parallel transient transfections in these cells comparing pGL4-Sep2.637, pGL4-Sep1357, and pGL4-Sep674 reporters with the empty pGL4 vector demonstrated that all three cloned putative seprase promoter fragments allowed for very high transcriptional activity in three of the five cell lines tested (Fig. 1C), validating their ability to function as true gene promoters. Even the most minimal 674-bp promoter had ∼45-, 100-, and 92-fold increases in activity above pGL4 in A375, LOX, and CCD-28SK cell lines, respectively. More importantly, the pattern of expression of exogenous luciferase transcribed off of the cloned seprase promoters was similar to the endogenous seprase mRNA level in that same cell line tested (moderate/high/low/high/none) (Fig. 1, B and C). It should be noted that we did not graph results for primary HEMa-LP melanocytes because of the inability to efficiently transfect promoter constructs in these cells as compared with cell lines used; however, the trend seemed to show moderate control promoter activity but no seprase promoter activity (data not shown).
Because differences in transfection efficiency were normalized and because we observed that reporter levels were expressed to essentially the same level as that of endogenous seprase mRNA in the very same cell type tested, we determined that the seprase gene was regulated at the transcriptional level in these cells. Basal transcriptional regulation of seprase was also found in six different human cell lines (26) in agreement with our findings that seprase is a transcriptionally regulated gene.
In addition, treatment of cell lines with a 100 μm concentration of the RNA polymerase II inhibitor 5,6-dichlorobenzimidazole resulted in significant and sustained decreases in the level of seprase mRNA in as little as 30 min of treatment with continual decreases throughout the entire time course of 12 h. In contrast, the β-actin housekeeping gene had no significant decrease in mRNA until 10 h of treatment (data not shown). Taken together, these results strongly suggest that the transcriptional activity of the seprase promoter, as opposed to mRNA stability, turnover, or negative regulation by siRNAs/microRNAs, is the driving force behind the differential level of endogenous seprase mRNA observed.
Invasive Potential of Melanoma/Melanocyte Cells
Previous studies from our laboratory using melanoma cell lines (several that were used in this study) demonstrated that extracellular gelatin degradation and cellular invasiveness were directly related to seprase enzymatic activity (4). Therefore, we hypothesized that in vitro invasion of melanoma cell lines through an extracellular matrix such as Matrigel should coincide with seprase expression. To this end, the in vitro invasive/metastatic potentials of melanoma/normal cells were examined using Matrigel invasion chambers. Representative images of invaded cells fixed to the bottom of invasion membranes are shown in Fig. 1D. A375 cells were moderately invasive as compared with LOX cells, which demonstrated a rather high invasive potential. SK-MEL-28 cells, which were devoid of seprase mRNA, showed very little invasive capability as was the case for non-transformed HEMa-LP melanocytes, which were unable to invade across the extracellular matrix (Fig. 1D).
To quantitatively assess the invasion of cells examined, experiments were performed in triplicate, and three representative fields from each were counted and analyzed. A375 cells invaded at an average of ∼18 cells/field of view as compared with the LOX cell line, which invaded ∼2.7-fold higher on average (p < 0.0001, n = 9) at ∼49 cells/field of view (Fig. 1E). SK-MEL-28 cells were largely incapable of invading (average of one cell/field of view), which was ∼18- and ∼49-fold less on average than A375 and LOX cells, respectively (p < 0.0001, n = 9 for all) (Fig. 1E). HEMa-LP melanocytes displayed no invasive/metastatic potential without any evidence of any cellular invasion (Fig. 1E). Strikingly, the invasive potential of the cells seemed to be corollary to the level of seprase expression with cells with a higher expression of seprase, possibly enabling a more robust invasive capability.
Stably Reducing/Abolishing Seprase Expression by RNA Interference in Seprase-expressing Melanoma Cell Lines Inhibits Their Invasive Phenotype in Vitro
LOX pGUS-NT and LOX pGUS-Sep KD (pGUS-SEP1384) are previously generated cell lines with stable expression of hairpins against either NT or seprase, respectively, in addition to expressing GFP. The hairpin against seprase was found to be very specific, only targeting the gene of interest (10, 16). A375 pGUS-NT and A375 pGUS-Sep KD cell lines were generated in a similar manner. Western blotting using protein lysates from all four cell lines demonstrated that indeed efficient knockdown of seprase was achieved for both A375 pGUS-Sep KD and LOX pGUS-Sep KD cells lines relative to their wild-type counterparts (Fig. 3, A and C).
FIGURE 3.
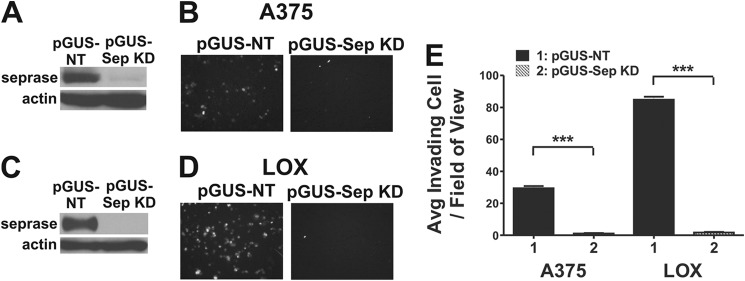
Stable RNAi of seprase in invasive melanoma cell lines inhibits their invasive phenotype in vitro. A, A375 cell lines were selected to stably express either a hairpin against a non-target (pGUS) or a hairpin against seprase (pGUS-Sep KD) in addition to expressing GFP. Western blotting was carried out using whole-cell extracts of A375 pGUS and A375 pGUS-Sep KD cells. Blots were probed with anti-seprase to confirm knockdown, and β-actin was used as a loading control. B, representative images of the Matrigel invasion assay after 12 h looking at GFP fluorescence. Magnification, 10×. C, same as A but with LOX pGUS and LOX pGUS-Sep KD cells (previously generated reagents). D, same as B but imaging LOX pGUS and LOX pGUS-Sep KD cells instead. E, analysis of the average (Avg) number of invading cells ±S.E. of a representative Matrigel invasion assay (***, p < 0.0001; n = 10). Error bars represent S.E.
The in vitro invasive potential of A375 and LOX seprase knockdown cell lines was examined using Matrigel invasion chambers. Representative images of invaded cells fixed to the bottom of invasion membranes are shown in Fig. 3, B and D. A375 pGUS-NT and LOX pGUS-NT cell lines readily invaded across the extracellular matrix, whereas both A375 pGUS-Sep KD and LOX pGUS-Sep KD cell lines had a diminished ability to do so. Cellular invasion of all four cell lines was quantitatively assessed, and a representative experiment was plotted in Fig. 3E. A375 pGUS-NT cells invaded at an average of ∼29 cells/field of view as compared with the A375 pGUS-Sep KD cell line, which invaded ∼29-fold less on average (p < 0.0001, n = 10) at ∼1 cell/field of view. LOX pGUS-NT cells invaded at an average of ∼84 cells/field of view as compared with the LOX pGUS-Sep KD cell line, which invaded at ∼1.7 cells/field of view (∼49-fold reduction in invasion on average) (p < 0.0001, n = 10). LOX pGUS-NT cells invaded ∼2.9-fold more on average as compared with the A375 pGUS-NT cell line.
Canonical TGF-β Signaling Pathway in A375 and LOX Melanoma Cell Lines
The differential expression of seprase in the closely matched A375 and LOX melanoma cell lines made the two cell lines very useful for examining the fine-tuned regulation of the seprase gene. Western blotting using protein lysates from both A375 and LOX cell lines revealed that all core proteins of the canonical TGF-β signaling machinery were expressed (Fig. 4A). Apparently equal levels of Smad2/3, Smad4, and TGF-β RI were expressed by each cell line. LOX cells expressed a higher level of TGF-β RII and had higher amounts of activated (phospho-) Smad2 indicative of higher TGF-β signaling that were consistent with the increased seprase mRNA/protein observed. In contrast, A375 cells expressed the TGF-β signaling transcriptional repressor c-Ski in a considerably higher amount as compared with LOX cells, which expressed negligible amounts of c-Ski (Fig. 4A).
FIGURE 4.

Analysis of the canonical TGF-β signaling pathway in invasive melanoma cell lines. A, Western blotting was performed using whole-cell extracts from A375 and LOX melanoma cell lines. The major signaling molecules in the canonical TGF-β pathway were analyzed for their expression using the specific antibodies indicated. B, TGF-β1 production by the indicated cell lines was assessed by ELISA. Cells were cultured in CCC media (in Medium 254 for HEMa-LP cells) at equal cell densities, washed three times in 1× PBS, and conditioned in serum-free (S.F.) CCC media for 24 h. Samples were acid-activated prior to ELISA to measure total TGF-β1 production in pg/ml (left panel). Bioactive TGF-β1 levels were also determined by skipping the acid activation step and are represented as pg/ml (right panel). Values were normalized to account for differences in cell number at the time of assay. Results depicted for each assessment are the mean ± S.D. of three ELISA assays (n = 9). Error bars represent S.D. C, activity of TGF-β signaling in A375 and LOX cells was determined by parallel transient transfection of the TGF-β-responsive reporter plasmid 4x-SBE-Luc into each cell line. Cells were untreated or treated with 2 ng/ml TGF-β1 for 6 h before luciferase activities were determined. Samples were normalized for transfection efficiency by co-transfection with pRL-TK vector. Reporter activity is expressed as relative luciferase units (RLUs). Results are the mean ± S.D. of triplicate samples from two of three experiments (n = 6). Error bars represent S.D. D, same as C but using (CAGA)9-MLP-Luc reporter. E, cells were treated or not with a specific TGF-β type I receptor inhibitor SB-431542 for 24 h, and seprase levels were determined by qRT-PCR. Values were normalized to β-actin. Results are the mean ± S.D. (n = 3 of one representative experiment of three conducted; ***, p < 0.001). Error bars represent S.D. F, A375 and LOX cell lines cultured in full CCC media were washed three times in PBS and treated or not with a neutralizing antibody against TGF-β1 in fresh CCC media for 24 h. Seprase levels were then determined by qRT-PCR. Values were normalized to β-actin. Results are the mean ± S.D. (n = 3 of one representative experiment of three conducted; **, p < 0.01; ***, p < 0.001; n.s., not significant). Error bars represent S.D. G, the in vitro invasive potential of A375 and LOX cell lines treated with or without 10 μm SB-431542 was examined using Matrigel invasion chambers. Cells were treated with the inhibitor from before serum starvation until the end point of the experiment. Cellular invasion was quantitatively assessed ±S.E., and shown is a representative experiment of three conducted (p < 0.0001, n = 10). Error bars represent S.E. P-Smad2, phospho-Smad2; TβR, TGF-β receptor.
Conditioned media from cells cultured in serum-free CCC media for 24 h were measured for total TGF-β1 (Fig. 4B, left) as well as activated TGF-β1 (Fig. 4B, right). LOX cells secreted a remarkable amount of TGF-β1, roughly 1500 pg/ml, which was ∼3-fold higher than A375 cells, ∼12-fold higher than SK-MEL-28 cells, and ∼97-fold higher than HEMa-LP cells. In addition, LOX cells produced ∼25 pg/ml bioactive TGF-β1, which was ∼3-fold higher than A375 cells, ∼9-fold higher than SK-MEL-28 cells, and ∼250-fold higher than HEMa-LP cells. These results are in agreement with the higher levels of activated R-Smad and lower levels of c-Ski found in LOX versus A375 cells, i.e. a more robust signal transduction.
To assess the functionality of TGF-β signaling, cell lines were tested for their ability to transactivate two different minimal TGF-β-responsive reporters in transient cell transfections upon stimulation with TGF-β1. As seen in Fig. 4, C and D, transcriptional activity was observed for both reporters in both seprase-expressing cell lines (untreated); the activity was ∼2-fold higher for each reporter in LOX versus A375 cells. This result is in agreement with the higher activity of TGF-β signaling detected in the LOX cell line. A stimulatory effect of TGF-β was observed with both reporters in both A375 and LOX cells upon addition of TGF-β1 for 6 h (∼4- and 2-fold increases in reporter activity, respectively) (Fig. 4, C and D). Transcriptional activity of reporters in treated conditions was essentially the same in each cell line, indicating that both cell lines are equally equipped to activate gene promoters in a TGF-β-dependent context.
Furthermore, we found that abrogating TGF-β signaling by either blocking the activity of TGF-β RI using the specific TGF-β type I receptor inhibitor SB-431542 (Fig. 4E) or depleting cells in culture of autocrine TGF-β1 using a neutralizing antibody against TGF-β (Fig. 4F) led to significant decreases in TGF-β-driven seprase transcription in both A375 and LOX cell lines.
Cell lines were treated with either DMSO or 10 μm SB-431542 for 24 h, and the resulting RNA was examined by qRT-PCR for seprase. We found that seprase mRNA levels were reduced ∼11-fold in A375 cells and ∼7-fold in LOX cells (p < 0.001, n = 3 for all) in SB-431542-treated versus untreated cells (Fig. 4E).
We also effectively blocked seprase transcription in both cell lines by using increasing concentrations (0.5–10 μg/ml) of a neutralizing antibody against TGF-β for 24 h. Cell lines were cultured and washed in PBS, and neutralizing antibody was added or not with fresh CCC media. A375 cells had significant reductions in seprase mRNA using as little as 0.5 μg/ml antibody (p < 0.01, n = 3). Levels were further reduced in cells treated with higher concentrations (p < 0.001, n = 3 for all) (Fig. 4F). Significant reductions in seprase mRNA were observed in LOX cells using 2 μg/ml antibody and were further reduced in cells treated with higher concentrations (p < 0.01, n = 3 for all) (Fig. 4F). Taken together, these results indicate that TGF-β signaling is a crucial pathway for seprase transcription in these metastatic melanoma cell lines.
Chemical Inhibition of TGF-β Signaling Using SB-431542 in Seprase-expressing Melanoma Cell Lines Inhibits Their Invasive Phenotype in Vitro
The in vitro invasive potential of A375 and LOX cell lines treated with or without 10 μm SB-431542 was examined using Matrigel invasion chambers. Cells were treated with the inhibitor from before serum starvation until the end point of the experiment. Cellular invasion was quantitatively assessed, and a representative experiment is plotted in Fig. 4G. A375 cells treated with DMSO invaded at an average of ∼19 cells/field of view as compared with A375 cells treated with SB-431542 that invaded ∼2.7-fold less on average at ∼7 cells/field of view (p < 0.0001, n = 10). LOX cells treated with DMSO invaded at an average of ∼45 cells/field of view as compared with LOX cells treated with SB-431542 that invaded ∼4-fold less on average at ∼11 cells/field of view (p < 0.0001, n = 10).
TGF-β1 Increases Endogenous Seprase mRNA Levels and Seprase Promoter Activity in Invasive Melanoma Cells
Treatment of A375 and LOX cell lines with TGF-β1 led to rapid and significant (p < 0.05, n = 6) increases in the level of seprase mRNA (∼2.5-fold) in 1 h of treatment for both cell lines (Fig. 5A). Seprase mRNA levels remained significantly higher (p < 0.05, n = 6 all time points) than basal expression and continually increased over time, displaying apparently similar seprase mRNA levels by 12–24 h of treatment (Fig. 5A). Strikingly, addition of TGF-β1 to both the non-invasive SK-MEL-28 cell line and non-transformed HEMa-LP cells resulted in no apparent increase in seprase mRNA at any time point, even up to 24 h of treatment (Fig. 5A).
FIGURE 5.

TGF-β1 increases endogenous seprase mRNA levels and seprase promoter activity in invasive melanoma cell lines. A, the cell lines indicated were serum-starved for 8 h and then untreated or treated with 2 ng/ml TGF-β1 over a 24-h time course. Relative seprase mRNA levels were determined by qRT-PCR for all time points as indicated (top panel). Seprase mRNA levels were normalized to β-actin levels for all samples for comparison. Results are the mean ± S.D. of triplicate samples from two of at least three experiments (n = 6; *, p < 0.05; ***, p < 0.001). Error bars represent S.D. B, the cell lines indicated were treated with identical conditions as described above, and their c-Ski protein expression was examined. Western blotting was carried out using protein lysates from the TGF-β1 time course experiment to assess c-Ski repressor protein levels in cell lines. (+)Cont represents lysate from SK-MEL-28 cells, and (−)Cont represents lysate from HEMa-LP cells for simple comparative purposes only. β-Actin was used as a loading control. C, the seprase promoter was assessed for TGF-β responsiveness in A375, LOX, and SK-MEL-28 cell lines by parallel transient transfection of pGL4-Sep2.637, pGL4-Sep1357, pGL4-Sep674, and pGL4 vector in each cell line. Cells were untreated or treated with 2 ng/ml TGF-β1 for 12 h before luciferase activities were determined. Samples were normalized for transfection efficiency by co-transfection with pRL-TK vector. Reporter activity is expressed as relative luciferase units (RLUs). Results are the mean ± S.D. of triplicate samples from two of three independent experiments (n = 6). Error bars represent S.D. D, A375 and LOX cell lines were serum-starved for 8 h and then untreated or treated with 20 ng/ml latent TGF-β1 over a 24-h time course. Relative seprase mRNA levels were determined by qRT-PCR and were normalized to β-actin levels for all samples for comparison. Results are the mean ± S.D. of triplicate samples from one of at least three experiments (n = 3; **, p < 0.001.). Error bars represent S.D.
Accompanying this increase in seprase mRNA was a concomitant abolishment of the c-Ski repressor protein in A375 cells (Fig. 5B). TGF-β-induced degradation of c-Ski in melanoma (including A375) as well as other cell lines has been described and is mediated by the ubiquitin-proteasomal degradation pathway (48–50). We infer that the removal of c-Ski, a known Smad transcriptional repressor, allows for this increased production of seprase mRNA, i.e. a higher amount of TGF-β-dependent transcription to take place. This would explain the differential seprase mRNA levels observed in the A375 and LOX cell lines and increases in seprase mRNA observed upon TGF-β treatment. Interestingly, SK-MEL-28 and HEMa-LP cells treated with TGF-β also had decreases in c-Ski repressor protein upon treatment (Fig. 5B), indicating that TGF-β signaling is intact and functioning in these cells despite their inability to up-regulate the seprase gene.
In addition, the LOX cell line was capable of processing latent TGF-β into bioactive TGF-β as treatment of cells with latent TGF-β1 led to significant increases in seprase mRNA by 12 h (p < 0.05, n = 3) that continued to increase upward of 24 h (p < 0.001, n = 3) of treatment (Fig. 5D). No significant increases in seprase mRNA were observed in A375 cells upon treatment with latent TGF-β1, indicating that these cells seemed to be incapable of processing latent TGF-β1 outside the cell. These observations could again explain the higher amount of TGF-β activity observed in the LOX cell line as compared with the A375 cell line.
To determine whether our cloned seprase promoter regions were TGF-β-responsive, parallel transient cell transfection experiments were conducted. We found that pGL4-Sep2.637, pGL4-Sep1357, and pGL4-Sep674 reporters all had robust transcriptional reporter activity as compared with pGL4 vector in both A375 and LOX cells but not in SK-MEL-28 cells and were all activated ∼1.5–2-fold higher in LOX versus A375 cells in the untreated condition (Fig. 5C). A stimulatory effect of TGF-β was observed for pGL4-Sep2.637, pGL4-Sep1357, and pGL4-Sep674 plasmids but not for pGL4 in both A375 and LOX cell lines in response to 12 h of treatment with TGF-β1 as compared with untreated cells (Fig. 5C). Seprase promoter plasmids were induced ∼6-fold in A375 cells and 4-fold in the LOX cell line in response to treatment. Reporters were also minimally induced in SK-MEL-28 cells upon TGF-β1 treatment to a similar level as that observed for the basal activity of reporters in A375 cells (Fig. 5C). Substantial increases in both endogenous seprase expression and human seprase promoter activity in response to TGF-β1 in invasive A375 and LOX cell lines strongly suggested that the seprase gene is regulated at the level of transcription in a TGF-β-dependent manner. Therefore, it became evident that the human seprase promoter must contain TGF-β-responsive regions.
Functional Analysis of the Human Seprase Promoter
Of the nine putative Smad3/4 cis-elements identified in the proximal 674-bp human seprase promoter (Fig. 2), five had consensus Smad-binding elements (SBEs) (45): sites 1, 2, 4, 7AB (two SBEs), and 9. Putative Smad sites 3, 6, and 8 were non-consensus but had the core (C/A)CAGA of the CAGA box element (46), and site 5 had only the core CAGA. Comparing the human, rat, and mouse 674-bp regions, we found that putative Smad sites 1, 5, 6, 7AB, and 8 were conserved across species (Fig. 2). Putative Smad site 1 and/or site 7AB, therefore, seemed likely to encompass the TGF-β-responsive region(s) as these sites were conserved across species and encompassed consensus SBE sequences.
WT pGL4-Sep674 and Smad site mutant reporters were assessed for transcriptional activity in transient cell transfection experiments in the LOX cell line. WT pGL4-Sep674 and pGL4-Sep674-mNon-site control (mutated at +64 to +67; no binding site) had comparable transcriptional activities that were well above pGL4 vector (∼50-fold) (Fig. 6A). The mutant TATA reporter had a ∼4-fold reduction in activity as compared with the WT promoter suggesting its function as a true TATA box (Fig. 6A). Interestingly, pGL4-Sep674-Smad promoter mutant sites 1, 2, 3, 4, 8, and 9 all consistently demonstrated no apparent loss in transcriptional activity as compared with pGL4-Sep674 (Fig. 6A). Smad site 5 and 6 mutants had modest albeit consistent reductions in promoter activity. Both of the pGL4-Sep674 Smad site 7 mutants (7A/7B) displayed ∼4-fold reductions in transcriptional activity as compared with pGL4-Sep674 (Fig. 6A), indicating that this region contains the major positive regulatory input for effective seprase promoter transactivation. All of the multiple binding site mutants in which Smad site 7 were mutated had reductions in activity similar to that of site 7AB mutant alone (Fig. 6B), suggesting that Smad site 7AB is necessary and sufficient to drive high level transcriptional activity in these melanoma cells. Similar results were obtained using the A375 cell line (data not shown).
FIGURE 6.
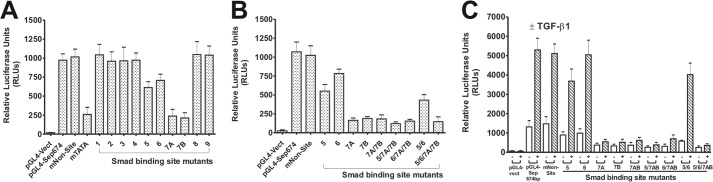
Functional analysis of putative SBEs within the human seprase promoter. A, site-directed mutagenesis was carried out to mutate all nine putative SBEs in the 674-bp (−469/+205) seprase promoter. To assess the transcriptional activity of resultant mutants, LOX cells were transfected with either the pGL4-Sep674 (WT) reporter construct or pGL4-Sep674 harboring individual mutations in SBEs 1–9, and luciferase assays were conducted. Controls were pGL4-Sep674 mNon-site and pGL4 vector (Vect). All samples were normalized for transfection efficiency by co-transfection with pRL-TK vector. Reporter activity is expressed as relative luciferase units (RLUs). Results are the mean ± S.D. of triplicate samples from one (representative) of three independent experiments. Error bars represent S.D. B, same as for A with the exception that only SBE mutants 5, 6, and 7 or double/triple/quadruple mutants of these SBEs were compared with WT. C, to map out the TGF-β-responsive region of the seprase promoter, LOX cells were transfected with either the pGL4-Sep674 (WT) reporter plasmid or SBE mutant 5, 6, or 7 or multiple SBE mutant versions. Controls were pGL4-Sep674 mNon-site and pGL4 vector. Cells were untreated or treated with 2 ng/ml TGF-β1 for 12 h before luciferase activities were determined. Samples were normalized for transfection efficiency by co-transfection with pRL-TK vector. Reporter activity is expressed as relative luciferase units (RLUs). Results are the mean ± S.D. of triplicate samples from one (representative) of three independent experiments. Error bars represent S.D.
To determine whether the TGF-β-induced increase in seprase promoter activity mapped to the Smad binding sites deemed important for overall transcription (sites 5, 6, and 7), we conducted parallel transient cell transfection experiments in LOX cells with or without TGF-β1 addition for 12 h. A stimulatory effect of TGF-β1 (∼4–5-fold) was observed for both WT pGL4-Sep674 and pGL4-Sep674-mNon-site promoters but not with pGL4 (Fig. 6C). The Smad site 5 mutant reporter was still induced very strongly in response to TGF-β1 (∼3.5-fold) albeit to a lesser extent than the WT promoter, suggestive of low TGF-β-responsiveness (Fig. 6C). The Smad site 6 mutant promoter was strongly activated (∼5-fold) upon TGF-β1 treatment very similarly to the WT promoter, thus ruling this site out as a TGF-β-responsive region.
The most dramatic effect was observed for the Smad site 7 promoter mutants as mutation of these sites (7A/7B/7AB) abolished the TGF-β-induced transcriptional activation of the seprase promoter, reducing levels to near basal activity (Fig. 6C). Both sites 7A and 7B are necessary for TGF-β-responsiveness as mutation of either abolished TGF-β-induced activation to the same degree as the site 7A/7B double mutant. All double/triple/quadruple mutants that contained the Smad site 7 mutation were incapable of transactivating the seprase promoter in response to TGF-β (Fig. 6C). Similar results were obtained in the A375 cell line (data not shown). The major TGF-β-responsive region in the human seprase promoter is therefore Smad site 7AB (+118 to +125) with the sequence 5′-AGACAGAC-3′.
TGF-β1-induced Binding of Smad3/4 to the TGF-β-responsive Region in the Seprase Promoter in Vivo
ChIP assays were conducted to determine Smad3, Smad4, and c-Ski binding to the TGF-β-responsive regions of the proximal seprase promoter. Cell lines were serum-starved for 8 h followed by readdition of CCC media and treated or not with 5 ng/ml TGF-β1 for an additional 8 h. The distal seprase promoter region (non-site) had no enrichment of Smad3/4 or c-Ski binding compared with IgG in any cell lines in either condition (Fig. 7, A, B, C, and D). Despite showing low TGF-β-responsiveness in reporter experiments, we found no enrichment of Smad 3/4 binding to the putative Smad site 5 in either treated or untreated cells as compared with the IgG control (Fig. 7, A and B). Therefore, we concluded that putative Smad site 5 is not a true Smad3/4 binding site and that the low level TGF-β-responsiveness observed is Smad-independent.
FIGURE 7.

Assessment of Smad3/4 and c-Ski binding to the TGF-β-responsive element of the human seprase promoter in invasive and non-invasive cells. A, A375 melanoma cells were serum-starved for 8 h followed by treatment with or without 5 ng/ml TGF-β1 in CCC media for an additional 8 h and then collected for ChIP analysis. Antibodies used for ChIP were anti-Smad2/3, anti-Smad4, ant-c-Ski, and anti-IgG. Promoter regions examined were putative SBE site 5 (low TGF-β responsiveness), putative SBE site 7 (high TGF-β responsiveness), and non-site (negative control; distal promoter region). Enrichment was assessed by real time PCR and is represented as percentage of input. Results are the mean ± S.D. of triplicate samples from two of three independent experiments. Error bars represent S.D. B, same as A but using the LOX cell line. C, same as A but using the SK-MEL-28 cell line. D, same as A but using primary HEMa-LP cells.
Putative Smad site 7AB was highly enriched in binding for Smad3 and Smad4 as compared with IgG control in non-stimulated A375 and LOX cell lines and more so in LOX cells by comparison but absent in SK-MEL-28 and HEMa-LP cells (Fig. 7, A, B, C, and D). Putative Smad site 7AB is thus a bona fide SBE both highly capable of binding Smads in vivo and of driving TGF-β-dependent transcription of the human seprase gene in invasive melanoma cell lines. Additional TGF-β1 treatment increased binding of both Smad3 and Smad4 to the SBE in both A375 and LOX cell lines, demonstrating that site 7AB is a TGF-β-inducible promoter in vivo (Fig. 7, A and B). An enrichment of c-Ski repressor protein binding to Smad site 7AB was found in A375 cells as compared with the IgG control in the untreated condition; however, no c-Ski binding was observed in LOX, SK-MEL-28, and HEMa-LP cells (Fig. 7, A, B, C, and D). Furthermore, c-Ski binding was lost (reduced to IgG levels) in A375 cells treated with TGF-β1 (Fig. 7A), consistent with Western data showing the TGF-β-driven abolishment of c-Ski in these cells.
ChIP results are in agreement with the differences in seprase expression observed in A375, LOX, SK-MEL-28, and HEMa-LP cells; i.e. a higher amount of Smad3/4 binding to the SBE (higher TGF-β activation) was seen in LOX cells as compared with A375 cells. Consistently, c-Ski repressor binding was found on the SBE in A375 cells but was absent in LOX cells. These experiments also demonstrated that complexes of c-Ski and Smads are formed on the SBE of the seprase promoter in vivo. We presume this because c-Ski has no intrinsic DNA binding capability and therefore binds to Smads bound to SBEs (51–53).
Genetic Inhibition of Seprase Transcription by Stable Overexpression of c-Ski
Upon binding to the seprase promoter, c-Ski could exert its inhibitory effects in a variety of ways such as through the stabilization of inactive Smad complexes on the SBE, by blocking Smad recruitment of transcriptional co-activators such as p300/CREB-binding protein, or by recruitment of nuclear receptor co-repressor complexes (51, 54–56). In addition, c-Ski protein could prevent transcription by direct interactions with Smad3 or Smad4 or by disruption of the formation of crucial R-Smad·co-Smad complexes (56, 57). To determine whether c-Ski was truly repressing seprase transcription, A375 and LOX cell lines were infected with either pBabe-Puro (BP) or pBabe-Puro/Ski virus, and stable lines were selected for further characterization.
Results of qRT-PCR indicated that c-Ski mRNA levels for both A375 BP-Ski and LOX BP-Ski cell lines were quite comparable (Fig. 8A). Endogenous c-Ski mRNA levels were much higher in the A375 cell line as compared with the LOX cell line (Fig. 8A). When we looked at corresponding c-Ski protein levels, we found that A375 BP-Ski cells expressed extremely high amounts as compared with LOX BP-Ski cells, which expressed far less (Fig. 8B). However, the level of c-Ski expressed in LOX BP-Ski cells was much higher than that in LOX BP cells and slightly higher than that in A375 BP cells. We carried out qRT-PCR to determine whether overexpression of c-Ski in A375 and LOX cell lines blocked seprase transcription. Indeed, both A375 BP-Ski and LOX BP-Ski cell lines had significant reductions in seprase mRNA levels as compared with their BP counterparts (Fig. 8C). Seprase mRNA levels in A375 BP-Ski cells were reduced ∼11-fold (p < 0.001, n = 6) to the basal state level, whereas seprase mRNA levels in LOX BP-Ski cells were reduced ∼1.9-fold (p = 0.0032, n = 6) (Fig. 8C). The amount of seprase mRNA in LOX BP-Ski cells was reduced comparably with the levels seen in A375 BP cells (∼1.5-fold difference). In addition, parallel transient transfections of WT pGL4-Sep674 in both A375 BP-Ski and LOX BP-Ski cell lines resulted in decreases in promoter activity as compared with BP cells (data not shown).
FIGURE 8.

c-Ski mediated repression of seprase transcription in A375 and LOX melanoma cell lines. A and B, A375 and LOX cell lines were selected to stably overexpress empty vector (BP) or exogenous c-Ski (BP-Ski). Relative c-Ski mRNA levels in these cell lines were determined by qRT-PCR, and values were normalized to the β-actin level (A). Results are the mean ± S.D. (n = 3). Error bars represent S.D. Western blotting was carried out using whole-cell extracts of LOX BP/BP-Ski cells and A375 BP/BP-Ski cells. Blots were probed with anti-c-Ski to confirm overexpression. β-Actin served as a loading control (B). C, effect of c-Ski overexpression on seprase mRNA levels was determined by qRT-PCR for seprase. All values were normalized to the β-actin level. Results are the mean ± S.D. of two qRT-PCR results conducted in triplicate (n = 6; **, p < 0.01; ***, p < 0.001). Error bars represent S.D. D, ChIP assays were conducted to determine Smad3 and c-Ski binding to the major TGF-β-responsive region (SBE site 7) in both A375 BP-Ski and LOX BP-Ski cell lines. Enrichment of binding was assessed by real time PCR and is represented as percentage of input. Results are the mean ± S.D. of triplicate samples from two of three independent experiments. Error bars represent S.D. E and F, to test whether c-Ski is degraded in the LOX cell line, LOX BP and BP-Ski cell lines were treated with the proteasomal inhibitor MG132 for 1 h. The effect of the stabilization of c-Ski on seprase mRNA levels was determined by qRT-PCR for seprase (E). All values were normalized to the β-actin level. Results are the mean ± S.D. of two quantitative RT-PCR results conducted in triplicate (***, p < 0.001). Error bars represent S.D. Western blotting was conducted to assess c-Ski protein levels. β-Actin was used as the loading control (F). G, Western blotting was carried out using whole-cell extracts of LKO1-GFP-KD (negative control for our purposes) and LKO1-SKI-KD (RNAi against c-Ski) stable A375 cell lines. Blots were probed with anti-c-Ski to confirm knockdown. β-Actin served as a loading control. H, the indicated cell lines were analyzed for seprase mRNA levels by qRT-PCR. All values were normalized to the β-actin level. Results are the mean ± S.D. of two quantitative RT-PCR results conducted in triplicate (*, p < 0.05). Error bars represent S.D.
To confirm that the c-Ski-mediated reduction in seprase mRNA was at the level of the seprase promoter, i.e. transcriptional repression, we conducted ChIP assays using A375 BP-Ski and LOX BP-Ski cell lines. A375 BP-Ski cells had enriched Smad3 binding to the seprase promoter SBE site 7 as compared with control IgG pulldown (Fig. 8D). SBE site 7 was also highly occupied by c-Ski protein as compared with IgG in these cells, and the amount of c-Ski binding was equal to that of Smad3 binding (Fig. 8D). These results indicate that on essentially every SBE that was bound by Smad3, the site was also bound by c-Ski, rendering the promoter inactive. LOX BP-Ski cells were also highly enriched for Smad3 binding on SBE site 7 as compared with the IgG control (Fig. 8D). In addition, c-Ski binding was enhanced ∼3.5-fold on the seprase promoter SBE as compared with IgG (Fig. 8D). The level of overexpression of c-Ski was high enough that we observed a previously unseen occupancy on the seprase promoter SBE by the repressor and a concomitant ∼1.9-fold reduction in seprase mRNA in LOX BP-Ski cells.
We next tested whether or not c-Ski protein was degraded in the LOX cell line via proteasomal degradation mediated by TGF-β signaling as was seen in other melanoma cell lines (48). If c-Ski protein was being degraded in such a manner, it would account for the differences found in the level of c-Ski mRNA and corresponding amount of c-Ski protein in LOX BP-Ski cells. To test this, a 10 μm concentration of the proteasome inhibitor MG132 was added to both LOX BP and LOX BP-Ski cell lines for 1 h. Accumulation of c-Ski protein upon MG132 treatment profoundly inhibited seprase transcription, reducing levels of seprase mRNA ∼13-fold in LOX BP-Ski cells as compared with LOX BP cells (p < 0.001, n = 6) (Fig. 8E). Western blotting showed extremely high levels of stabilized c-Ski protein in the LOX BP-Ski overexpressor (Fig. 8F) similar to levels found in A375 BP-Ski cells and much more in line with the c-Ski mRNA level produced in these cells. These results suggested that the LOX cell line, which produces an abundance of TGF-β1, has all the necessary machinery for constant and efficient TGF-β-induced c-Ski degradation and, therefore, seprase transcription.
RNAi-mediated Knockdown of c-Ski in A375 Cells
To complement c-Ski overexpression studies, loss-of-function experiments in A375 cells were also conducted. Western blotting using protein lysates from A375 LKO1-GFP-KD (negative control) and A375 LKO1-SKI-KD cells demonstrated that efficient knockdown of c-Ski was achieved (Fig. 8G). Results of qRT-PCR indicated an ∼2-fold increase (p < 0.05, n = 6) in the seprase mRNA level in A375 LKO1-SKI-KD cells as compared with A375 LKO1-GFP-KD cells (Fig. 8H). These results are in agreement with the findings of Fig. 5, A and B, demonstrating that upon TGF-β treatment the negative regulator c-Ski is degraded, resulting in an increase in seprase transcription.
DISCUSSION
In this study, we cloned the human seprase promoter and identified key features of the core promoter such as a TATA box 36 bp upstream of the transcriptional start site, a 209-bp 5′-UTR, and numerous putative cis-regulatory elements. Zhang et al. (26) were unable to identify the TATA box in the mouse seprase promoter as it is not conserved between mouse and human, which they aligned, but is in fact conserved between rat and human as we found. In addition, we classified the transcriptional start (+1) site based on the Incyte complete full-length human seprase cDNA sequence as opposed to the most abundant transcript found in HeLa cells (26) and therefore were able to locate the TATA box in the expected 30–40-bp range upstream of +1.
It is also noteworthy to mention that although Zhang et al. (26) cloned the mouse seprase promoter the region they identified as important for basal seprase transcription has 80% sequence homology between the human and mouse seprase gene and contains a conserved EGR1 binding site. In our studies, we identified a crucial Smad3/4 SBE and determined its role in TGF-β-dependent seprase transcription in invasive melanoma cell lines, validating several independent studies using fibroblasts showing that TGF-β1 induced seprase protein expression (29, 30).
We provide evidence that the seprase gene is transcriptionally regulated in a pair of invasive human malignant melanoma cell lines with differential levels of seprase mRNA. A375 cells had a moderate level of seprase mRNA, whereas the LOX cell line had an ∼3-fold higher amount. Non-invasive SK-MEL-28 and primary HEMa-LP cells had little to no seprase mRNA by comparison. Seprase promoter reporters had ∼2.5-fold higher promoter activity in the LOX cell line than in the A375 cell line and ∼27-fold higher promoter activity than in SK-MEL-28 cells. In addition, HaCat keratinocytes expressed no detectable seprase mRNA and had no seprase promoter activity. CCD-28SK fibroblast cells expressed high levels of seprase mRNA and had high seprase promoter activity. Taken together, these results suggested that the observed seprase transcriptional regulation occurs in a variety of cell types, including differentially in invasive versus non-invasive melanoma cell lines.
Several studies have demonstrated the increased expression and secretion of TGF-β isoforms in melanoma cell lines in comparison with normal melanocytes (58–62). In addition, human metastatic melanoma cell lines were found to be more resistant to TGF-β-dependent growth inhibition as compared with cells from primary tumors (60). Despite resistance to TGF-β-induced growth arrest, TGF-β-resistant melanoma cell lines still exhibit intact Smad-dependent regulation of gene expression in response to TGF-β that occurs in an autocrine manner (63). In agreement with these observations, we found both A375 and LOX melanoma cell lines to be highly invasive and that both cell lines secreted very high amounts of TGF-β1 as compared with non-invasive SK-MEL-28 and primary HEMa-LP cells. LOX cells produced ∼3-fold more of the ligand than A375 cells, ∼12-fold more than SK-MEL-28 cells, and ∼97-fold more than HEMa-LP cells.
In addition to producing more of the ligand, LOX cells also expressed ∼4-fold more of the TGF-β type II receptor protein than A375 cells. LOX cells, which produce higher amounts of TGF-β1 and express higher levels of the TGF-β type II receptor as compared with A375 cells, would in theory have a more robust autocrine signal transduction capability. In accordance with these observations, the LOX cell line had higher levels of phospho-Smad2, suggesting that TGF-β signaling is more active in LOX cells as compared with A375 cells. Additionally, we found a difference in the level of the c-Ski transcriptional repressor protein, which was expressed in a far greater abundance in A375 cells as compared with LOX cells.
We also found that both A375 and LOX cells had functional TGF-β signaling as determined by their ability to transactivate two different minimal Smad-responsive reporters, validating that each cell line could signal in an autocrine manner. LOX cells induced higher expression of the reporters than A375 cells, although both cells transactivated the reporters equally well in response to TGF-β1 treatment. SK-MEL-28 cells had very low level reporter activity in the basal state and were induced in response to TGF-β1 treatment (data not shown). A 674-bp region of the seprase promoter (−469 to +205) was sufficient to drive high level expression of the reporter in cell lines tested and was also found to be highly responsive to TGF-β1 in A375 and LOX cells but not in SK-MEL-28 cells, which only saw minimal increases in reporter activity.
In agreement with reporter results, treatment of A375 and LOX cells with TGF-β1 for 1 h led to rapid and significant increases in the level of endogenous seprase mRNA and a decrease in the amount of c-Ski repressor, which were observed for all time points tested. Very strikingly, however, non-invasive SK-MEL-28 and HEMa-LP cells saw no such increases in seprase mRNA level even upward of 24 h of treatment with TGF-β1. This, despite the fact that TGF-β was able to abolish the expression of c-Ski in these cells, indicates that although TGF-β signaling is functioning in these cells it is incapable of transactivating or targeting the seprase promoter. Neither positive (Smads) nor negative (c-Ski) regulators were able to bind the seprase promoter in vivo as determined in ChIP assays. These results are highly intriguing and lead us to hypothesize that either there are additional factors (repressors/activators) present or absent in invasive versus non-invasive cells or that the seprase promoter is accessible in invasive but not non-invasive melanoma cells perhaps through one form of epigenetic regulation/silencing or another. Using EMBOSS Cpgplot, we were unable to locate any putative CpG island near the proximal seprase promoter, although we did locate a putative CpG island in a non-coding region, −19421 to −19126 relative to the human seprase gene transcriptional start site. It is possible that the methylation status of this putative CpG island alters the binding or not of some long distance enhancer/repressor protein crucial to seprase transcription. Future studies need to be conducted to test either hypothesis.
Because TGF-β1 signaling ultimately results in the activation of the Smad transcription factors, which are then able to translocate to the nucleus and alter gene expression (32–34), we interrogated nine different putative Smad-binding elements in the cloned seprase promoter by mutagenesis and subsequently determined promoter activities of the resulting mutants as compared with the WT seprase promoter. Using this approach, we mapped the TGF-β-responsive region of the promoter to the +118 to +125 portion of the gene promoter, which encompassed two putative and consecutive SBEs (site 7AB; 5′-AGACAGAC-3′). Mutation of either one these SBEs reduced the activity of the promoter to similar low/basal levels, suggesting that they are functionally redundant sites. Furthermore, mutation of the SBEs rendered the promoter completely insensitive to TGF-β1 stimulation, indicating that SBE site 7AB is both necessary and sufficient for TGF-β-dependent seprase transcription.
A significant amount of Smad3 and Smad4 binding to SBE site 7AB was observed in A375 and LOX cells as both cells had active TGF-β signaling and expressed seprase mRNA. We found no Smad3/4 binding in either SK-MEL-28 or HEMa-LP cells in full agreement with their inability to transcribe seprase mRNA. In addition, there was an enhanced binding of the Smads to SBE site 7AB in LOX cells as compared with A375 cells, suggesting increased transcription of seprase in LOX cells. Consistently, we found binding of the c-Ski repressor protein to SBE site 7AB in A375 cells but not in LOX cells. The binding of c-Ski was not enriched to the same degree as Smad3/4 in these cells, suggesting that not all seprase gene promoters occupied by Smad3/4 on SBE site 7AB were equally occupied by c-Ski. The very low level expression of the c-Ski repressor and lack of binding to SBE site 7AB in LOX cells would explain the higher seprase mRNA produced in LOX cells than in A375 cells. Interestingly, we observed no c-Ski binding to SBE site 7AB in either SK-MEL-28 or HEMa-LP cells despite the fact that SK-MEL-28 cells express c-Ski to a similar level as seen in A375 cells.
Addition of exogenous TGF-β1 to both A375 and LOX cells caused increases in binding of Smad3 and Smad4 to SBE site 7AB as compared with the untreated counterparts. We found no such binding of Smad3 or Smad4 to SBE site 7AB in either SK-MEL-28 or HEMa-LP cells in the presence of exogenous TGF-β, indicating that even with functional TGF-β signaling initiated the seprase promoter was incapable of being targeted by Smads in these non-invasive cells. Together, our results demonstrated that the seprase promoter was TGF-β-inducible in invasive but not non-invasive melanoma cells.
In addition, c-Ski repressor protein levels were abolished in response to TGF-β1 stimulation in all cell lines tested, and c-Ski binding to the seprase promoter was severely diminished in A375 cells. Overexpression of c-Ski caused significant decreases in both the level of endogenous seprase mRNA and seprase promoter activity in both A375 and LOX cell lines. In addition, c-Ski binding to SBE site 7AB was enhanced, demonstrating that the seprase promoter was directly targeted by this repressor to inhibit TGF-β-mediated gene transcription. The results suggested that c-Ski was predominantly repressing the seprase promoter through either the recruitment of additional corepressors, blockade of transcriptional activators, or the formation of inactive Smad complexes on the SBE (44, 51, 54, 55). If c-Ski prevented or interfered with Smad complex formation (56, 57) or prevented R-Smad phosphorylation (64), we would expect a more diminished binding of Smad3/4 to the SBE as a result of the inability of the Smads to enter the nucleus and/or bind to DNA, but this was not observed. Similar to A375 cells, LOX cells were capable of TGF-β-induced ubiquitin-dependent proteasomal degradation of c-Ski as addition of MG132 stabilized c-Ski protein significantly in LOX c-Ski-overexpressing cells and caused a more severe repression of seprase transcription accordingly. Complimentary to these findings, stable RNAi against c-Ski in A375 cells allowed for a more robust signal transduction and therefore increased the level of seprase transcription accordingly.
Our findings of an increased level of seprase transcription (seprase mRNA) in the LOX cell line as compared with the A375 cell line is in full agreement with a previous study from our laboratory demonstrating increased seprase protein expression, invadopodium formation, gelatinase activity, and resulting invasiveness in LOX versus A375 cells (65). In accordance with these observations, abolishing seprase expression via RNAi inhibited the in vitro invasive capability of A375 and LOX melanoma cell lines quite profoundly as did blocking TGF-β signaling using the chemical inhibitor SB-431542. These findings are in line with previous studies from our laboratory (4) and others (66), indicating that the level of seprase expression and resulting seprase activity are crucial for melanoma cell migration and/or invasion.
In conclusion, we found that seprase, which was cloned from the LOX human metastatic melanoma cell line (5, 6) and is associated with increased cell invasiveness (4), is transcriptionally up-regulated in invasive but not non-invasive human melanoma cells or primary melanocytes via the canonical TGF-β signaling pathway. Although non-invasive melanocytes/melanoma cells have apparently functional TGF-β signaling as determined by their ability to degrade the negative regulator c-Ski in the presence of exogenous TGF-β, the seprase promoter was incapable of being targeted by Smads in these cells. Therefore, it is not only the increased production of TGF-β1 by invasive melanoma cells as compared with non-invasive melanocytes/melanoma cells but also the apparent accessibility of the seprase promoter that enables efficient and constitutive transcription of the gene.
Acknowledgments
We thank Dr. K. Luo for pBabe-Puro and pBabe-Puro/Ski plasmids and Dr. M. Sadelain for GPG29 cells. We also thank S. Tulley's dissertation committee (in particular Dr. M. Hayman) for helpful advice, review, and discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA0039077 and R01EB002065 (to W.-T. C.).
- FAPα
- fibroblast activation protein-α
- R-Smad
- receptor-regulated Smad
- SBE
- Smad-binding element
- HEMa-LP
- human epidermal melanocytes isolated from lightly pigmented skin
- CCC
- cancer cell culture
- KD
- knockdown
- NT
- non-target
- qRT-PCR
- quantitative real time RT-PCR
- TGF-β R
- TGF-β receptor
- CREB
- cAMP-response element-binding protein
- BP
- pBabe-Puro
- SKI
- Sloan-Kettering Institute protein.
REFERENCES
- 1. O'Brien P., O'Connor B. F. (2008) Seprase: an overview of an important matrix serine protease. Biochim. Biophys. Acta 1784, 1130–1145 [DOI] [PubMed] [Google Scholar]
- 2. Scanlan M. J., Raj B. K., Calvo B., Garin-Chesa P., Sanz-Moncasi M. P., Healey J. H., Old L. J., Rettig W. J. (1994) Molecular cloning of fibroblast activation protein α, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. U.S.A. 91, 5657–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mathew S., Scanlan M. J., Mohan Raj B. K., Murty V. V., Garin-Chesa P., Old L. J., Rettig W. J., Chaganti R. S. (1995) The gene for fibroblast activation protein α (FAP), a putative cell surface-bound serine protease expressed in cancer stroma and wound healing, maps to chromosome band 2q23. Genomics 25, 335–337 [DOI] [PubMed] [Google Scholar]
- 4. Aoyama A., Chen W. T. (1990) A 170-kDa membrane-bound protease is associated with the expression of invasiveness by human malignant melanoma cells. Proc. Natl. Acad. Sci. U.S.A. 87, 8296–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldstein L. A., Ghersi G., Piñeiro-Sánchez M. L., Salamone M., Yeh Y., Flessate D., Chen W. T. (1997) Molecular cloning of seprase: a serine integral membrane protease from human melanoma. Biochim. Biophys. Acta 1361, 11–19 [DOI] [PubMed] [Google Scholar]
- 6. Piñeiro-Sánchez M. L., Goldstein L. A., Dodt J., Howard L., Yeh Y., Tran H., Argraves W. S., Chen W. T. (1997) Identification of the 170-kDa melanoma membrane-bound gelatinase (seprase) as a serine integral membrane protease. J. Biol. Chem. 272, 7595–7601 [DOI] [PubMed] [Google Scholar]
- 7. Garin-Chesa P., Old L. J., Rettig W. J. (1990) Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. U.S.A. 87, 7235–7239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park J. E., Lenter M. C., Zimmermann R. N., Garin-Chesa P., Old L. J., Rettig W. J. (1999) Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 274, 36505–36512 [DOI] [PubMed] [Google Scholar]
- 9. Rettig W. J., Chesa P. G., Beresford H. R., Feickert H. J., Jennings M. T., Cohen J., Oettgen H. F., Old L. J. (1986) Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res. 46, 6406–6412 [PubMed] [Google Scholar]
- 10. Chen D., Kennedy A., Wang J. Y., Zeng W., Zhao Q., Pearl M., Zhang M., Suo Z., Nesland J. M., Qiao Y., Ng A. K., Hirashima N., Yamane T., Mori Y., Mitsumata M., Ghersi G., Chen W. T. (2006) Activation of EDTA-resistant gelatinases in malignant human tumors. Cancer Res. 66, 9977–9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goscinski M. A., Suo Z., Flørenes V. A., Vlatkovic L., Nesland J. M., Giercksky K. E. (2008) FAP-α and uPA show different expression patterns in premalignant and malignant esophageal lesions. Ultrastruct. Pathol. 32, 89–96 [DOI] [PubMed] [Google Scholar]
- 12. Huang Y., Simms A. E., Mazur A., Wang S., León N. R., Jones B., Aziz N., Kelly T. (2011) Fibroblast activation protein-alpha promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin. Exp. Metastasis 28, 567–579 [DOI] [PubMed] [Google Scholar]
- 13. Huber M. A., Kraut N., Park J. E., Schubert R. D., Rettig W. J., Peter R. U., Garin-Chesa P. (2003) Fibroblast activation protein: differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Invest. Dermatol. 120, 182–188 [DOI] [PubMed] [Google Scholar]
- 14. Iwasa S., Jin X., Okada K., Mitsumata M., Ooi A. (2003) Increased expression of seprase, a membrane-type serine protease, is associated with lymph node metastasis in human colorectal cancer. Cancer Lett. 199, 91–98 [DOI] [PubMed] [Google Scholar]
- 15. Jin X., Iwasa S., Okada K., Mitsumata M., Ooi A. (2003) Expression patterns of seprase, a membrane serine protease, in cervical carcinoma and cervical intraepithelial neoplasm. Anticancer Res. 23, 3195–3198 [PubMed] [Google Scholar]
- 16. Kennedy A., Dong H., Chen D., Chen W. T. (2009) Elevation of seprase expression and promotion of an invasive phenotype by collagenous matrices in ovarian tumor cells. Int. J. Cancer 124, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang R. F., Zhang L. H., Shan L. H., Sun W. G., Chai C. C., Wu H. M., Ibla J. C., Wang L. F., Liu J. R. (2013) Effects of the fibroblast activation protein on the invasion and migration of gastric cancer. Exp. Mol. Pathol. 95, 350–356 [PubMed] [Google Scholar]
- 18. Yang W., Han W., Ye S., Liu D., Wu J., Liu H., Li C., Chen H. (2013) Fibroblast activation protein-α promotes ovarian cancer cell proliferation and invasion via extracellular and intracellular signaling mechanisms. Exp. Mol. Pathol. 95, 105–110 [DOI] [PubMed] [Google Scholar]
- 19. Kelly T., Kechelava S., Rozypal T. L., West K. W., Korourian S. (1998) Seprase, a membrane-bound protease, is overexpressed by invasive ductal carcinoma cells of human breast cancers. Mod. Pathol. 11, 855–863 [PubMed] [Google Scholar]
- 20. Zhang M. Z., Qiao Y. H., Nesland J. M., Trope C., Kennedy A., Chen W. T., Suo Z. H. (2007) Expression of seprase in effusions from patients with epithelial ovarian carcinoma. Chin. Med. J. 120, 663–668 [PubMed] [Google Scholar]
- 21. Henry L. R., Lee H. O., Lee J. S., Klein-Szanto A., Watts P., Ross E. A., Chen W. T., Cheng J. D. (2007) Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 13, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 22. Wikberg M. L., Edin S., Lundberg I. V., Van Guelpen B., Dahlin A. M., Rutegård J., Stenling R., Oberg A., Palmqvist R. (2013) High intratumoral expression of fibroblast activation protein (FAP) in colon cancer is associated with poorer patient prognosis. Tumour Biol. 34, 1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan D., Liu B., Liu K., Zhu G., Dai Z., Xie Y. (2013) Overexpression of fibroblast activation protein and its clinical implications in patients with osteosarcoma. J. Surg. Oncol. 108, 157–162 [DOI] [PubMed] [Google Scholar]
- 24. Cohen S. J., Alpaugh R. K., Palazzo I., Meropol N. J., Rogatko A., Xu Z., Hoffman J. P., Weiner L. M., Cheng J. D. (2008) Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 37, 154–158 [DOI] [PubMed] [Google Scholar]
- 25. Mori Y., Kono K., Matsumoto Y., Fujii H., Yamane T., Mitsumata M., Chen W. T. (2004) The expression of a type II transmembrane serine protease (seprase) in human gastric carcinoma. Oncology 67, 411–419 [DOI] [PubMed] [Google Scholar]
- 26. Zhang J., Valianou M., Cheng J. D. (2010) Identification and characterization of the promoter of fibroblast activation protein. Front. Biosci. (Elite Ed.) 2, 1154–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dolznig H., Schweifer N., Puri C., Kraut N., Rettig W. J., Kerjaschki D., Garin-Chesa P. (2005) Characterization of cancer stroma markers: in silico analysis of an mRNA expression database for fibroblast activation protein and endosialin. Cancer Immun. 5, 10. [PubMed] [Google Scholar]
- 28. Milner J. M., Kevorkian L., Young D. A., Jones D., Wait R., Donell S. T., Barksby E., Patterson A. M., Middleton J., Cravatt B. F., Clark I. M., Rowan A. D., Cawston T. E. (2006) Fibroblast activation protein α is expressed by chondrocytes following a pro-inflammatory stimulus and is elevated in osteoarthritis. Arthritis Res. Ther. 8, R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rettig W. J., Su S. L., Fortunato S. R., Scanlan M. J., Raj B. K., Garin-Chesa P., Healey J. H., Old L. J. (1994) Fibroblast activation protein: purification, epitope mapping and induction by growth factors. Int. J. Cancer 58, 385–392 [DOI] [PubMed] [Google Scholar]
- 30. Denys H., Derycke L., Hendrix A., Westbroek W., Gheldof A., Narine K., Pauwels P., Gespach C., Bracke M., De Wever O. (2008) Differential impact of TGF-β and EGF on fibroblast differentiation and invasion reciprocally promotes colon cancer cell invasion. Cancer Lett. 266, 263–274 [DOI] [PubMed] [Google Scholar]
- 31. Chen H., Yang W. W., Wen Q. T., Xu L., Chen M. (2009) TGF-β induces fibroblast activation protein expression; fibroblast activation protein expression increases the proliferation, adhesion, and migration of HO-8910PM [corrected]. Exp. Mol. Pathol. 87, 189–194 [DOI] [PubMed] [Google Scholar]
- 32. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 33. Feng X. H., Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 34. Massagué J., Seoane J., Wotton D. (2005) Smad transcription factors. Genes Dev. 19, 2783–2810 [DOI] [PubMed] [Google Scholar]
- 35. Kang Y. (2006) Pro-metastasis function of TGFβ mediated by the Smad pathway. J. Cell. Biochem. 98, 1380–1390 [DOI] [PubMed] [Google Scholar]
- 36. Levy L., Hill C. S. (2006) Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 17, 41–58 [DOI] [PubMed] [Google Scholar]
- 37. Korpal M., Kang Y. (2010) Targeting the transforming growth factor-β signalling pathway in metastatic cancer. Eur. J. Cancer 46, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 38. Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Derynck R., Akhurst R. J., Balmain A. (2001) TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 29, 117–129 [DOI] [PubMed] [Google Scholar]
- 40. Boukamp P., Petrussevska R. T., Breitkreutz D., Hornung J., Markham A., Fusenig N. E. (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fodstad O., Aamdal S., McMenamin M., Nesland J. M., Pihl A. (1988) A new experimental metastasis model in athymic nude mice, the human malignant melanoma LOX. Int. J. Cancer 41, 442–449 [DOI] [PubMed] [Google Scholar]
- 42. Ory D. S., Neugeboren B. A., Mulligan R. C. (1996) A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U.S.A. 93, 11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., Naldini L. (1998) A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. He J., Tegen S. B., Krawitz A. R., Martin G. S., Luo K. (2003) The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J. Biol. Chem. 278, 30540–30547 [DOI] [PubMed] [Google Scholar]
- 45. Zawel L., Dai J. L., Buckhaults P., Zhou S., Kinzler K. W., Vogelstein B., Kern S. E. (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 [DOI] [PubMed] [Google Scholar]
- 46. Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J. M. (1998) Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Freudenberg J. A., Chen W. T. (2007) Induction of Smad1 by MT1-MMP contributes to tumor growth. Int. J. Cancer 121, 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le Scolan E., Zhu Q., Wang L., Bandyopadhyay A., Javelaud D., Mauviel A., Sun L., Luo K. (2008) Transforming growth factor-β suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 68, 3277–3285 [DOI] [PubMed] [Google Scholar]
- 49. Nagano Y., Mavrakis K. J., Lee K. L., Fujii T., Koinuma D., Sase H., Yuki K., Isogaya K., Saitoh M., Imamura T., Episkopou V., Miyazono K., Miyazawa K. (2007) Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-β signaling. J. Biol. Chem. 282, 20492–20501 [DOI] [PubMed] [Google Scholar]
- 50. Sun Y., Liu X., Ng-Eaton E., Lodish H. F., Weinberg R. A. (1999) SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc. Natl. Acad. Sci. U.S.A. 96, 12442–12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luo K., Stroschein S. L., Wang W., Chen D., Martens E., Zhou S., Zhou Q. (1999) The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev. 13, 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagase T., Mizuguchi G., Nomura N., Ishizaki R., Ueno Y., Ishii S. (1990) Requirement of protein co-factor for the DNA-binding function of the human ski proto-oncogene product. Nucleic Acids Res. 18, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nicol R., Stavnezer E. (1998) Transcriptional repression by v-Ski and c-Ski mediated by a specific DNA binding site. J. Biol. Chem. 273, 3588–3597 [DOI] [PubMed] [Google Scholar]
- 54. Akiyoshi S., Inoue H., Hanai J., Kusanagi K., Nemoto N., Miyazono K., Kawabata M. (1999) c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with Smads. J. Biol. Chem. 274, 35269–35277 [DOI] [PubMed] [Google Scholar]
- 55. Suzuki H., Yagi K., Kondo M., Kato M., Miyazono K., Miyazawa K. (2004) c-Ski inhibits the TGF-β signaling pathway through stabilization of inactive Smad complexes on Smad-binding elements. Oncogene 23, 5068–5076 [DOI] [PubMed] [Google Scholar]
- 56. Wu J. W., Krawitz A. R., Chai J., Li W., Zhang F., Luo K., Shi Y. (2002) Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-β signaling. Cell 111, 357–367 [DOI] [PubMed] [Google Scholar]
- 57. Ueki N., Hayman M. J. (2003) Direct interaction of Ski with either Smad3 or Smad4 is necessary and sufficient for Ski-mediated repression of transforming growth factor-β signaling. J. Biol. Chem. 278, 32489–32492 [DOI] [PubMed] [Google Scholar]
- 58. Albino A. P., Davis B. M., Nanus D. M. (1991) Induction of growth factor RNA expression in human malignant melanoma: markers of transformation. Cancer Res. 51, 4815–4820 [PubMed] [Google Scholar]
- 59. Krasagakis K., Garbe C., Schrier P. I., Orfanos C. E. (1994) Paracrine and autocrine regulation of human melanocyte and melanoma cell growth by transforming growth factor β in vitro. Anticancer Res. 14, 2565–2571 [PubMed] [Google Scholar]
- 60. Krasagakis K., Krüger-Krasagakes S., Fimmel S., Eberle J., Thölke D., von der Ohe M., Mansmann U., Orfanos C. E. (1999) Desensitization of melanoma cells to autocrine TGF-β isoforms. J. Cell. Physiol. 178, 179–187 [DOI] [PubMed] [Google Scholar]
- 61. Rodeck U., Bossler A., Graeven U., Fox F. E., Nowell P. C., Knabbe C., Kari C. (1994) Transforming growth factor β production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res. 54, 575–581 [PubMed] [Google Scholar]
- 62. Rodeck U., Melber K., Kath R., Menssen H. D., Varello M., Atkinson B., Herlyn M. (1991) Constitutive expression of multiple growth factor genes by melanoma cells but not normal melanocytes. J. Invest. Dermatol. 97, 20–26 [DOI] [PubMed] [Google Scholar]
- 63. Rodeck U., Nishiyama T., Mauviel A. (1999) Independent regulation of growth and SMAD-mediated transcription by transforming growth factor β in human melanoma cells. Cancer Res. 59, 547–550 [PubMed] [Google Scholar]
- 64. Prunier C., Pessah M., Ferrand N., Seo S. R., Howe P., Atfi A. (2003) The oncoprotein Ski acts as an antagonist of transforming growth factor-β signaling by suppressing Smad2 phosphorylation. J. Biol. Chem. 278, 26249–26257 [DOI] [PubMed] [Google Scholar]
- 65. Monsky W. L., Lin C. Y., Aoyama A., Kelly T., Akiyama S. K., Mueller S. C., Chen W. T. (1994) A potential marker protease of invasiveness, seprase, is localized on invadopodia of human malignant melanoma cells. Cancer Res. 54, 5702–5710 [PubMed] [Google Scholar]
- 66. Fazakas C., Wilhelm I., Nagyoszi P., Farkas A. E., Haskó J., Molnár J., Bauer H., Bauer H. C., Ayaydin F., Dung N. T., Siklós L., Krizbai I. A. (2011) Transmigration of melanoma cells through the blood-brain barrier: role of endothelial tight junctions and melanoma-released serine proteases. PLoS One 6, e20758. [DOI] [PMC free article] [PubMed] [Google Scholar]