

Background: Sodium channels underlie neuronal excitability and are regulated by seizures.
Results: Mass spectrometric analysis of brain sodium channels revealed novel phosphorylation and methylation sites that decreased and increased, respectively, after seizures. Inducing methylation increased sodium channel activity.
Conclusion: Reciprocal phosphorylation and methylation after seizures will alter sodium channel function.
Significance: Such regulation would impact neuronal excitability.
Keywords: Electrophysiology, Epilepsy, Ion Channels, Mass Spectrometry (MS), Proteomics
Abstract
Voltage-gated sodium (Nav) channels initiate action potentials in brain neurons and are primary therapeutic targets for anti-epileptic drugs controlling neuronal hyperexcitability in epilepsy. The molecular mechanisms underlying abnormal Nav channel expression, localization, and function during development of epilepsy are poorly understood but can potentially result from altered posttranslational modifications (PTMs). For example, phosphorylation regulates Nav channel gating, and has been proposed to contribute to acquired insensitivity to anti-epileptic drugs exhibited by Nav channels in epileptic neurons. However, whether changes in specific brain Nav channel PTMs occur acutely in response to seizures has not been established. Here, we show changes in PTMs of the major brain Nav channel, Nav1.2, after acute kainate-induced seizures. Mass spectrometry-based proteomic analyses of Nav1.2 purified from the brains of control and seizure animals revealed a significant down-regulation of phosphorylation at nine sites, primarily located in the interdomain I-II linker, the region of Nav1.2 crucial for phosphorylation-dependent regulation of activity. Interestingly, Nav1.2 in the seizure samples contained methylated arginine (MeArg) at three sites. These MeArgs were adjacent to down-regulated sites of phosphorylation, and Nav1.2 methylation increased after seizure. Phosphorylation and MeArg were not found together on the same tryptic peptide, suggesting reciprocal regulation of these two PTMs. Coexpression of Nav1.2 with the primary brain arginine methyltransferase PRMT8 led to a surprising 3-fold increase in Nav1.2 current. Reciprocal regulation of phosphorylation and MeArg of Nav1.2 may underlie changes in neuronal Nav channel function in response to seizures and also contribute to physiological modulation of neuronal excitability.
Introduction
Voltage-gated sodium (Nav)2 channels mediate the rapid influx of sodium that underlies action potential initiation, propagation, and neuronal excitability. Nav channels are composed of a large principal voltage-sensing and pore-forming α subunit of ∼2,000 amino acids that contains 24 transmembrane segments. The α subunit polypeptide comprises four internally repeated homologous domains (I–IV) containing the transmembrane and extracellular segments, and linked by cytoplasmic linker domains, as well as cytoplasmic N and C termini (1). The intracellular domains are targets for posttranslational modification (PTM) by phosphorylation at multiple sites (2, 3). The mammalian Nav channel gene family encodes 10 distinct α subunits, four of which (Nav1.1, Nav1.2, Nav1.3, and Nav1.6) are highly expressed in the mammalian brain (4). Among these, Nav1.2 is the most highly expressed, and it is found localized in neuronal axons and nerve terminals, where it controls conductance of axonal action potentials and neurotransmitter release in presynaptic terminals (4).
Nav channels, including Nav1.2, are modulated through changes in phosphorylation state, primarily on the large cytoplasmic linker domains between the first and second internally repeated domains (i.e. the interdomain (ID) I-II linker) (2). Nav channels are phosphorylated by various protein kinases whose impact is to modulate Nav channel activity and gating and, as a consequence, cellular excitability (2, 5). Recent mass spectrometry (MS)-based proteomic analyses of Nav1.2 purified from rat brain (3) or present in whole mouse brain phosphoproteome fractions (reviewed in Refs. 6 and 7) have identified >60 in vivo phosphorylation sites on brain Nav1.2, many more than have been identified on any other Nav channel. Increased Nav1.2 phosphorylation in the ID I-II linker region is generally associated with a reduction in Nav current (2), in the ID II-III linker region changes in channel localization (8), and in the ID III-IV linker region modulation of inactivation (9, 10).
Epilepsy is a complex neurological disorder that affects ∼65 million people in the world (nearly 3 million in the United States; see the Epilepsy Foundation Web site). The pathogenesis of epilepsy or epileptogenesis is complex and has not been clearly defined, but it generally involves an imbalance between excitatory and inhibitory neurotransmission in multiple brain structures (11). Changes in the expression, localization, and function of a number of ion channels, including Nav1.2 (12, 13), occur in the period following the initial acute seizures and may contribute to the resultant epileptogenesis, and at least some of these are mediated through altered PTMs (14). Nav channels, including Nav1.2, are also mutated in several forms of inherited epilepsy (15). Nav channels are targets for many of the most commonly used antiepileptic drugs (AEDs) (16), yet neurons in the epileptic brain display resistance to certain Nav channel-specific AEDs (17–20). Pharmacoresistance may initially arise in response to acute seizures, and with a time course consistent with changes in Nav channel PTM and inconsistent with de novo expression of distinct Nav channel isoforms (21–24). Acute changes in Nav channel gating, primarily an enhancement of the sustained component, also occur in response to seizure (22, 25–27). The signaling pathways triggered by a brain insult and leading to these acquired changes in ion channel function are not known and represent possible targets for AED development.
Here we use monoclonal antibody (mAb)-based immunopurification (IP) and MS analyses to provide the first evidence of aberrantly altered in vivo PTMs of rat Nav1.2 protein upon acute seizures induced by kainate (KA) treatment. These include reciprocal changes in phosphorylation and arginine methylation of Nav1.2 in the ID I-II linker domain that is crucial to channel modulation. Discovery of seizure-associated changes in Nav channel PTMs is crucial to a detailed understanding of epileptogenesis and the associated changes in gating and pharmacosensitivity of neuronal Nav channels as well as identification of novel pathways for therapeutic modulation.
EXPERIMENTAL PROCEDURES
Preparation of Seizure Model and Rat Brain Membrane Proteins
All animal use procedures were in strict accordance with the Guide for the Care and Use of Laboratory Animals described by the National Institutes of Health and approved by the University of California Davis Institutional Animal Care and Use Committee. Acute seizures were induced in adult (8–12 weeks, 250 g) male rats by systemic KA administration at a dose of 10 mg/kg (28). Seizure progression was assessed by visual observation of the behavioral seizure stage according to Racine's classification (29). A full tonic-clonic behavioral seizure, with loss of postural control, was considered as a class 5 motor seizure. Rats exhibiting stage 5 seizures were sacrificed by decapitation and rapid preparation of a crude rat brain membrane (RBM) fraction (30) in ice-cold buffer containing protease and phosphatase inhibitors (28).
IP and Reciprocal IP of Nav Channels
We immunopurified Nav1.2 from RBM using Nav1.2-specific IgG2a mouse mAb K69/3 (3), obtained from the University of California Davis/National Institutes of Health NeuroMab Facility. Frozen or fresh samples of RBM were individually solubilized at 1 mg of protein/ml in lysis buffer (1% Triton X-100, 0.15 m NaCl, 1 mm EDTA, 10 mm sodium azide, 10 mm Tris-HCl, pH 8.0, 2 mm NaF, 1.5 μg/ml aprotinin, 10 μg/ml antipain, 10 μg/ml leupeptin, 0.1 mg/ml benzamidine, 1 mm PMSF) on a rotator at 4 °C for 30 min. The detergent-insoluble fraction was removed by centrifugation at 4 °C/16,000 × g/30 min. Nav1.2 was immunopurified by incubation of the detergent-soluble RBM fraction for 13 h at 4 °C with K69/3 mAb cross-linked to protein G-agarose beads. Beads were then washed seven times with lysis buffer, and the IP product was eluted from the beads by incubation in 5% SDS-containing reducing sample buffer at 37 °C for 15 min. The eluted Nav1.2 protein was subjected to 7.5% SDS-PAGE, and efficient IP was confirmed by immunoblotting analysis. Reciprocal IP of Nav1.2 was performed with anti-mono- and di-MeArg 7E6 IgG1 mAb (Abcam, catalogue no. ab5394) and solubilized RBM. For sequential IP experiments, Nav1.2 was first purified with K69/3 mAb beads, and Nav1.2 protein was eluted with 0.1 m glycine buffer, pH 3.0, followed by immediate neutralization with 0.1 m Tris-HCl buffer (pH 8.0) containing 1% Triton X-100, 0.15 m NaCl, and 1 mm EDTA. Serial IP was performed as for initial IP, using K58/35 IgG1 mAb as a pan-Nav channel positive control (3), anti-Kv2.1 channel K89/34 IgG1 mAb as negative control, and anti-MeArg 7E6 IgG1 mAb for MeArg Nav1.2, followed by immunoblot analysis with anti-pan-Nav channel K58/35 mAb.
SDS-PAGE and Immunoblotting
For LC-MS analyses, Nav channel IPs were separated on 10% polyacrylamide-SDS gels and visualized with colloidal Coomassie staining. For immunoblotting analysis, samples were electrotransferred to pure nitrocellulose membranes for 3 h at 4 °C. The membrane was blocked with 4% nonfat dry milk and 0.1% Tween 20 in 1× TBS buffer (pH 8.0) for 30 min and incubated overnight in primary mAb solution diluted in blocking buffer. Immunoblots were washed in blocking buffer and then incubated with horseradish peroxidase-conjugated secondary antibody diluted in blocking buffer for 1 h at room temperature and, after serial washes with TBS buffer, incubated with ECL solution and imaged on a FluorChem Q imager (Cell Biosciences).
In-gel Digestion and Mass Spectrometry
For MS analyses, the Nav channel band of 250 kDa was excised from Coomassie-stained gels and diced into small pieces. The gel bands were destained with 50% acetonitrile or methanol, 50 mm ammonium bicarbonate, and then washed extensively with distilled water by vortexing. After dehydrating with 50% acetonitrile, 50 mm ammonium bicarbonate buffer and 100% acetonitrile solution, the protein was reduced in 10 mm dithiothreitol at 56 °C for 1 h and alkylated in 5 mm iodoacetamide at room temperature for 30 min. Gel pieces were then washed for 10 min in 50 mm ammonium bicarbonate; dehydrated in 50% acetonitrile, 50 mm ammonium bicarbonate for 10 min; and dried to complete dryness. Dried gel pieces were swollen with trypsin solution (Promega, Madison, WI) at 12.5 ng/ml in 50 mm ammonium bicarbonate and incubated overnight at 37 °C. Digested peptide mixtures were extracted three times with 10% formic acid for 15 min on ice and 100% acetonitrile for 5 min at room temperature. Extracted peptides were desalted and stored on StageTip C18 at 4 °C (or room temperature) until LC-MS/MS. Peptides were loaded on a homemade C18 trap column (12 mm × 75 μm) and separated on a C18 analytical column (120 mm × 75 μm) on an Eksigent nano-LC system and analyzed by LTQ-FT MS. Mass spectra were acquired in a data-dependent manner, with a duty cycle that included one full survey FT scan with 100,000 resolution and seven subsequent consecutive tandem mass scans with 400-ms ion-filling time. All mass spectra were processed with Sorcerer (SEQUEST), Trans-Proteomics Pipeline (X!Tandem), MS-GF+, and MaxQuant software against a target and decoy database (IPI rat version 3.72 and contaminant sequences) or two Nav1.2 protein sequences (rat Nav1.2 and Nav1.2 isoform 2 protein). The false discovery rate was below 1%.
Label-free Quantification and Statistical Data Analysis
Label-free quantification was performed with two sets of LC-MS/MS data: three technical replicates of in-gel digestion from the first pair of RBM samples and duplicates from the second pair of RBM samples (i.e. control and KA-treated seizure rat brains). Ion chromatograms for all Nav1.2 peptide precursors were extracted with narrow precursor mass window (e.g. 10 ppm) by Qualbrowser software (Thermo Finnigan), and peak areas were calculated by the ICIS algorithm considering retention time at the point of identification. Normalization for the area of each Nav1.2 peptide was performed with the summed area of total identified Nav1.2 peptides (n = 107). Quantitative significance (ANOVA and principle component analysis) was performed by Perseus software (Max-Planck Institute of Biochemistry, Department of Proteomics and Signal Transduction, Munich). Statistical significance between the means of two samples was estimated by Student's t test.
Electrophysiology
Human embryonic kidney tsA-201 cells in DMEM/Ham's F-12 with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 100 units/ml penicillin and streptomycin (Invitrogen) were grown at 37 °C and 10% CO2. Cells were transiently transfected with cDNAs encoding Nav1.2 (2 μg) and GFP (0.5 μg) or Nav1.2 (2 μg) and GFP-tagged PRMT8 (2 μg) using TransIT-LT1 (Mirus Bio LLC, Madison, WI).
Currents were recorded in the whole-cell configuration using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The pipette solutions contained 170 mm N-methyl-d-glucamine, 5 mm NaCl, 4 mm MgCl2, 0.1 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, 12 mm Tris-phosphocreatine, 2 mm Tris-ATP, 0.2 mm sodium-GTP, and 40 mm HEPES, adjusted to pH 7.2 with H2SO4. External solution contained 140 mm NaCl, 5.4 mm CsCl, 1.8 mm CaCl2, 1 mm MgCl2, and 10 mm HEPES, adjusted to pH 7.4 with NaOH. Recording pipettes were pulled from borosilicate glass transfer tubes and fire-polished to resistances of 2–4 megaohms. Data were acquired using Pulse software (Heka, Lambrecht, Germany) controlling an ITC18 DA/AD interface (Instrutech, Great Neck, NY). All analyses were performed using IGOR Pro (Wavemetrics, Lake Oswego, OR). Standard P/4 subtraction of linear leak and capacitance was used. Series resistance was typically 6–8 megaohms, of which >80% was compensated electronically.
The voltage dependence of activation was measured from a holding potential of −100 mV. Cells were depolarized to potentials from −100 to +100 mV in 10-mV increments, and peak inward currents were measured. Conductance (G)-voltage relationships were determined from peak current (I) versus voltage relationships as G = I/(V − VRev), where V was the test potential, and VRev was the extrapolated reversal potential. The voltage dependence of inactivation was measured from a holding potential of −100 mV. Cells were depolarized for 100 ms to potentials from −100 to −15 mV in 5-mV increments followed by test pulses to 0 mV. Recovery from inactivation was examined by applying a 20-ms conditioning pulse to 0 mV from a holding potential of −100 mV, followed by a recovery interval of variable duration (Δ1 ms) and a test pulse to 0 mV. Voltage dependence of slow inactivation was measured by holding the membrane potential for 5 s at conditioning voltages from −100 mV to 0 mV (at 10-mV increments) before stepping for 20 ms to −100 mV to recover channels in the fast inactivated state followed by a test pulse at 0 mV for 10 ms.
RESULTS
Identification of in Vivo Phosphorylation Sites on Nav1.2
To discover acute seizure-responsive PTMs in rat brain, we performed MS analysis on Nav1.2 purified from the brains of control rats and rats subjected to KA-induced seizures. We first determined that Kv2.1 potassium channels in the brain samples from KA-seized rats exhibited the altered electrophoretic mobility that had been seen in previous studies of this seizure model (28) and that samples from control and KA-seized rats had comparable levels of Nav1.2 (Fig. 1A). We isolated Nav1.2 protein from control and KA-induced seizure samples by target-specific IP using the Nav1.2-specific mouse mAb K69/3 rather than PTM-specific enrichment techniques (e.g. phosphopeptide or glycol capture methods). This approach allowed us to analyze in vivo Nav1.2 PTMs independent of the nature of the PTM. Effective detergent solubilization (Fig. 1B) and IP (Fig. 1C) of Nav1.2 was confirmed by immunoblotting of detergent-soluble and -insoluble fractions and purified and depleted fractions, respectively. The purified Mr ∼250,000 Nav1.2 α subunit protein was further purified by excision from Coomassie Brilliant Blue-stained SDS-PAGE (Fig. 1D). The Nav1.2 α subunit gel band from control and seizure samples was subjected to trypsinization, and extracted peptides were subjected to label-free quantitative MS analysis. To increase the identification efficiency for low abundant peptides containing PTMs, such as phosphorylation, we applied two mass spectrometric options: longer ion accumulation (400 ms) and multistage activation for tandem MS (specifically useful for phosphopeptide identification).
FIGURE 1.
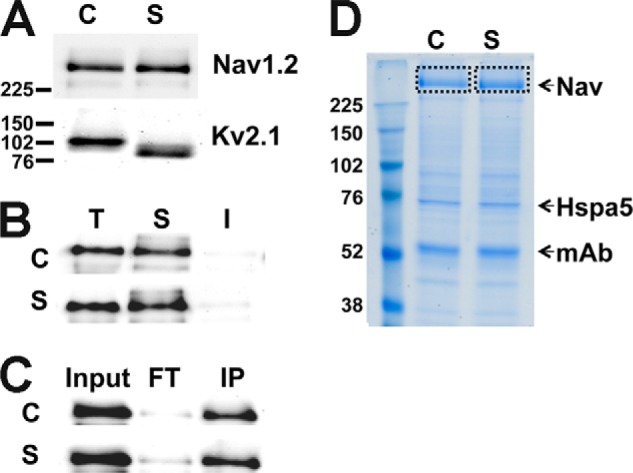
Immunopurification of rat brain Nav1.2 from control and seizure animals. A, immunoblot analysis of Nav1.2 and Kv2.1 in control (C) and seizure (S) RBM samples (10 μg of protein/lane). B, solubilization of Nav1.2 in 1% Triton X-100 detergent extract. T, total RBM starting material; S, detergent-soluble fraction; I, detergent-insoluble fraction. C, immunoblot analysis of Nav1.2 IP. Input, 20 μg of protein; FT, flow-through fraction from immunopurification; IP, IP fraction. Fractions were each 10 μg of protein. D, colloidal Coomassie Blue staining of IP products from control and seizure brain. Nav, Nav channel band excised for analysis. Hspa5, IgG binding protein. mAb, mAb IgG heavy chain. Numbers to the left of panels refer to Mr standards.
In these analyses, we identified a total of 264 Nav1.2 peptides. Among the five described alternative splice variant isoforms of rat brain Nav1.2, we observed only two isoforms, with 11 splice variant isoform-specific peptides from the canonical Nav1.2 isoform (SWISS-PROT P04775), and five splice variant isoform-specific peptides from an alternative isoform (IPI00476429.2), which we refer to hereafter as isoform 2. Isoform-specific peptides from the three other alternative splice variant isoforms of rat brain Nav1.2 (SWISS-PROT: F1LN1, F1LLV9, F1M7F6) were not detected. The overall coverage of the Nav1.2 sequence was ∼50%, with >80% coverage of cytoplasmic domains. Of the 107 unique Nav1.2 peptides derived from cytoplasmic regions, including six peptides from Nav1.2 isoform 2, 11 peptides were derived from the N terminus, 45 from the ID I-II linker, five from the ID II-III linker, four from the ID III-IV linker, and 20 from the cytoplasmic C-terminal region (see Fig. 2 for the Nav1.2 membrane topology). In addition, we identified 13 peptides from the pore domains, four from transmembrane domains, three from the extracellular loops, and two from the voltage sensor region (Fig. 2).
FIGURE 2.

Mapping of in vivo multiple PTMs on rat brain Nav1.2. Closed circles, phosphosites on Ser/Thr/Tyr; open boxes, MeArg. **, novel phosphorylation sites identified here; *, phosphorylation sites known previously only in mice and identified here in rats.
We next investigated the nature and extent of Nav1.2 phosphorylation in these samples. Our previous MS analyses of in vivo phosphorylation sites on Nav1.2 purified from rat brain had identified 15 phosphorylation sites (3). Global mouse brain phosphoproteome analyses have also identified a partially overlapping set of in vivo Nav1.2 phosphorylation sites, which together yielded a total 61 distinct phosphorylation sites (reviewed in Refs. 6 and 7).
To increase the sensitivity and reliability of phosphosite identification in our analyses, we searched our data set against the entire rat protein sequence database using four search engines: SEQUEST, X!Tandem, MaxQuant (31), and MS-GF+ (32). We also performed a targeted MaxQuant software-based analysis against the Nav1.2 and Nav1.2 isoform 2 protein sequences (false discovery rate <0.01). As shown in Fig. 2 and Table 1, using these approaches, we identified a total of 33 in vivo phosphorylation sites on Nav1.2 purified from rat brain. This set includes 14 phosphorylation sites known from previous studies of rat brain Nav1.2 (Ser(P)-4, Ser(P)-468, Ser(P)-471, Ser(P)-484, Ser(P)-554, iso-Ser(P)-582, Ser(P)-589, Ser(P)-687, Ser(P)-688, Ser(P)-721, Ser(P)-1930, Ser(P)-1966, Thr(P)-1969/1970, and Ser(P)-1971; no asterisk in Fig. 2), 10 Nav1.2 phosphorylation sites previously known only in mouse brain (Thr(P)-113, Ser(P)-488, iso-Ser(P)-543, Ser(P)-553, Ser(P)-568, Tyr(P)-689, Ser(P)-692, Ser(P)-706, Ser(P)-709, and Thr(P)-712; one asterisk in Fig. 2), and nine novel sites (Ser(P)-104, Thr(P)-106, Ser(P)-107, Ser(P)-494(495), Ser(P)-665, Ser(P)-686, Ser(P)-1076, Tyr(P)-1429, and Ser(P)-1977; two asterisks in Fig. 2). In this study, of the 33 in vivo phosphorylation sites identified on Nav1.2, 26 are on Ser, five are on threonine, and two are on tyrosine (closed circles in Fig. 2). All phosphorylation sites were found on cytoplasmic domains except for Tyr(P)-1429, which is predicted to be in the S5-S6 P-loop region of domain III (Fig. 2).
TABLE 1.
Summary of identified Nav1.2 peptides containing PTMs
| Position | Residue | PTM | Novel ID | Search engine and databasea |
Confidence (no. of hits/5 search engines) | ||||
|---|---|---|---|---|---|---|---|---|---|
| S | X | G | M(1) | M(2) | |||||
| 4 | Ser | Phosphorylation | × | × | × | Medium (3/5) | |||
| 104 | Ser | Phosphorylation | Yes | × | Low (1/5) | ||||
| 106 | Thr | Phosphorylation | Yes | × | Low (1/5) | ||||
| 107 | Ser | Phosphorylation | Yes | × | Low (1/5) | ||||
| 113 | Thr | Phosphorylation | × | Low (1/5) | |||||
| 468 | Ser | Phosphorylation | × | × | Medium (3/5) | ||||
| 471 | Ser | Phosphorylation | × | × | × | × | × | High (5/5) | |
| 484 | Ser | Phosphorylation | × | × | × | × | × | High (5/5) | |
| 488 | Ser | Phosphorylation | × | × | Medium (3/5) | ||||
| 494(495) | Ser(Ser) | Phosphorylation | Yes | × | Low (1/5) | ||||
| 553 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 554 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 568 | Ser | Phosphorylation | × | × | × | Medium (3/5) | |||
| 543 (isoform 2) | Ser | Phosphorylation | × | × | × | Medium (3/5) | |||
| 582 (isoform 2) | Ser | Phosphorylation | × | × | × | Medium (3/5) | |||
| 589 | Ser | Phosphorylation | × | × | × | × | × | High (5/5) | |
| 665 | Ser | Phosphorylation | Yes | × | Low (1/5) | ||||
| 686 | Ser | Phosphorylation | Yes | × | × | × | × | High (4/5) | |
| 687 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 688 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 689 | Tyr | Phosphorylation | × | × | × | Medium (3/5) | |||
| 692 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 706 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 709 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 712 | Thr | Phosphorylation | × | × | × | Medium (3/5) | |||
| 721 | Ser | Phosphorylation | × | × | × | × | High (4/5) | ||
| 1076 | Ser | Phosphorylation | Yes | × | × | × | × | High (4/5) | |
| 1429 | Tyr | Phosphorylation | Yes | × | × | × | × | High (4/5) | |
| 1930 | Ser | Phosphorylation | × | × | Medium (3/5) | ||||
| 1966 | Thr | Phosphorylation | × | × | × | × | × | High (5/5) | |
| 1969(1970) | Thr(Thr) | Phosphorylation | × | Low (1/5) | |||||
| 1971 | Ser | Phosphorylation | × | × | × | Medium (3/5) | |||
| 1977 | Ser | Phosphorylation | Yes | × | Low (1/5) | ||||
| 472 | Arg | Methylation | Yes | × | × | × | × | × | High (5/5) |
| 563 | Arg | Methylation | Yes | × | × | × | Medium (3/5) | ||
| 570 | Arg | Methylation | Yes | × | × | × | Medium (3/5) | ||
a S, searched with rat protein sequences (IPI version 3.72) by SEQUEST; X, searched with rat protein sequences (IPI version 3.72) by X!Tandem; G, searched with rat protein sequences (IPI version 3.72) by MS-GF+; M(1), searched with rat protein sequences (IPI version 3.72) by MaxQuant; M(2), searched with rat Nav1.2 protein sequences by MaxQuant.
Arginine Methylation of Nav1.2
Surprisingly, we found three MeArg sites, Arg-472, Arg-563, and Arg-570, in multiple tryptic peptides derived from the ID I-II linker region of Nav1.2 (open boxes in Fig. 2). Because MeArg has not been commonly identified as an in vivo PTM of membrane proteins, we validated MeArg on Nav1.2 using a number of independent approaches. We first performed IP experiments on a lysate prepared from a crude rat brain membrane fraction using an anti-MeArg mAb and probed the resultant MeArg protein fraction for the presence of Nav1.2 protein by immunoblot. As shown in Fig. 3A, IPs performed with the anti-MeArg IgG1 mAb contained Nav1.2 protein. However, no Nav1.2 was detected in negative control IPs performed with irrelevant anti-Kv2.1 IgG1 mAb or with no mAb (Fig. 3A). To verify that the MeArg yielding the specific IP with the anti-MeArg mAb was on Nav1.2 and not an associated protein, we performed a sequential IP experiment, first purifying Nav1.2 with an anti-Nav1.2 mAb and then performing a second IP reaction on the eluted Nav1.2 protein. As shown in Fig. 3B, the sequential IPs performed on purified Nav1.2 with a pan-Nav IgG1 mAb and with the anti-MeArg IgG1 mAb, but not with irrelevant anti-Kv2.1 IgG1 mAb, yielded Nav1.2 signals. These results indicate that rat brain Nav1.2 is modified with MeArg. We next confirmed MeArg at Nav1.2 MeArg-563 by comparison of the tandem mass spectrum of the native Nav1.2 tryptic peptide with that of a synthesized standard peptide 552FSSPHQSLLSIRGSLFSPR570 (Mr = 2129.145 Da; MeArg at position 563 in boldface and underlined; mirror image in Fig. 3C). We note that this peptide has a consensus RGX motif (RGS) sequence for MeArg at Arg-563. These data suggest the existence of a novel MeArg on Nav1.2.
FIGURE 3.

Biochemical confirmation of Arg methylation on Nav1.2. A, IP of Nav1.2 from three independent RBM samples using anti-MeArg mAb. B, sequential IP of purified Nav1.2 with anti-pan-Nav (K58/35), anti-Kv2.1 (K89/34; negative control), or anti-MeArg (me-R) IgG1 mAbs. All immunoblots were probed for Nav1.2. C, mirrored mass spectra of seizure rat brain Nav1.2 peptide with MeArg at MeArg-563 (red) and corresponding synthetic peptide (blue).
Quantification of Nav1.2 Peptides Using Label-free Quantification
For statistical analysis, each sample was analyzed on the LC-MS platform with multiple LC-MS/MS runs. Nav1.2 peptides were quantified using an MS1 intensity-based label-free quantification method. The procedure was as follows: 1) we used Qualbrowser software to manually select extracted ion chromatograms (XICs) for the charge-free Nav1.2 peptides with a 10-ppm mass window; 2) we assigned the appropriate XIC to an exact identification time point; and 3) we measured the area of the XIC using an ICIS peak algorithm in cases where the peak had a quantifiable shape. When an Nav1.2 peptide was identified at least once in six LC-MS/MS runs, we considered it for label-free quantification. We obtained 134 quantifiable XICs from total of 264 unique peptides from the Nav1.2 protein, which were merged into a charge state-free set of 107 unique peptides (see supplemental Table 1). We normalized the abundance of each Nav1.2 peptide with the total sum abundance. Alternatively, we confirmed the normalization using the abundance of several unmodified peptides of Nav1.2 as internal standards (I.S.) (see Fig. 5, right box).
FIGURE 5.
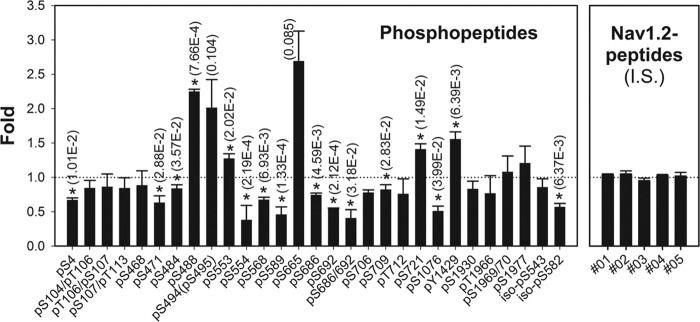
Stoichiometry of Nav1.2 phosphopeptides observed by label-free quantification. Bars indicate fold changes of phosphopeptides in seizure samples relative to control samples, normalized against unmodified internal standard (I.S.) peptide levels shown on the right. *, significant (p < 0.05) changes by ANOVA analysis with individual p values shown. Error bars, S.E.
To determine the peptides with quantitative significance, we performed ANOVA (p value calculation) for all 107 Nav1.2 peptides. Alternatively, to extract key components exhibiting more significant differences, we performed principal component analysis (Fig. 4). As shown in Fig. 4A, principal component analysis discriminated between control and seizure samples mainly by component 1 (81.2%, p value = 4.57E−3). As shown in Fig. 4B, combinatorial analysis of ANOVA significance with principle component analysis was able to highlight key PTMs showing significant changes in response to acute seizures (8 of 10 closed black circles; ANOVA p value <0.01 and higher/lower than ±0.1 of component 1). Of these, three phosphorylation sites in the ID I-II linker region (Ser(P)-554, -589, and -692) were decreased in the seizure sample, whereas the Ser(P)-488 site and the corresponding unmodified peptide (amino acids 473–492) were increased in the seizure sample.
FIGURE 4.
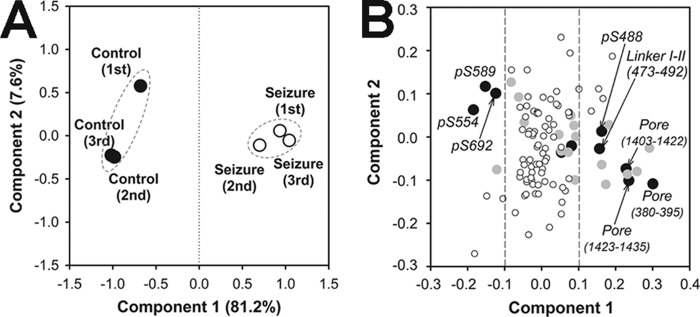
Principle component analyses of Nav1.2 peptides quantified by label-free quantification. A, sample group separation. Closed circles, control samples; open circles, seizure samples. B, peptide component separation. Black circles (n = 10), ANOVA p value < 0.001; gray circles (n = 18), 0.001 ≤ p value < 0.01; white circles (n = 79), p value ≥ 0.01.
Altered Stoichiometry of Nav1.2 Phosphorylation in Response to Acute Seizures
As confirmed by immunoblot analysis and as expected due to the short period of the acute seizure, the expression level of rat brain Nav1.2 polypeptide was not different between the control and acute seizure samples (Fig. 1A). To determine seizure-induced PTM changes of Nav1.2, we applied isoform-specific mAb-based purification to obtain similar amounts of Nav1.2 from control and seizure rat brain membrane fractions. We then used precursor-quantifiable high resolution MS (r = 100,000) to reveal the stoichiometry of multiple peptides containing in vivo Nav1.2 PTMs. We calculated the relative stoichiometry of 29 phosphopeptides in the control and acute seizure samples (Fig. 5), using five internal standard (I.S.) peptides as a control. The levels of 16 phosphopeptides were significantly changed (p value <0.05) in response to acute seizures. As shown in Fig. 5, most of these phosphopeptides were down-regulated in the seizure sample relative to the control (12 peptides, ranging between 0.38- and 0.83-fold control levels), whereas only four phosphopeptides were up-regulated (ranging between 1.27- and 2.25-fold control levels; note that the large increases in Ser(P)-494/495 and Ser(P)-665 were not significant due to the large coefficient of variation in control samples). We also found phosphorylation at Ser-543 and Ser-582 sites in Nav1.2 isoform 2, corresponding to the Ser-540 and Asn-579 sites, respectively, of canonical rat Nav1.2. Overall, the magnitude of changes in the stoichiometry of phosphorylation at individual sites between the control and seizure samples was not large, in general being ∼2-fold. However, the number of phosphorylation sites showing significantly altered stoichiometry was high (14 sites in 16 peptides).
Reciprocal Regulation of Nav1.2 Arg Methylation and Phosphorylation in Response to Acute Seizures
Three MeArg sites (MeArg-472, -563, and -570) were found near phosphosites (Ser(P)-468, -471, -553, -554, and -568) located in the ID I-II linker, a region especially critical to Nav1.2 modulation. Interestingly, MeArg peptides were within the same tryptic peptides containing these phosphorylation sites (Table 2). In each case, peptides were not detected that were simultaneously modified with both phosphorylation and MeArg, as we found (underlined residues indicate the sites with PTMs): QQEEAQAAAAAASAESR (MeR472) and QQEEAQAAAAAASAESR (Ser(P)-468, Ser(P)-471), as well as FSSPHQSLLSIRGSLFSPR (MeArg-563 and MeArg-570) and FSSPHQSLLSIRGSLFSPR (Ser(P)-553, Ser(P)-554, Ser(P)-568). Moreover, our quantitative analyses revealed that these two distinct forms of PTMs were reciprocally regulated in response to seizure (Fig. 6). Two peptides (MeArg-472 and MeArg-563) with mono-Me were increased in the seizure sample, whereas the corresponding peptides with phosphorylation (Ser(P)-471, Ser(P)-554, and Ser(P)-568) were found decreased in the seizure sample. This mutual exclusivity and reciprocal presence of these PTMs and their proximity to one another suggest that the presence of one PTM may negatively influence the other.
TABLE 2.
Mutually exclusive identification of protein methylation and phosphorylation
Residues in boldface type and underlined indicate sites with PTMs.
| Positions | Peptide sequence | PTM | Residues |
|---|---|---|---|
| 456–472 | QQEEAQAAAAAASAESR | MeArg | Arg-472 |
| 456–472 | QQEEAQAAAAAASAESR | Ser(P) | Ser-468, Ser-471 |
| 552–563 | FSSPHQSLLSIRGSLFSPR | MeArg | Arg-563, Arg-570 |
| 552–563 | FSSPHQSLLSIRGSLFSPR | Ser(P) | Ser-553, Ser-554, Ser-568 |
FIGURE 6.
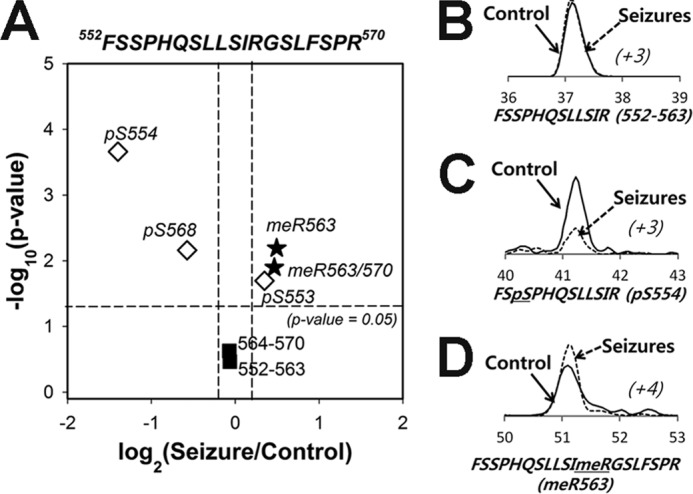
Phosphorylation (pS554) and Arg methylation (MeR563) in the Nav1.2 ID I-II linker region are reciprocally regulated in control and seizure samples. A, volcano plot of Nav1.2 peptides from the region 552–570 containing phosphorylation and MeArg. B–D, examples of label-free quantification of peptides that are either unmodified (B), phosphorylated at Ser(P)-554 (C), or methylated at MeArg-563 (D) between control (solid line) and seizure (dashed line) samples. The x axis shows the retention time (min) of peptides on C18-column liquid chromatography, and parentheses with italic type indicate the charge state of each peptide.
Effects of Increased Arginine Methylation in Nav Channel Function
The results detailed above suggest a reciprocal relationship between phosphorylation and arginine methylation of Nav1.2 in mammalian brain. Because phosphorylation is known to have diverse effects on Nav1.2 function, primarily to reduce the overall amplitude of Nav1.2 currents but also to impact gating, we next determined the impact of increased arginine methylation. We compared the amplitude and biophysical properties of recombinant Nav1.2 channels expressed in heterologous human embryonic kidney tsA-201 cells in the presence and absence of coexpressed PRMT8, the major arginine methyltransferase expressed in the brain (33). We found that coexpression of PRMT8 led to a significant increase in Nav1.2 currents (Fig. 7). This is seen in both sets of current traces from cells with and without PRMT8 coexpression (Fig. 7A) and also in data pooled from numerous cells (Fig. 7, C and D). Overall, cells coexpressing PRMT8 exhibited an ∼3-fold increase in Nav1.2 current relative to cells expressing Nav1.2 alone (3.1-fold, n = 6 for Nav1.2 alone, n = 10 for Nav1.2 + PRMT8, p < 0.05; Fig. 7D). No significant differences were observed in the voltage dependence of activation and inactivation (Fig. 7F) or the voltage dependence of slow inactivation (Fig. 7H) in cells expressing Nav1.2 alone versus Nav1.2 plus PRMT8. The kinetics of recovery from inactivation were enhanced 1.5-fold with PRMT8 coexpression (Fig. 7G). Expression of PRMT8 did not yield any increase in Nav currents in cells in the absence of Nav1.2 coexpression (Fig. 7, B and E), showing that PRMT8 was acting on coexpressed Nav1.2 and not any endogenous Nav channels that may be expressed in tsA-201 cells.
FIGURE 7.

Coexpression of PRMT8 increases Nav1.2 currents without effects on voltage-dependent properties. A, representative families of Nav1.2 current without and with coexpression of PRMT8. Currents in response to steps from −100 to +100 mV in 20-mV increments are shown. B, average currents from tsA-201 cells expressing GFP alone or PRMT8 alone at 0 mV, n = 5 for each. C, mean current-voltage (I-V) relationships of peak currents. Currents were elicited in response to voltage steps to the indicated potentials in 10-mV steps from a holding potential of −100 mV. Peak current is plotted as a function of test pulse potential. Note that the y axis is in nA. D, percentage change in peak amplitude of Nav1.2 current at 0 mV associated with coexpression of PRMT8. E, average of maximal inward currents from tsA-201 cells expressing GFP alone or PRMT8 alone at 0 mV. Note that the y axis is in pA. F, voltage dependence of the activation and voltage dependence of fast inactivation for cells expressing Nav1.2 alone or with coexpression of PRMT8. Conductance-voltage relationships (circles) were determined from I-V relationships as in B as G = I/(V − VRev), where V is the test potential, and VRev is the extrapolated reversal potential. The voltage dependence of inactivation (squares) was determined using 100-ms-long prepulses to the indicated potentials, followed by a test pulse to 0 mV. Normalized peak test pulse current is plotted as a function of prepulse potential. G, recovery from fast inactivation. Nav1.2, τ = 4.59 ± 0.289 ms, n = 10; Nav1.2 + PRMT8, τ = 3.05 ± 0.284 ms, n = 12; p = 0.001. H, voltage dependence of slow inactivation (Nav1.2, n = 6; Nav1.2 with PRMT8, n = 10). Error bars, S.E.
DISCUSSION
Here we show, using MS-based analyses, that Nav1.2 purified from rat brain exhibits altered phosphorylation and methylation in response to acute seizures, and that phosphorylation and MeArg are mutually exclusive PTMs that are reciprocally regulated on the same Nav1.2 peptides.
Nav1.2 is the most abundant Nav channel in the mammalian brain, where it is found primarily on axons and in nerve terminals (4). Numerous chromosomal duplications, nonsense, and missense mutations in the human Nav1.2-coding SCN2A gene have been associated with a variety of neurological disorders, including a variety of epilepsies as well as autism spectrum disorder (34). These and other findings underscore the importance of Nav1.2 in normal brain function, as a major Nav channel important in regulating hyperexcitability associated with epilepsy, and as an important target of AEDs (34). A number of classical biochemical studies and more recent MS-based proteomic analyses have revealed a remarkable degree of phosphorylation on Nav1.2 relative to other Nav channels (reviewed in Refs. 6 and 7). Prior to this study, of the prominent brain Nav channels, 61 in vivo phosphorylation sites had been identified on Nav1.2 versus 28 on Nav1.1 and 13 on Nav1.6. Here we confirm 23 of these sites and provide nine novel in vivo Nav1.2 phosphorylation sites to bring the total to 70 known sites. The role of phosphorylation at many of these sites remains to be established. However, the majority of these sites (39 of the 71) are found in the ID I-II linker region that is known to be crucial to functional modulation of Nav1.2 (reviewed in Refs. 2 and 5), and a large number of the remaining sites (13 of the 69) are on the cytoplasmic C terminus, which is critical to modulation by binding to calmodulin and G protein subunits (34). We did not detect any of the phosphorylation sites in the ID III-IV linker region (e.g. Tyr-1497, Tyr-1498, and Ser-1506) that have previously been shown to be important in modulation of Nav1.2 inactivation gating (35). This region contains multiple trypsin cleavage sites, making it prone to poor MS detection due to the short length and high charge state (≥4) of the cleavage fragments.
The proteomics analysis presented here reveals that 12 of these Nav1.2 phosphorylation sites exhibit significant (p < 0.05) down-regulation in response to acute seizures: Ser(P)-4, -471, -484, -554, -568, -589, -686, -692, -586/692, -709, -1076, and -582 on isoform 2. The overall changes in the stoichiometry of phosphorylation in response to acute seizures were small (∼2-fold); however, it should be noted that these values are from whole brain samples, and regional changes in areas most affected by acute KA-induced seizures (e.g. hippocampus) (36) could potentially be much larger. Interestingly, the bulk (10 of 12) of these regulated phosphorylation sites are located in the ID I-II linker region, the major modulatory region of Nav channels. Previous studies have shown a reduction in Nav current with increased phosphorylation in this region, either by PKA and/or PKC activation or through protein phosphatase activation (5). The reduced phosphorylation in this region in response to acute seizures seen here should enhance Nav current in affected neurons. Increased Nav channel activity, the appearance of a persistent Nav current, and acquired AED insensitivity are seen in neurons obtained from the brains of animal models of epilepsy (25, 27, 37, 38), including KA-induced seizures, and in brain tissue from epilepsy patients (22). Enhancement of ion channel activity is associated with the initial period following seizures and may promote subsequent epileptogenesis (39). Although some of these changes may be due to altered Nav channel gene expression, as has been shown in both KA-seized rats (12, 40) and epilepsy patients (41), a recent study suggests that altered Nav channel function first appears in the period immediately following acute seizures (42). Studies in cultured rat cortical neurons suggest that altered PTM of Nav channels occurs within 5 min of stimulus and underlies changes in Nav channel gating as well as altered sensitivity to AEDs (21, 23). Because biosynthesis and trafficking of neuronal Nav1.2 are quite slow, with cell surface Nav1.2 appearing on the cell surface ∼8 h after translation (43), it is unlikely that increased Nav channel gene expression, translation, and trafficking could yield the rapid changes in Nav current seen in response to seizures. As such, modulation of existing pools of the major brain Nav channel Nav1.2 through changes in PTMs represents a plausible mechanism for the rapid changes in Nav channel activity seen in neurons in response to seizures and other stimuli that impact neuronal excitability.
Our studies reveal MeArg as a novel form of in vivo PTM on a brain Nav channel. Protein methylation is a common PTM for nuclear proteins, and few published reports exist of methylation of plasma membrane proteins. The identification of MeArg on Nav1.2 further increases the already appreciated complexity whereby Nav channels can be dynamically regulated by PTMs, including phosphorylation (5), palmitoylation (44), and glycosylation (45). The emergence of Nav1.2 as a platform for signal integration through a diverse array of signaling pathways that impact membrane excitability is key to understanding the means by which neuronal activity is regulated by physiological and pathophysiological stimuli. We note that the MeArg and phosphorylated Ser residues identified here in Nav1.2 purified from rat brain are well conserved in most human and rat brain Nav channels, suggesting that this could represent a conserved mode of Nav channel regulation.
The mutually exclusive nature of Nav1.2 phosphorylation and MeArg on two different peptides in the intracellular ID I-II linker (Fig. 6 and Table 2) suggests reciprocal regulation of these in vivo PTMs in neurons. Although little is known of the coordinated regulation of protein phosphorylation and methylation in membrane proteins, there exists a rich literature on these PTMs in nuclear proteins, especially histones (46). These studies reveal that in many cases, protein phosphorylation and methylation are mutually exclusive and that the molecular mechanisms for this opposition are varied. One set of mechanisms acts in cis, such that modification of the target protein with one class of PTMs inhibits the acquisition or enhances the loss of the other type of PTM on the same target protein (46). In the case of Nav1.2, this would lead to a model whereby methylation of Arg-472 could negatively impact phosphorylation at Ser-471 or Ser-468 and methylation at Arg-563 and Arg-570 for phosphorylation at Ser-553, Ser-554, and Ser-568. It is interesting to speculate that because basic residues, such as Arg, are crucial components of the consensus sequence for phosphorylation by PKA, PKC, CDK5, etc. (47), the presence of MeArg could directly prevent phosphorylation at the adjacent Ser residues.
It should also be noted that another known mechanism for opposing cross-talk between protein phosphorylation and methylation can occur in trans via negative regulation of PRMT activity by phosphorylation, as in the case of inhibition of PRMT5 by JAK2 phosphorylation during myeloproliferation and erythroid differentiation (48). Reciprocal regulation of protein kinase activity by Arg methylation has also been proposed (49). Although we have not identified the PRMT that methylates Nav1.2 in vivo, the likely candidate is PRMT8, a membrane-associated PRMT specifically expressed in the brain (33) where it is found at high levels in neurons (50, 51). It is not known whether PRMT8 is regulated by phosphorylation as are other PRMTs. One emerging theme from these studies is that protein methylation may be generally more stable than phosphorylation (52), suggesting that once methylation is acquired, it may have a long lasting impact on target protein function.
We observed increased amplitude of Nav1.2 currents in heterologous cells upon PRMT8 coexpression (Fig. 7). The recombinant cardiac Nav1.5 Nav channel isoform expressed in heterologous HEK293 cells is also methylated (53), and coexpression of Nav1.5 with PRMT3 and PRMT5 in heterologous cells leads to increased Nav1.5 currents (54). These results are consistent with increased methylation resulting in enhanced amplitude of Nav current. In the case of Nav1.5, the authors presented data supporting increased cell surface expression of Nav1.5 channels as a mechanism contributing to the increased Nav current (54). We found in samples from mammalian brain that increased arginine methylation was associated with decreases in Nav1.2 phosphorylation. Because increased phosphorylation in the ID I-II linker region of Nav1.2 can acutely suppress Nav1.2 currents (reviewed in Ref. 5), it is possible that decreases in phosphorylation associated with increased Nav1.2 methylation upon PRMT8 coexpression may contribute to the observed increase in current. We speculate that the increased methylation and decreased phosphorylation of mammalian brain Nav1.2 that we observed in response to acute seizures in vivo underlie the acquired increases in Nav current in neurons following a seizure or other pro-epileptic insult. We also suggest that the enzymes catalyzing the reactions that lead to these alterations in PTMs, and the components of upstream signaling pathways leading from the initial insult to the disease state, may represent attractive targets for therapeutic modulation of Nav1.2 to reduce the altered neuronal excitability that occurs in epileptic neurons. Coupling of phosphorylation and methylation also represents an attractive candidate mechanism for effective physiological regulation of neuronal excitability.
Supplementary Material
Acknowledgments
We thank Dr. Brett Phinney and the staff of the University of California Davis Proteomics Facility for assistance and discussion and for providing Eksigent nano-LC and LTQ-FT mass spectrometry. We thank Dr. Mark T. Bedford of M.D. Anderson Cancer Center for the generous gift of the PRMT8 plasmid. We thank Rachel Silverman for expert technical assistance in generation of seizure animals, and we thank Dr. Danielle Mandikian, Ashleigh Evans, Dr. Oscar Cerda, and Steve Wiler for technical assistance and helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 NS64428 (to J. S. T. and T. S.) and R01 NS25704 (to William A. Catterall).

This article contains supplemental Table 1.
- Nav
- voltage-gated sodium
- AED
- anti-epileptic drug
- ID
- interdomain
- IP
- immunopurification
- KA
- kainate
- MeArg
- methylated arginine
- PTM
- posttranslational modification
- XIC
- extracted ion chromatogram
- RBM
- rat brain membrane
- FT
- Fourier transform
- PTM
- posttranslational modification
- ANOVA
- analysis of variance
- PRMT
- protein arginine methyltransferase.
REFERENCES
- 1. Catterall W. A., Goldin A. L., Waxman S. G. (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409 [DOI] [PubMed] [Google Scholar]
- 2. Cantrell A. R., Catterall W. A. (2001) Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat. Rev. Neurosci. 2, 397–407 [DOI] [PubMed] [Google Scholar]
- 3. Berendt F. J., Park K. S., Trimmer J. S. (2010) Multisite phosphorylation of voltage-gated sodium channel α subunits from rat brain. J. Proteome Res. 9, 1976–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vacher H., Mohapatra D. P., Trimmer J. S. (2008) Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheuer T. (2011) Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 22, 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baek J. H., Cerda O., Trimmer J. S. (2011) Mass spectrometry-based phosphoproteomics reveals multisite phosphorylation on mammalian brain voltage-gated sodium and potassium channels. Semin. Cell Dev. Biol. 22, 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trimmer J. S., Misonou H. (2014) Phosphorylation of voltage-gated ion channels. in Handbook of Ion Channels (Zheng J., Trudeau M. C., eds) CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 8. Bréchet A., Fache M. P., Brachet A., Ferracci G., Baude A., Irondelle M., Pereira S., Leterrier C., Dargent B. (2008) Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 183, 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. West J. W., Numann R., Murphy B. J., Scheuer T., Catterall W. A. (1991) A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science 254, 866–868 [DOI] [PubMed] [Google Scholar]
- 10. Chen Y., Yu F. H., Surmeier D. J., Scheuer T., Catterall W. A. (2006) Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron 49, 409–420 [DOI] [PubMed] [Google Scholar]
- 11. Ben-Ari Y. (2006) Seizures beget seizures: the quest for GABA as a key player. Crit. Rev. Neurobiol. 18, 135–144 [DOI] [PubMed] [Google Scholar]
- 12. Qiao X., Werkman T. R., Gorter J. A., Wadman W. J., van Vliet E. A. (2013) Expression of sodium channel α subunits 1.1, 1.2 and 1.6 in rat hippocampus after kainic acid-induced epilepsy. Epilepsy Res. 106, 17–28 [DOI] [PubMed] [Google Scholar]
- 13. Guo F., Xu X., Cai J., Hu H., Sun W., He G., Shao D., Wang L., Chen T., Shaw C., Zhu T., Hao L. (2013) The up-regulation of voltage-gated sodium channels subtypes coincides with an increased sodium current in hippocampal neuronal culture model. Neurochem. Int. 62, 287–295 [DOI] [PubMed] [Google Scholar]
- 14. Jung S., Bullis J. B., Lau I. H., Jones T. D., Warner L. N., Poolos N. P. (2010) Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signaling. J. Neurosci. 30, 6678–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Williams C. A., Battaglia A. (2013) Molecular biology of epilepsy genes. Exp. Neurol. 244, 51–58 [DOI] [PubMed] [Google Scholar]
- 16. White H. S., Smith M. D., Wilcox K. S. (2007) Mechanisms of action of antiepileptic drugs. Int. Rev. Neurobiol. 81, 85–110 [DOI] [PubMed] [Google Scholar]
- 17. Vreugdenhil M., Wadman W. J. (1999) Modulation of sodium currents in rat CA1 neurons by carbamazepine and valproate after kindling epileptogenesis. Epilepsia 40, 1512–1522 [DOI] [PubMed] [Google Scholar]
- 18. Remy S., Urban B. W., Elger C. E., Beck H. (2003) Anticonvulsant pharmacology of voltage-gated Na+ channels in hippocampal neurons of control and chronically epileptic rats. Eur. J. Neurosci. 17, 2648–2658 [DOI] [PubMed] [Google Scholar]
- 19. Schaub C., Uebachs M., Beck H. (2007) Diminished response of CA1 neurons to antiepileptic drugs in chronic epilepsy. Epilepsia 48, 1339–1350 [DOI] [PubMed] [Google Scholar]
- 20. Jeub M., Beck H., Siep E., Rüschenschmidt C., Speckmann E. J., Ebert U., Potschka H., Freichel C., Reissmüller E., Löscher W. (2002) Effect of phenytoin on sodium and calcium currents in hippocampal CA1 neurons of phenytoin-resistant kindled rats. Neuropharmacology 42, 107–116 [DOI] [PubMed] [Google Scholar]
- 21. Curia G., Aracri P., Sancini G., Mantegazza M., Avanzini G., Franceschetti S. (2004) Protein-kinase C-dependent phosphorylation inhibits the effect of the antiepileptic drug topiramate on the persistent fraction of sodium currents. Neuroscience 127, 63–68 [DOI] [PubMed] [Google Scholar]
- 22. Vreugdenhil M., Hoogland G., van Veelen C. W., Wadman W. J. (2004) Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur. J. Neurosci. 19, 2769–2778 [DOI] [PubMed] [Google Scholar]
- 23. Curia G., Aracri P., Colombo E., Scalmani P., Mantegazza M., Avanzini G., Franceschetti S. (2007) Phosphorylation of sodium channels mediated by protein kinase-C modulates inhibition by topiramate of tetrodotoxin-sensitive transient sodium current. Br. J. Pharmacol. 150, 792–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beck H. (2007) Plasticity of antiepileptic drug targets. Epilepsia 48, Suppl. 1, 14–18 [DOI] [PubMed] [Google Scholar]
- 25. Agrawal N., Alonso A., Ragsdale D. S. (2003) Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia 44, 1601–1604 [DOI] [PubMed] [Google Scholar]
- 26. Hargus N. J., Merrick E. C., Nigam A., Kalmar C. L., Baheti A. R., Bertram E. H., 3rd, Patel M. K. (2011) Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol. Dis. 41, 361–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ellerkmann R. K., Remy S., Chen J., Sochivko D., Elger C. E., Urban B. W., Becker A., Beck H. (2003) Molecular and functional changes in voltage-dependent Na+ channels following pilocarpine-induced status epilepticus in rat dentate granule cells. Neuroscience 119, 323–333 [DOI] [PubMed] [Google Scholar]
- 28. Misonou H., Mohapatra D. P., Park E. W., Leung V., Zhen D., Misonou K., Anderson A. E., Trimmer J. S. (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718 [DOI] [PubMed] [Google Scholar]
- 29. Racine R., Okujava V., Chipashvili S. (1972) Modification of seizure activity by electrical stimulation. 3. Mechanisms. Electroencephalogr. Clin. Neurophysiol. 32, 295–299 [DOI] [PubMed] [Google Scholar]
- 30. Trimmer J. S. (1991) Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 10764–10768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 32. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J., Pevzner P. A. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee J., Sayegh J., Daniel J., Clarke S., Bedford M. T. (2005) PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem. 280, 32890–32896 [DOI] [PubMed] [Google Scholar]
- 34. Catterall W. A. (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590, 2577–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beacham D., Ahn M., Catterall W. A., Scheuer T. (2007) Sites and molecular mechanisms of modulation of Na(v)1.2 channels by Fyn tyrosine kinase. J. Neurosci. 27, 11543–11551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Velisek L. (2006) Models of chemically-induced acute seizures. in Models of Seizures and Epilepsy (Pitkanen A., Schwartzkroin P. A., Moshe S. L. eds) pp. 127–152, Elsevier Academic Press, San Diego [Google Scholar]
- 37. Ketelaars S. O., Gorter J. A., van Vliet E. A., Lopes da Silva F. H., Wadman W. J. (2001) Sodium currents in isolated rat CA1 pyramidal and dentate granule neurones in the post-status epilepticus model of epilepsy. Neuroscience 105, 109–120 [DOI] [PubMed] [Google Scholar]
- 38. Vreugdenhil M., Faas G. C., Wadman W. J. (1998) Sodium currents in isolated rat CA1 neurons after kindling epileptogenesis. Neuroscience 86, 99–107 [DOI] [PubMed] [Google Scholar]
- 39. Rakhade S. N., Jensen F. E. (2009) Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol. 5, 380–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bartolomei F., Gastaldi M., Massacrier A., Planells R., Nicolas S., Cau P. (1997) Changes in the mRNAs encoding subtypes I, II and III sodium channel α subunits following kainate-induced seizures in rat brain. J. Neurocytol. 26, 667–678 [DOI] [PubMed] [Google Scholar]
- 41. Whitaker W. R., Faull R. L., Dragunow M., Mee E. W., Emson P. C., Clare J. J. (2001) Changes in the mRNAs encoding voltage-gated sodium channel types II and III in human epileptic hippocampus. Neuroscience 106, 275–285 [DOI] [PubMed] [Google Scholar]
- 42. Minge D., Bähring R. (2011) Acute alterations of somatodendritic action potential dynamics in hippocampal CA1 pyramidal cells after kainate-induced status epilepticus in mice. PLoS One 6, e26664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt J. W., Catterall W. A. (1986) Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46, 437–444 [DOI] [PubMed] [Google Scholar]
- 44. Bosmans F., Milescu M., Swartz K. J. (2011) Palmitoylation influences the function and pharmacology of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 108, 20213–20218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Isaev D., Isaeva E., Shatskih T., Zhao Q., Smits N. C., Shworak N. W., Khazipov R., Holmes G. L. (2007) Role of extracellular sialic acid in regulation of neuronal and network excitability in the rat hippocampus. J. Neurosci. 27, 11587–11594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Badeaux A. I., Shi Y. (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat. Rev. Mol. Cell Biol. 14, 211–224 [PubMed] [Google Scholar]
- 47. Ubersax J. A., Ferrell J. E., Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 48. Liu F., Zhao X., Perna F., Wang L., Koppikar P., Abdel-Wahab O., Harr M. W., Levine R. L., Xu H., Tefferi A., Deblasio A., Hatlen M., Menendez S., Nimer S. D. (2011) JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 19, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kanade S. R., Eckert R. L. (2012) Protein arginine methyltransferase 5 (PRMT5) signaling suppresses protein kinase Cδ- and p38δ-dependent signaling and keratinocyte differentiation. J. Biol. Chem. 287, 7313–7323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taneda T., Miyata S., Kousaka A., Inoue K., Koyama Y., Mori Y., Tohyama M. (2007) Specific regional distribution of protein arginine methyltransferase 8 (PRMT8) in the mouse brain. Brain Res. 1155, 1–9 [DOI] [PubMed] [Google Scholar]
- 51. Kousaka A., Mori Y., Koyama Y., Taneda T., Miyata S., Tohyama M. (2009) The distribution and characterization of endogenous protein arginine N-methyltransferase 8 in mouse CNS. Neuroscience 163, 1146–1157 [DOI] [PubMed] [Google Scholar]
- 52. Barth T. K., Imhof A. (2010) Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem. Sci. 35, 618–626 [DOI] [PubMed] [Google Scholar]
- 53. Beltran-Alvarez P., Pagans S., Brugada R. (2011) The cardiac sodium channel is post-translationally modified by arginine methylation. J. Proteome Res. 10, 3712–3719 [DOI] [PubMed] [Google Scholar]
- 54. Beltran-Alvarez P., Espejo A., Schmauder R., Beltran C., Mrowka R., Linke T., Batlle M., Pérez-Villa F., Pérez G. J., Scornik F. S., Benndorf K., Pagans S., Zimmer T., Brugada R. (2013) Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Lett. 587, 3159–3165 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.