

Background: Monocyte accumulation contributes to inflammatory disease progression.
Results: ASMCs overexpressing V3 resist monocyte adhesion by promoting elastogenesis, depleting hyaluronan, and reducing VCAM1, via differentially regulating TGFβ-, EGF-, and NFκB-signaling pathways.
Conclusion: V3 expression by ASMCs generates a microenvironment resistant to monocyte adhesion.
Significance: Modifying ECM components via V3 expression alters monocyte adhesion, which suggests therapeutic possibilities to treat inflammation.
Keywords: Elastin, Epidermal Growth Factor Receptor (EGFR), Extracellular Matrix, Hyaluronate, Monocytes, NFkappaB, Transforming Growth Factor Beta (TGFbeta), Vascular Smooth Muscle Cells, Versican
Abstract
Monocyte/macrophage accumulation plays a critical role during progression of cardiovascular diseases, such as atherosclerosis. Our previous studies demonstrated that retrovirally mediated expression of the versican V3 splice variant (V3) by arterial smooth muscle cells (ASMCs) decreases monocyte adhesion in vitro and macrophage accumulation in a model of lipid-induced neointimal formation in vivo. We now demonstrate that V3-expressing ASMCs resist monocyte adhesion by altering the composition of the microenvironment surrounding the cells by affecting multiple signaling pathways. Reduction of monocyte adhesion to V3-expressing ASMCs is due to the generation of an extracellular matrix enriched in elastic fibers and depleted in hyaluronan, and reduction of the proinflammatory cell surface vascular cell adhesion molecule 1 (VCAM1). Blocking these changes reverses the protective effect of V3 on monocyte adhesion. The enhanced elastogenesis induced by V3 expression is mediated by TGFβ signaling, whereas the reduction in hyaluronan cable formation induced by V3 expression is mediated by the blockade of epidermal growth factor receptor and NFκB activation pathways. In addition, expression of V3 by ASMCs induced a marked decrease in NFκB-responsive proinflammatory cell surface molecules that mediate monocyte adhesion, such as VCAM1. Overall, these results indicate that V3 expression by ASMCs creates a microenvironment resistant to monocyte adhesion via differentially regulating multiple signaling pathways.
Introduction
Monocyte/macrophage accumulation plays a critical role during progression of cardiovascular diseases, such as atherosclerosis (1–3). Previously, we demonstrated that controlled expression of the V3 isoform of versican (V3)2 by arterial smooth muscle cells (ASMCs), which lack glycosaminoglycan binding domains, reduced monocyte adhesion in vitro and monocyte/macrophage accumulation in vivo (4). However, the mechanism by which V3 expression reduces monocyte adhesion was not known.
In the present study, we have demonstrated that V3-expressing ASMCs resist monocyte adhesion not only by generating an extracellular matrix (ECM) enriched in elastic fibers and depleted in hyaluronan, but also by reducing proinflammatory cell adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM1). The enhanced elastogenesis induced by V3 expression was mediated by transforming growth factor β (TGFβ) signaling in ASMCs. In addition to these effects on elastic fibers, V3-expressing ASMCs had fewer hyaluronan cable structures that support monocyte accumulation in the ECM (5–15). This reduction in hyaluronan cable formation was mediated by the blockade of epidermal growth factor receptor (EGFR) and nuclear factor κB (NFκB) activation. Furthermore, expression of V3 by ASMCs induced a marked reduction in NFκB-responsive proinflammatory cell surface molecules that mediate monocyte adhesion, such as VCAM1, both at transcript and protein levels. Importantly, V3-expressing ASMCs had reduced levels of VCAM1-dependent monocyte adhesion compared with control cells. These data suggest that V3 expression by ASMCs generates a microenvironment resistant to monocyte adhesion by affecting the ECM components as well as cell adhesion molecules via differentially regulating multiple key signaling pathways.
MATERIALS AND METHODS
Cell Culture
Fischer rat ASMCs were obtained and cultured as described previously (16). Retrovirally transduced cells were maintained in Dulbecco's high glucose modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, glutamine, and penicillin-streptomycin (Invitrogen). Cells were used between 4 and 8 passages after the initial transduction.
Expression of V3 in ASMCs
The V3 splice variant of versican was expressed in Fischer rat ASMCs using retroviral vectors, as described previously (17–19). Briefly, the rat V3 sequence was inserted into the BamHI site of the retroviral vector LXSN (courtesy of Dr. A. D. Miller, Fred Hutchinson Cancer Research Center, Seattle, WA). The retroviral vector containing the V3 gene (LV3SN) as well as the empty control vector (LXSN) were used to infect Fischer rat ASMCs using PA317 packaging cells as described previously (16, 18).
Quantitative Polymerase Chain Reaction (qPCR)
DNA-free RNA was obtained from cell layers using the Total RNA Isolation Kit (Agilent Technologies), and cDNA was prepared from the isolated RNA by reverse transcription using random primers with the High-Capacity cDNA Archive Kit from Applied Biosystems. Real-time PCR was carried out using an Applied Biosystems Prism 7500 sequence detection system and the SYBR Green or TaqMan Universal PCR Master Mix reagent kit from Applied Biosystems as directed by the manufacturer. Applied Biosystems gene expression assays used with TaqMan reagents were as follows: Rn01499783_m1, tropoelastin; Mm01336252_m1, fibulin 5; Rn01493752_m1, Has1; Rn00565774_m1, Has2; Mm00515092_m1, Has3; Mm00476206_m1, hyaluronidase 1; and Mm01230689_g1, hyaluronidase 2. Primers used with SYBR Green reagents were as follows: GAGAACTGTGGCACCACGCAGTC (forward) and AGGATCCAGCTCCACTCGCTCTG (reverse) for intercellular adhesion molecule 1 (ICAM1); TCGGCTCGCAGTTGCGAAGT (forward) and GGCGGGTATTACCAAGGAGGATGC (reverse) for VCAM1.
For each group, assays were run on samples isolated from 2–4 replicate dishes. mRNA levels were then expressed as estimated copy numbers of mRNA using the relative standard curve method (Applied Biosystems). Levels of tropoelastin and fibulin 5 mRNA were quantified, and the degree of inhibition was determined by comparing mRNA levels in the shRNA-treated groups with those in negative control shRNA-treated groups.
Immunofluorescence Staining
Control or V3-expressing ASMCs were allowed to deposit ECM for 2 weeks postconfluence. Cells were fixed in 3.7% paraformaldehyde, blocked in 2% BSA (bovine serum albumin, Fraction V heat shock, Roche Applied Science) and 2% normal donkey serum (Jackson Immunoresearch) in PBS, prior to staining for tropoelastin (1:1000; rabbit polyclonal antiserum against bovine tropoelastin, a kind gift from Dr. Robert Mecham (St. Louis, MO)), followed by Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:500; Invitrogen). Nuclei of cells were visualized by TO-PRO-3 staining (1:1000; Invitrogen). In some experiments, human U937 promyelomonocytic cells (ATCC, Manassas, VA) were allowed to adhere to ECM deposited by control or V3-expressing ASMCs for 1 h at 4 °C. The cells were fixed and blocked as described above, prior to staining for the monocyte marker CD68 (1:100 dilution; mouse monoclonal KP-1 antibody against human CD68 (Abcam)) and tropoelastin (1:1000), followed by Alexa Fluor 488-conjugated donkey anti-mouse IgG and Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:500; Invitrogen) and nuclear counterstaining with TO-PRO-3 staining (1:1000; Invitrogen). Images were obtained and merged on a Leica SP5 confocal microscope.
Hyaluronan cable structures formed after tunicamycin treatment were immunostained and visualized as described previously (20). Briefly, control or V3-expressing ASMCs cultured on glass coverslips for 10 days were growth-arrested for 48 h in 0.1% FBS-containing culture medium followed by 24-h treatment with the endoplasmic reticulum (ER) stress inducer tunicamycin (5 μg/ml) in 10% FBS-containing culture medium. Some cells were treated with an EGFR inhibitor (5 μm; AG1478, Calbiochem) or NFκB activation inhibitor (5 μm; 6-amino-4-(4-phenoxyphenylethylamino)quinazoline, Calbiochem) for 1 h prior to tunicamycin stimulation to determine the role of EGFR- or NFκB-dependent signaling pathways in generating ER stress-induced hyaluronan cable structures. Coverslips were then fixed in acid-formalin-ethanol (3.7% paraformaldehyde, 5% acetic acid, and 70% ethanol, v/v/v), followed by rinsing in PBS. Cells were stained for hyaluronan using biotinylated hyaluronan-binding protein (21) followed by streptavidin Alexa Fluor 488 in PBS containing 1% BSA. Nuclei were stained with DAPI. Images were acquired and merged on a Leica DMIRB inverted microscope, which was equipped for fluorescent epi-illumination, using a Diagnostic Instruments Pursuit 4.0-megapixel chilled color CCD camera and Spot software, version 4.5.9.1.
TGFβ-blocking Experiments
Control or V3-expressing ASMCs were treated with pharmacological inhibitors against TGFBRI (SB431542, Calbiochem), IGF1R (AG1024, Calbiochem), or EGFR2 (AG825, Calbiochem) at 5 μm for 6 days. In some experiments, ASMCs were treated with blocking antibodies against all isoforms of TGFβ (10 μg/ml; AB-100, R&D Systems), TGFβ1 (1 μg/ml; AF-101, R&D Systems), or TGFβ3 (1 μg/ml; AF-243, R&D Systems) for 6 days. The levels of tropoelastin and fibulin 5 mRNA with or without the treatments were examined by using qPCR as described above.
Tropoelastin and Fibulin 5 Knockdown
To determine whether monocyte adhesion is dependent on elastic fiber accumulation in ECM deposited by ASMCs, tropoelastin, a soluble monomeric subunit of elastic fiber, or fibulin 5, a scaffold protein of microfibrils critical in elastic fiber formation (22–24), was knocked down using lentiviral shRNA (Santa Cruz Biotechnology, Inc.). Briefly, control or V3-expressing ASMCs were infected with lentiviral particles containing tropoelastin shRNA sequences (CUGCAUCCAAAGCUGCUAA, CCACUAUCAACCUGGUUCA, and CACAUGCAGUACUGUAAUC) or fibulin 5 (CCUAUUCCUGUACGUGUAA, GGUCUUGCCAAGAUGUGAA, and CUCUAUCUCUUGCUGCAUU) at a multiplicity of infection of 0.5. Lentiviral particles containing nonsense shRNA sequence (Santa Cruz Biotechnology, Inc.) were used as a negative control. ASMCs expressing lentiviral shRNA were then selected by puromycin resistance. Knockdown efficiency of shRNA was determined by measuring tropoelastin and fibulin 5 mRNA levels using real-time PCR as described above.
Monocyte Adhesion Assay on the ECM Generated by ASMCs
Monocyte binding to ECM generated by control LXSN or ASMCs expressing V3 was assayed as described previously (10, 14) with some modifications. Control or V3-expressing ASMCs were grown for 2 weeks. The human monocytic cell line, U937 (ATCC), was labeled with 5 μg/ml calcein AM (Invitrogen) in the dark for 45 min at 37 °C and washed to remove unincorporated calcein. The labeled U937 cells suspended in ice-cold RPMI medium 1640 without phenol red (Invitrogen) were added to ASMCs in 96-well plates (3 × 105 cells/well) and allowed to bind for 90 min at 4 °C. Non-bound monocytic cells were removed by washing with cold RPMI medium. Monocyte binding was measured by exciting the calcein incorporated into the monocytes at 485 nm and reading fluorescence at 530 nm using a Fusion Series Universal Microplate Analyzer (PerkinElmer Life Sciences) (14).
Some ASMCs were treated with the ER stress inducer tunicamycin (5 μg/ml) in culture medium for 18 h at 37 °C prior to assaying for monocyte adhesion (12). ECM generated by control or V3-expressing ASMCs was treated with Streptomyces hyaluronidase (1 unit/ml; Seikagaku) at 37 °C for 30 min prior to adding monocytes to determine the role of hyaluronan in monocyte binding.
Some cells were treated with an EGFR inhibitor (5 μm; AG1478, Calbiochem) or NFκB activation inhibitor (5 μm; 6-amino-4-(4-phenoxyphenylethylamino)quinazoline, Calbiochem) for 1 h prior to tunicamycin stimulation to determine the role of EGFR- or NFκB-dependent signaling pathways in hyaluronan-dependent monocyte adhesion.
Monocyte Adhesion on Elastin-coated Plates
To address whether elastin resists monocyte adhesion in a dose-response manner, 96-well non-tissue culture-treated polystyrene plates were coated with 0, 10, and 100 μg/ml soluble elastin (extracted from bovine ligament by hot oxalic acid, Elastin Products Co.). The monocyte adhesion assay was performed on these elastin-coated plates as described above. In another experiment, the monocyte adhesion assay was performed on tissue culture-treated polystyrene plates coated with 20 μg/ml type I rat tail collagen (BD Biosciences), soluble elastin, or κ-elastin (Elastin Products Co.).
Western Blotting
For evaluating the impact of V3 expression on EGFR-dependent signaling pathways, control or V3-expressing ASMCs cultured for 24 h were growth-arrested in culture medium containing 0.1% FBS for 48 h, followed by stimulation with or without 25 ng/ml EGF (Sigma-Aldrich) for 2 min. At these time points, the levels of pEGFR were greatest in control ASMCs after EGF stimulation in the initial time course experiments. Total cell lysates were harvested in ice-cold cell lysis buffer (50 mm HEPES, 150 mm NaCl, 1% Nonidet P-40, 5 mm NaF, 2 mm Na3VO4, supplemented with protease inhibitor mixture (P8340, Sigma-Aldrich)), followed by sonication for 30 s. Total protein concentration was determined by BCA assay as directed by the manufacturer (Thermo Scientific). Total cell lysates (20 μg/lane) were run on 8% SDS-PAGE and transferred to a nitrocellulose membrane at 10 V constant voltage overnight at 4 °C. Membranes were fixed with 0.05% glutaraldehyde for 10 min, prior to blocking in 2% BSA in Tris-buffered saline with 0.5% Tween 20 (pH 7.4) and incubated with anti-phospho-EGFR (Tyr-1068; Cell Signaling), anti-EGFR (Cell Signaling), anti-phospho-p85/p55 (Tyr-458/Tyr-199) regulatory subunit of PI3K (Cell Signaling), anti-phospho-NFκB p65 (Ser-536; Cell Signaling), anti-NFκB p65 (Cell Signaling) or anti-β-actin (Sigma-Aldrich) at 1:1000 dilutions, followed by incubations with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibodies (1:80,000; Jackson Immunoresearch) and with a chemiluminescent substrate West Dura (Pierce). In some experiments, cytoplasmic proteins were harvested from control or V3-expressing ASMCs using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific) according to manufacturer's protocols. Cytoplasmic proteins (20 μg/lane) were resolved on 8% SDS-PAGE and transferred to nitrocellulose membranes as described above, followed by incubation with anti-EGFR (Cell Signaling), anti-p85 PI3K (1:1000 dilutions; Cell Signaling), anti-NFκB p65 (1:1000; Cell Signaling), anti-ICAM1 (1:500; Sigma-Aldrich), anti-VCAM1 (1:200; Santa Cruz Biotechnology), or anti-β-actin (1:1000; Abcam) followed by incubations with HRP-conjugated anti-rabbit secondary antibodies (1:80,000; Jackson Immunoresearch), HRP-conjugated anti-goat secondary antibodies (1:10,000), or HRP-conjugated anti-mouse secondary antibodies (1:10,000) and with a chemiluminescent substrate West Dura (Pierce). For reprobing, membranes were stripped by Restore Western blot stripping buffer (Pierce) as directed by manufacturer, followed by washing in Tris-buffered saline with 0.5% Tween 20 and blocking as described above. Densitometry was performed using ImageJ version 1.44.
Hyaluronan Quantification
Hyaluronan secretion into the medium and accumulation in the cell layer were quantified by a competitive enzyme-linked sorbent assay (ELSA), as previously described (25). Briefly, conditioned media and cell layers from ASMC cultures were digested with Pronase (500 μg/ml) overnight at 37 °C, followed by heat inactivation at 100 °C for 10 min. Digested media and cell layers were mixed with biotinylated hyaluronan-binding protein for 1 h at room temperature and added to microtiter plates coated with BSA-conjugated hyaluronan. The plates were then incubated with peroxidase-labeled streptavidin for 20 min and incubated with peroxidase substrate (0.03% H2O2, 0.5 mg/ml 2,2′-azino-bis(3-ethylbenzthiazoline)6-sulfonic acid in 0.1 m sodium citrate, pH 4.2). The absorbance at 405 nm was measured, which is inversely proportional to the level of hyaluronan in the sample that was calculated from a standard curve.
Lymphocyte Function-associated Antigen 1 (LFA1) and Very Late Antigen 4 (VLA4) Blocking Experiment
Calcein-labeled U937 cells were incubated in the presence of anti-integrin αL (anti-LFA1, 4 μg/ml; clone HI111, Biolegend) or anti-integrin α4 (anti-VLA4, 4 μg/ml; clone 2B4, R&D Systems) in ice-cold RPMI medium for 1 h. These antibodies were previously demonstrated as blocking interactions between LFA1 and ICAM1 (26) or between VLA4 and VCAM1 (27). These cells were then used in monocyte adhesion assays as described above.
Statistical Analysis
All data are expressed as the average ± S.E. unless otherwise specified. Differences were identified by two-tailed Student's t tests for the comparison of two groups and by one-way analysis of variance (ANOVA) followed by Tukey's post hoc tests for the comparison of three or more groups and were regarded significant if p was <0.05.
RESULTS
V3 Expression Enhances Elastic Fiber Formation and mRNA Expression of Tropoelastin and Fibulin 5
To determine whether V3 expression affects elastic fiber components at transcript levels, tropoelastin and fibulin 5 mRNA levels were determined in samples obtained from four independent transductions of empty vector (LXSN) control and V3 vector (LV3SN)-transduced ASMC lines. Elastic fiber deposition was examined by immunofluorescence using anti-tropoelastin antibodies after 3 weeks of culture. Consistent with our previous studies (19, 28), V3 expression by ASMCs significantly increased mRNA levels of tropoelastin as well as fibulin 5, a component of microfibrils critical in elastic fiber formation (Fig. 1, A and B) (22–24). Fibulin 1 and fibrillin 1 mRNA levels did not differ between V3-expressing ASMCs and control ASMCs (data not shown). The ECM generated by V3-expressing ASMCs (V3-generated ECM) was significantly enriched in elastic fibers compared with that generated by control ASMCs, as determined by immunocytochemistry (Fig. 1, C and D). These findings indicate that enhanced elastogenesis induced by V3 expression involves increasing tropoelastin and fibulin 5 at transcript levels.
FIGURE 1.
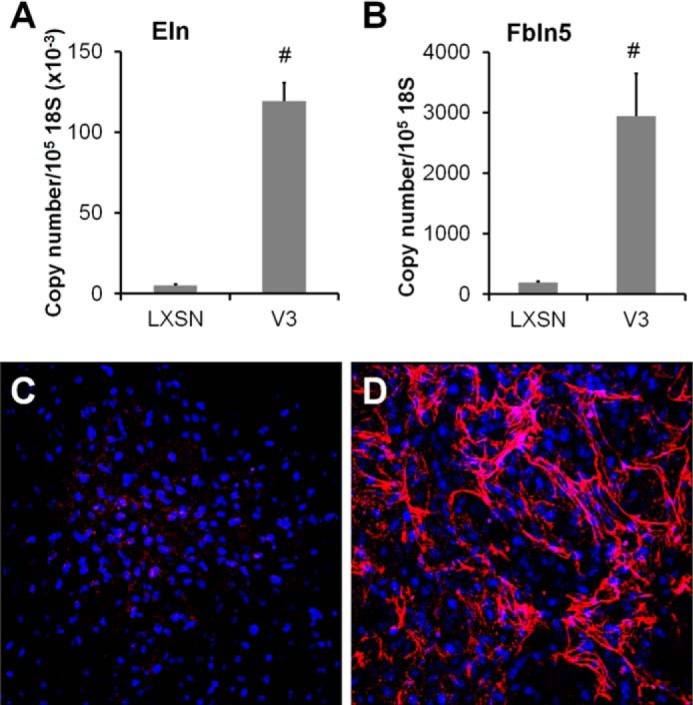
Expression of V3 by ASMCs enhances elastogenesis. A and B, total RNA was obtained from control or V3-expressing ASMCs, followed by qPCR as described under “Materials and Methods.” Tropoelastin (A) and fibulin 5 (B) mRNA levels were determined in samples obtained from four independent transductions of empty vector (LXSN) control and V3 vector (LV3SN)-transduced ASMC lines cultured for 2 weeks. #, p < 0.05 in Student's t test. C and D, elastic fiber accumulation in control LXSN (C) or V3-generated ECM (D) after 3 weeks of culture was examined by immunostaining against tropoelastin (red). Cell nuclei staining is shown in blue. Immunofluorescence images are representative of three independent experiments. Error bars, S.E.
ECM Generated by V3-expressing ASMCs Resists Monocyte Adhesion in an Elastin-dependent Manner
The ECM formed by V3-expressing ASMCs had reduced capacity to bind monocytes compared with control ASMCs expressing only the empty vector LXSN (Fig. 2A). To evaluate whether the accumulation of elastic fibers affected monocyte adhesion, the distribution of elastin and adherent monocytes was examined by immunofluorescence against the monocyte marker CD68 and tropoelastin. The number of adherent monocytes was greater in control ECM (green staining in Fig. 2, B and C) that contained less elastin (red staining in Fig. 2, B–E) than in V3-generated ECM (Fig. 2, D and E). Also, adherent monocytes on the V3-generated ECM were located in areas with fewer elastic fibers (Fig. 2, D and E).
FIGURE 2.
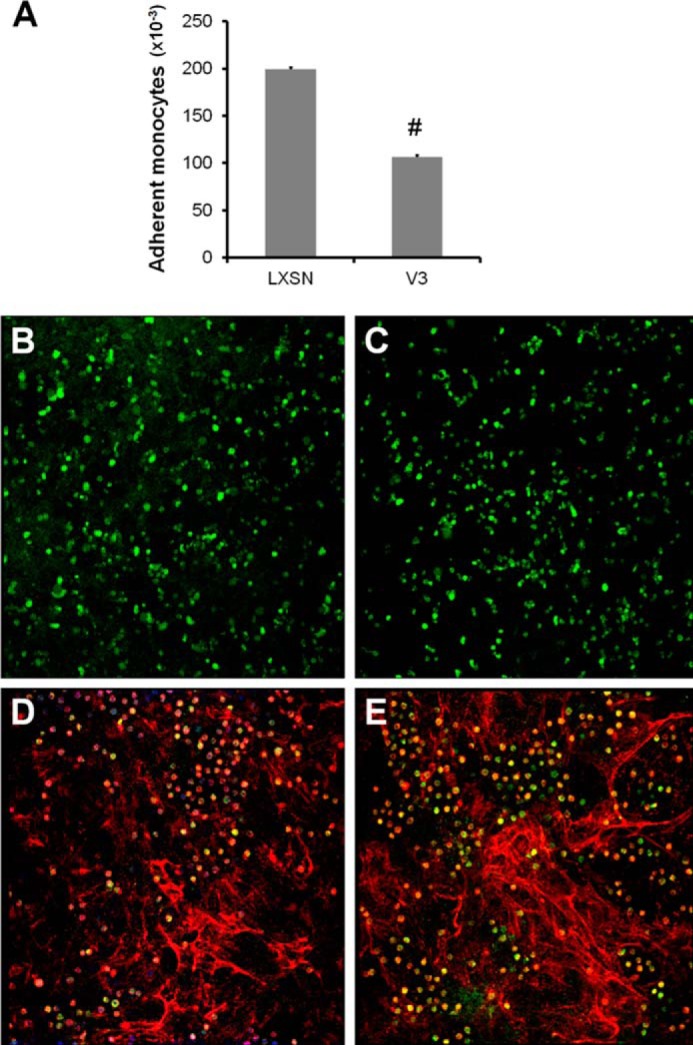
Elastic fiber-enriched ECM generated by V3-expressing ASMCs negatively correlates with monocyte adhesion. Control or V3-expressing ASMCs were allowed to deposit ECM for 2 weeks postconfluence. Human U937 promyelomonocytic cells (ATCC) were allowed to adhere to ECM deposited by control or V3-expressing ASMCs, followed by immunofluorescent staining against the monocyte marker CD68, tropoelastin, and nuclei (TO-PRO-3), as described under “Materials and Methods.” A, monocyte adhesion was performed as described under “Materials and Methods” in an independent experiment and quantified by examining fluorescently labeled monocytes adhering to the ECM. The graphs are representative of three independent experiments. B and C, control ECM had numerous adherent monocytes (green), whereas tropoelastin staining (red) was minimal. D and E, V3-generated ECM had areas that had abundant elastic fibers that had fewer adherent monocytes, indicating a negative correlation between elastin and monocyte adhesion. Immunofluorescent images are representative of three independent experiments. Error bars, S.E.
To determine whether V3-generated ECM resists monocyte adhesion in an elastic fiber-dependent manner, tropoelastin or fibulin 5 was knocked down in control and V3-expressing ASMCs using lentiviral shRNA. Elastin shRNA knockdown efficiency was 43 ± 1 and 50 ± 9% in control and V3-expressing ASMCs, respectively, whereas the efficiency of shRNA-mediated knockdown of fibulin 5 mRNA was 51 ± 3 and 47 ± 5%, in control and V3-expressing ASMCs, respectively. As shown in Fig. 3A, monocyte adhesion on V3-generated ECM treated with control shRNA was significantly lower than adhesion to LXSN-generated ECM (#, p < 0.05, 65 ± 2% of control shRNA-treated LXSN-ECM). Knocking down either tropoelastin or fibulin 5 significantly increased monocyte adhesion on V3-generated ECM (Fig. 3A, 77 ± 2 and 83 ± 3% of control shRNA-treated LXSN-ECM). In order to test whether elastin resisted monocyte adhesion, the effect on monocyte adhesion of coating non-tissue culture-treated polystyrene plates with varying concentrations of elastin was determined as shown in Fig. 3B. Monocyte adhesion was significantly reduced when the plates were coated with 10 and 100 μg/ml soluble elastin when compared with untreated polystyrene surfaces, whereas 1 μg/ml elastin did not significantly affect the number of adherent monocytes. When performed on tissue culture-treated polystyrene plates coated with collagen I, κ-elastin, or soluble elastin, fewer monocytes adhered to κ-elastin or soluble elastin-coated plates compared with collagen I-coated plates, indicating that elastin negatively regulates monocyte adhesion compared with other ECM molecules, such as collagen I (data not shown). These findings indicate that the elastin-enriched ECM generated by V3-expressing ASMCs is a poor substrate for monocyte adhesion and is partially responsible for the antiadhesive effect of V3 expression on monocyte adhesion.
FIGURE 3.

V3-generated ECM resisted monocyte adhesion in an elastin-dependent manner. A, control or V3-expressing ASMCs were infected with lentiviral particles containing nonsense control shRNA or shRNA sequences specific for tropoelastin or fibulin 5 (Santa Cruz Biotechnology) at a multiplicity of infection of 0.5 and were selected by puromycin resistance. These cells were grown for 2 weeks, followed by monocyte adhesion assays as described under “Materials and Methods.” Fewer monocytes adhered to the V3-generated ECM than to control LXSN ECM after treatment with negative control shRNA (ctrl shRNA; #, p < 0.05 in one-way ANOVA followed by Tukey's post hoc tests). Monocyte adhesion on V3-generated ECM significantly increased when treated with shRNA against tropoelastin or fibulin 5 compared with control shRNA-treated V3-generated ECM (ELN or FBLN5 shRNA; *, p < 0.05 in one-way ANOVA followed by Tukey's post hoc tests). B, the effect of elastin on monocyte adhesion was tested by quantifying adherent monocytes on untreated polystyrene plates coated with various concentrations of soluble elastin, as described under “Materials and Methods.” Monocyte adhesion significantly decreased when surfaces were coated with 10 or 100 μg/ml elastin (#, p < 0.05 compared with untreated surfaces). Error bars, S.E.
Enhanced Tropoelastin and Fibulin 5 mRNA Expression in V3-expressing ASMCs Is Dependent on TGFβ Signaling Pathways
TGFβ up-regulates elastin at the transcript level by affecting tropoelastin promoter activity and by enhancing tropoelastin transcript stability (29–32). To determine whether the TGFβ signaling pathway is responsible for the increase in tropoelastin transcript levels induced by V3 expression, tropoelastin mRNA levels were examined in control and V3-expressing ASMCs with or without the presence of a pharmacological inhibitor against TGFβ signaling, SB431542, or neutralizing antibodies against TGFβ. As shown in Fig. 4A, SB431542 completely abolished the increase in tropoelastin expression in V3-expressing ASMCs, whereas pharmacological inhibitors against IGF1R (AG1024) or EGFR2 (AG825) had only minimal effects. Consistently, neutralizing antibodies against pan-TGFβ or TGFβ1 also significantly reduced tropoelastin expression, whereas antibodies against TGFβ3 had no effect (Fig. 4B). The increase in fibulin 5 mRNA expression induced by V3 expression was also dependent on TGFβ signaling. The pharmacological inhibitor SB431542 (Fig. 4C) and blocking antibodies against pan-TGFβ or TGFβ1 (Fig. 4D) had effects similar to those shown for tropoelastin expression. These findings show that the enhanced expression of tropoelastin and fibulin 5 in V3-expressing ASMCs is mediated by TGFβ signaling.
FIGURE 4.

Blocking TGFβ signaling inhibits elastogenic phenotype of V3-expressing ASMCs. A, control or V3-expressing ASMCs were treated with pharmacological inhibitors against TGFBRI (SB431542, Calbiochem), IGF1R (AG1024, Calbiochem), or EGFR2 (AG825, Calbiochem) at 5 μm for 6 days. Tropoelastin mRNA levels were examined from total RNA isolated from control and V3-expressing ASMCs by qPCR. B, control or V3-expressing ASMCs were treated with blocking antibodies against all isoforms of TGFβ (Pan TGFβ Ab; 10 μg/ml, AB-100, R&D Systems), TGFβ1 (TGFβ1 Ab; 1 μg/ml, AF-101, R&D Systems), or TGFβ3 (TGFβ3 Ab; 1 μg/ml, AF-243, R&D Systems) for 6 days. Tropoelastin mRNA levels were examined as described above. C and D, fibulin 5 mRNA levels were examined from total RNA isolated from control and V3-expressing ASMCs treated with or without pharmacological inhibitors (C) or blocking antibodies (D) described in A and B. *, p < 0.05 in one-way ANOVA followed by Tukey's post hoc tests. Error bars, S.E.
Expression of V3 by ASMCs Reduces Hyaluronan Accumulation
Because inhibition of elastin deposition only partially reversed the inhibition of monocyte adhesion to the ECM deposited by V3-expressing ASMCs, experiments were designed to determine whether V3 induces changes in hyaluronan that are responsible for affecting monocyte adhesion. When the total amount of hyaluronan in the control and V3-generated ECM was quantitated by ELSA, the V3-generated ECM had reduced levels of hyaluronan in the cell layer and in the medium (Fig. 5A) compared with the ECM generated by control cells. Moreover, V3-expressing ASMCs had reduced levels of hyaluronan synthase 1 (Has1) mRNA, whereas Has2 and -3 mRNA did not change significantly (Fig. 5B). Hyaluronidase 1 or 2 mRNA levels did not change significantly (data not shown). These data indicate that the decrease in hyaluronan accumulation in V3-expressing ASMCs is due to decreased synthesis of hyaluronan.
FIGURE 5.
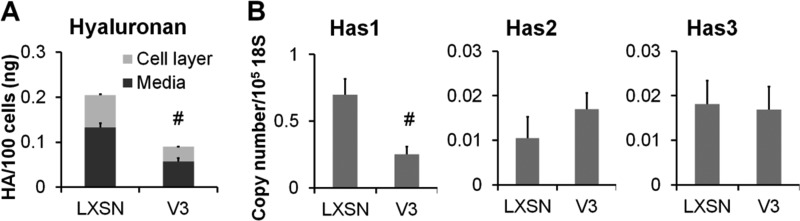
V3-expressing ASMCs had reduced hyaluronan accumulation. A, hyaluronan accumulated in the cell layers and media of control or V3-expressing ASMCs cultured for 2 weeks and quantified by ELSA as described under “Materials and Methods.” #, p < 0.05 in Student's t test. B, relative copy numbers of Has1, Has2, and Has3 mRNA levels were determined from samples obtained from four independent transductions of LXSN control and V3-transduced ASMC lines cultured for 2 weeks by qPCR as described under “Materials and Methods.” #, p < 0.05 in Student's t test. Error bars, S.E.
Expression of V3 Attenuates EGFR- and NFκB-dependent Signaling Pathways
Hyaluronan synthesis is regulated by a number of signaling pathways, including the EGFR-PI3K/ERK signaling network (33–37). Moreover, hyaluronan synthases have NFκB binding regions in their promoters that are regulated by NFκB agonists, such as interleukin 1β (IL-1β) and tumor necrosis factor α (TNFα) (25, 38–40). Previous studies have shown that V3 expression in melanoma cells interferes with the CD44/EGFR signaling pathways (41, 42). Therefore, the effect of V3 expression by ASMCs on EGF-induced signaling was examined. Western blot analysis of the activation of EGF-induced signaling pathways revealed that activation of EGFR and downstream signaling mediators PI3K and NFκB p65 was attenuated in V3-expressing ASMCs (Fig. 6A). Levels of total PI3K p85, but not EGFR or NFκB p65, were significantly decreased in V3-expressing ASMCs (Fig. 6B), suggesting that the altered signaling may be partially due to decreases in the levels of signaling mediators.
FIGURE 6.

Expression of V3 by ASMCs blocks EGFR signaling. A, control or V3-expressing ASMCs grown for 24 h followed by growth arrest in culture medium containing 0.1% FBS for 48 h were stimulated with 25 ng/ml EGF for 2 min. Total cell lysates collected from unstimulated or EGF-stimulated cells were examined by Western blotting against phospho-EGFR (pEGFR) (Tyr-1068), anti-phospho-p85/p55 (Tyr-458/Tyr-199) PI3K (pPI3K), anti-phospho-NFκB p65 (Ser-536) (pNFκB), and β-actin, as described above. Levels of protein bands were normalized by loading control β-actin by densitometry. Normalized densitometry values of protein bands were quantified from three independent experiments. *, p < 0.05 in Student's t test compared with EGF-stimulated LXSN controls. B, levels of total EGFR, p85 PI3K, and p65 NFκB were examined in three independent isolates of cytoplasmic protein harvested from control or V3-expressing ASMCs. Levels of protein bands were normalized by loading control β-actin by densitometry. *, p < 0.05 in Student's t test compared with LXSN controls. Error bars, S.E.
ECM Generated by V3-expressing ASMCs Reduces Hyaluronan-dependent Monocyte Adhesion
Hyaluronan cable-like structures generated by inflammatory stimuli, such as viral infection or ER stress, support monocyte adhesion (5–15). Formation of hyaluronan cable structures induced by tunicamycin stimulation in control ASMCs (Fig. 7A) was blocked in V3-expressing ASMCs (Fig. 7B). Likewise, hyaluronan-dependent monocyte adhesion present in control ECM induced by ER stress was abolished on the V3-generated ECM (Fig. 7C).
FIGURE 7.
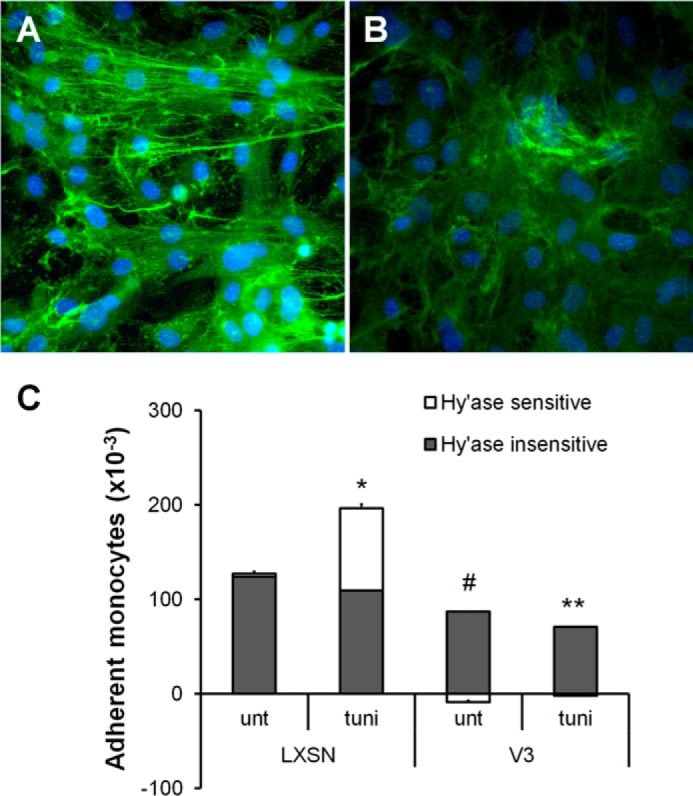
V3-expressing ASMCs had reduced hyaluronan-dependent monocyte adhesion induced by ER stress. A and B, control (A) or V3-expressing ASMCs (B) were cultured for 2 weeks, followed by 48-h serum starvation and 24-h treatment with the ER stress inducer tunicamycin. Cells were fixed in acid-formalin-ethanol and stained for hyaluronan using biotinylated hyaluronan-binding protein followed by strepavidin Alexa Fluor 488. Nuclei were stained with DAPI. C, monocyte adhesion assays were performed on control or V3-expressing ASMCs cultured for 2 weeks, serum-starved for 48 h, and treated with tunicamycin for 24 h, as described under “Materials and Methods.” *, p < 0.05 in Student's t test comparing hyaluronan-dependent monocyte adhesion on tunicamycin-treated control cells to untreated control cells. #, p < 0.05 in Student's t test comparing hyaluronan-dependent monocyte adhesion on control and V3-generated ECM. **, p < 0.05 in Student's t test comparing hyaluronan-dependent monocyte adhesion on V3 to LXSN with tunicamycin treatments. Error bars, S.E.
Because expression of V3 represses activation of EGFR and NFκB in ASMCs, formation of hyaluronan cables induced by ER stress was examined when the EGFR and NFκB-dependent signaling pathways were inhibited using pharmacological inhibitors (AG1478 or 6-amino-4-(4-phenoxyphenylethylamino) quinazoline, respectively). Cable formation induced in control cultures by ER stress (Fig. 8A) was repressed in the presence of either EGFR or NFκB inhibitors (Fig. 8, B and C). V3-expressing ASMCs did not generate hyaluronan cables with tunicamycin stimulation (Fig. 8D). Moreover, in control cultures, hyaluronan-dependent monocyte adhesion induced by ER stress was abolished by inhibition of EGFR or NFκB (Fig. 8E). Overall, these findings indicate that V3 expression by ASMCs blocks signaling pathways mediating hyaluronan synthesis, including EGFR and NFκB signaling pathways, and that the ER stress-induced hyaluronan cable formation, supporting monocyte adhesion, is absent in the V3-generated ECM.
FIGURE 8.
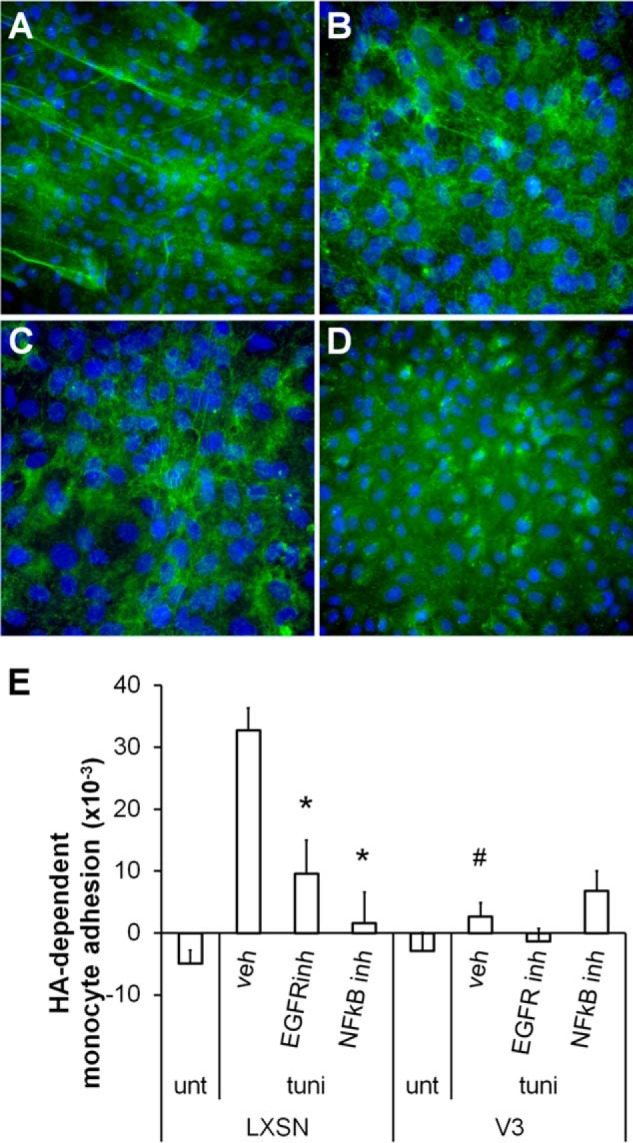
Blocking EGFR or NFκB signaling reduced hyaluronan-dependent monocyte adhesion induced by ER stress. A–D, control or V3-expressing ASMCs cultured for 2 weeks, followed by 48-h serum starvation, were pretreated with inhibitors against EGFR (5 μm; AG1478) or NFκB (5 μm; 6-amino-4-(4-phenoxyphenylethylamino)quinazoline) for 1 h prior to 24-h tunicamycin stimulation. Hyaluronan was visualized by biotinylated hyaluronan-binding protein, as described above. In control ASMCs, ER stress-induced hyaluronan cable structure formation (A) was abolished by pharmacological inhibitors against EGFR (B) or NFκB (C). Formation of hyaluronan cable structures induced by ER stress was absent in V3-expressing ASMCs (D). E, hyaluronan-dependent monocyte adhesion in control ASMCs induced by ER stress was significantly reduced by inhibiting EGFR or NFκB. *, p < 0.05 in one-way ANOVA followed by Tukey's post hoc tests comparing controls and treatments with different inhibitors. #, p < 0.05 in Student's t test comparing monocyte adhesion on control and V3-generated ECM. Error bars, S.E.
VCAM1-dependent Monocyte Adhesion Is Reduced on V3-expressing ASMCs
Although our data show that V3 expression alters components of the ECM that affect monocyte adhesion, it was not known whether V3 also modulates cell surface molecules critical for monocyte adhesion. Because V3 expression attenuates activation of NFκB p65, we examined a number of NFκB-responsive proinflammatory cell adhesion molecules that mediate monocyte adhesion, such as VCAM1 and ICAM1. V3-expressing ASMCs had significantly less VCAM1 and ICAM1 at transcript levels (Fig. 9A), and VCAM1 was significantly reduced at protein levels (Fig. 9B). To examine whether V3 expression affects monocyte adhesion mediated by these cell surface receptors, the ligands of VCAM1 or ICAM1, VLA4, or LFA1 on monocytes, respectively, were blocked by neutralizing antibodies prior to the monocyte adhesion assay. Blocking VLA4, but not LFA1, on monocytes significantly reduced the number of adherent cells on both control and V3-expressing ASMCs, indicating that VLA4-VCAM1 interaction was the major factor for supporting monocyte adhesion on the ASMCs (Fig. 9C). Importantly, the degree of reduction in monocyte adhesion induced by blocking VLA4 was significantly less in V3-expressing ASMCs than in control cells, indicating that the V3-expressing ASMCs support reduced amounts of VCAM1-dependent monocyte binding (Fig. 9D). These data demonstrate that reduced monocyte adhesion on V3-expressing ASMCs was due to a decrease in VCAM1, a critical component supporting monocyte adhesion on the ASMCs.
FIGURE 9.

V3-expressing ASMCs had reduced VCAM1 expression and VCAM1-dependent monocyte adhesion. A and B, expression of VCAM1 and ICAM1 in control and V3-expressing ASMCs cultured for 7 days was detected at mRNA (A) and protein (B) levels using real-time PCR and Western blotting as described under “Materials and Methods.” The data presented are representative of three independent experiments. *, p < 0.05 in Student's t test compared with control LXSN. C, control or V3-expressing ASMCs cultured for 1 week. Prior to monocyte adhesion assays, monocytes were incubated in the presence of function-blocking anti-LFA1 (4 μg/ml) or anti-VLA4 (4 μg/ml) or both antibodies (2 μg/ml each) in ice-cold RPMI medium for 1 h. These cells were added to control or V3-expressing ASMCs. Blocking VLA4, but not LFA1, significantly reduced monocyte adhesion (*, p < 0.05). unt, untreated. D, the degree of VCAM1- or ICAM1-dependent monocyte adhesion was calculated by subtracting the number of adherent monocytes after blocking VLA4 or LFA1 from the number of adherent monocytes without antibody treatment. VCAM1-dependent monocyte adhesion was significantly reduced in V3-expressing ASMCs (#, p < 0.05). Error bars, S.E.
DISCUSSION
In the present study, we have demonstrated that V3-expressing ASMCs resist monocyte adhesion by affecting multiple components within the microenvironment, first by generating an ECM enriched in elastic fibers and depleted in hyaluronan and, second, by reducing expression of proinflammatory cell adhesion molecules, such as VCAM1. These effects are mediated by the differential regulation of TGFβ-, EGFR-, and NFκB-dependent signaling pathways.
Elastic Fibers and Monocyte Adhesion
Interaction of ECM macromolecules critically regulates leukocyte trafficking and differentiation during inflammation in a complex manner (43–45). Upon adhesion to ECM molecules, such as collagen and fibronectin, β1 integrin facilitates leukocyte infiltration into the site of injury (43–45). In contrast, some ECM structures are poor substrates for leukocyte accumulation and activation, such as elastin (46, 47). Elastic laminae extracted from rat aorta are resistant to leukocyte adhesion and transmigration in vivo and in vitro compared with the collagen-enriched adventitia extracted from the same aorta (46, 47). Deficiency in fibulin 2 and fibulin 5 in animals results in disrupted elastic laminae accompanied by an increase in vascular adhesion molecules and tissue factor expression as well as thrombus formation after carotid artery ligation injury (23), suggesting that the elastin-enriched ECM present in healthy aortas protects against vascular injury. Previously, we have demonstrated that expression of the V3 isoform of versican in ASMCs generates an ECM enriched in elastic fibers and that this ECM had reduced capacity to bind monocytes (4, 18, 19, 28). We now show a causal relationship between changes in elastin accumulation and monocyte adhesion such that knocking down tropoelastin or fibulin 5, a component of microfibrils critical for elastic fiber formation (22–24), partially reverses the inhibition of monocyte adhesion caused by V3 expression.
Expression of V3 Enhances Elastogenesis by Modulating TGFβ-dependent Signaling Pathways
V3 is the smallest splice variant of versican and lacks the glycosaminoglycan binding domain and thus contains no chondroitin sulfate chains (48). Previous studies have demonstrated that chondroitin sulfate interferes with elastin deposition through blocking the interaction of the 67-kDa elastin-binding protein with the cell surface (49–53). In addition, we previously showed that V3 induction of elastic fibers was due to decreases in the V0/V1 chondroitin sulfate-containing isoforms of versican (17) and also involved stimulation of tropoelastin mRNA expression (19). We now show that increased tropoelastin and fibulin 5 expression caused by V3 expression is regulated through TGFβ signaling. TGFβ is one of the key cytokines that up-regulates tropoelastin by affecting tropoelastin promoter activity and by enhancing tropoelastin transcript stability (29–32). Our finding that fibulin 5, a key component for proper elastic fiber formation (22–24), is also up-regulated by V3 through TGFβ signaling in these ASMCs suggests that V3 expression may also be affecting microfibrillar protein accumulation and influencing the role of fibulin 5 in scaffold formation during elastic fiber deposition (28).
The mechanisms by which V3 affects TGFβ signaling are currently under investigation. The G3 domain within V3 can interact with the β1 integrin (54–56), which has been shown to interact with and activate latent TGFβ (57, 58). We have shown previously that V3-expressing ASMCs reduce pericellular cell coat enriched in hyaluronan (18) as well as reduce the amount of chondroitin sulfate-bearing proteoglycans, such as the larger versican isoforms V0/V1 (4). Based on these findings, it is possible that V3 may directly or indirectly alter the mechanical environment by interacting with the β1 integrin or by reducing pericellular matrix enriched in hyaluronan and chondroitin sulfate-bearing isoforms of versican. This altered mechanical environment or integrin signaling may result in elevated contractile force exerted by cells, which in turn activates latent TGFβ (59–67).
The Impact of V3 on Hyaluronan Accumulation and Hyaluronan-dependent Monocyte Adhesion
Because our findings suggest that the antiadhesive effect of elastin may only partially explain the decrease in monocyte adhesion on V3-generated ECM, we explored whether V3 expression in ASMCs affects other ECM components, such as hyaluronan because hyaluronan modulates elastic fiber deposition as well as monocyte adhesion (5–15, 68, 69). Indeed, our data show that hyaluronan accumulation as well as Has1 levels are reduced by V3 expression. Interestingly, a previous study by our group has shown that the ECM produced by ASMCs overexpressing Has1 bound more monocytes than ECMs produced by ASMCs overexpressing Has2 and Has3 (70). Such results implicate Has1 in this monocyte-adhesive ECM phenotype. Hyaluronan synthesis is regulated by a number of pathways, including the EGFR and NFκB signaling pathways (71). Hyaluronan triggers the CD44-EGFR signaling pathway, which up-regulates hyaluronan synthesis in a positive feedback mechanism (33–37). Moreover, hyaluronan synthases have NFκB binding regions in their promoters and are up-regulated by NFκB agonists, such as IL-1β and TNFα (25, 38–40). The data presented here demonstrate that V3 expression blocks the activation of EGFR and downstream NFκB in ASMCs, which is consistent with previous studies showing that V3 expression in cancer cells blocked the CD44-EGFR signaling pathway (41, 42). Furthermore, V3-expressing ASMCs had reduced levels of PI3K p85 downstream of EGFR, indicating that the blockade of EGFR-dependent signaling may be partially due to decreases in signaling mediators. Because V3 has EGF-like motifs that have been shown to affect EGF-dependent signaling pathways (72–74), the blockade of EGFR-dependent signaling might be mediated by the G3 domain within V3. These possibilities are currently under investigation.
Hyaluronan accumulation in the ECM has also been shown to promote monocyte accumulation when cells are stimulated by inflammatory conditions, such as viral infection or ER stress (5–15). This involves remodeling hyaluronan into cable-like structures cross-linked with other molecules interacting with hyaluronan, such as inter-α-trypsin inhibitor (IαI), tumor necrosis factor α-stimulated gene 6 (TSG-6), and versican (6, 10–13). Recently, we have shown that blocking the hyaluronan-binding region of versican during ECM formation significantly reduced hyaluronan-dependent monocyte binding to the ECM generated by poly(I:C)-stimulated lung fibroblasts (14), indicating that versican bound to hyaluronan has a role in regulating monocyte adhesion. Our data now show that hyaluronan-dependent monocyte binding as well as hyaluronan cable formation induced by ER stress were significantly reduced by V3 expression, suggesting that V3 may function as a dominant negative isoform against the larger isoforms such as V0/V1 and that this may further affect hyaluronan-dependent monocyte binding to ASMCs by reducing hyaluronan accumulation. Whether V3 blocks other signaling pathways induced by ER stress is not yet clear and is under investigation.
Reduction of VCAM1-dependent Monocyte Adhesion by V3 Expression
Components other than those present in the ECM, such as cell surface molecules, including ICAM1 and VCAM1, critically mediate monocyte adhesion and migration in stroma during inflammatory events (75). In addition to the ECM components that affect monocyte adhesion, our study now shows that V3 expression also interferes with expression of proinflammatory cell adhesion molecules, such as VCAM1, which are critical in leukocyte and myeloid cell activation and accumulation. VCAM1 expression is regulated by NFκB (76–78), and given that V3 expression led to decreased activation of NFκB, the reduction in VCAM1 expression is probably due to a reduction of NFκB activation in V3-expressing ASMCs. Previously, ECM molecules, such as V0/V1 versican and hyaluronan oligomers, have been shown to bind and activate Toll-like receptors, which trigger NFκB signaling pathways (79, 80). Interestingly, hyaluronan has previously been shown to increase VCAM1 expression via activating NFκB signaling (81, 82). Thus, the attenuation of NFκB signaling pathways in V3-expressing ASMCs might be due to reduced levels of V0/V1 versican and hyaluronan. VCAM1 interacts with VLA4 expressed on monocytes, and when these interactions were blocked by VLA4-blocking antibodies, monocyte adhesion to ASMCs was significantly reduced, indicating that the integrin-dependent adhesion was also impacted by V3 expression. We further showed that monocyte adhesion mediated via VCAM1-VLA4 interactions was indeed reduced in V3-expressing ASMCs, confirming that the V3 expression reduced monocyte adhesion via blocking VCAM1 expression. The mechanisms by which V3 affects NFκB-dependent signaling are currently under investigation.
Although our findings indicate that expression of V3 by ASMCs differentially regulates a number of signaling pathways, the precise mechanism(s) by which V3 influences these pathways is not clear. One possibility may be related to the cell shape changes created by V3 expression (18). V3 expression by ASMCs causes significant cell flattening, and cell shape changes have been shown to contribute to multiple gene expression patterns affecting cell phenotype (83–85). Compositional differences and mechanical properties of this newly remodeled microenvironment created by V3 expression probably play a role in creating this anti-monocyte-adhesive phenotype. These findings provide novel insight into factors controlling the generation of a microenvironment that modulates key events in inflammation, which could offer potential therapeutic targets in the treatment of inflammatory diseases in the future.
Acknowledgments
We thank Hyunsoo Lim for efforts in generating some of the preliminary data for the manuscript and Dr. Virginia Green for editing and preparation of the manuscript. We especially thank Drs. Michael Kinsella, Susan Perigo, and Mervyn Merrilees for helpful discussions and critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants EB012558, HL098067, HL106967, and HL018645 (to T. N. W.). This work was also supported by American Heart Association Grant 09POST2050065 (to I. K.).
- V3
- V3 isoform of versican
- ASMC
- arterial smooth muscle cell
- ECM
- extracellular matrix
- VCAM1
- vascular cell adhesion molecule 1
- EGFR
- epidermal growth factor receptor
- NFκB
- nuclear factor κB
- qPCR
- quantitative polymerase chain reaction
- ICAM1
- intercellular adhesion molecule 1
- ER
- endoplasmic reticulum
- ELSA
- enzyme-linked sorbent assay
- LFA1
- lymphocyte function-associated antigen 1
- VLA4
- very late antigen 4
- ANOVA
- analysis of variance
- Has1
- -2, and -3, hyaluronan synthase 1, 2, and 3, respectively.
REFERENCES
- 1. Ross R. (1999) Atherosclerosis: an inflammatory disease. N. Engl. J. Med. 340, 115–126 [DOI] [PubMed] [Google Scholar]
- 2. Hansson G. K. (2005) Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 [DOI] [PubMed] [Google Scholar]
- 3. Libby P. (2002) Inflammation in atherosclerosis. Nature 420, 868–874 [DOI] [PubMed] [Google Scholar]
- 4. Merrilees M. J., Beaumont B. W., Braun K. R., Thomas A. C., Kang I., Hinek A., Passi A., Wight T. N. (2011) Neointima formed by arterial smooth muscle cells expressing versican variant v3 is resistant to lipid and macrophage accumulation. Arterioscler. Thromb. Vasc. Biol. 31, 1309–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lauer M. E., Mukhopadhyay D., Fulop C., de la Motte C. A., Majors A. K., Hascall V. C. (2009) Primary murine airway smooth muscle cells exposed to poly(I,C) or tunicamycin synthesize a leukocyte-adhesive hyaluronan matrix. J. Biol. Chem. 284, 5299–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lauer M. E., Fulop C., Mukhopadhyay D., Comhair S., Erzurum S. C., Hascall V. C. (2009) Airway smooth muscle cells synthesize hyaluronan cable structures independent of inter-α-inhibitor heavy chain attachment. J. Biol. Chem. 284, 5313–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Selbi W., de la Motte C. A., Hascall V. C., Day A. J., Bowen T., Phillips A. O. (2006) Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells. Kidney Int. 70, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 8. Jones S., Jones S., Phillips A. O. (2001) Regulation of renal proximal tubular epithelial cell hyaluronan generation: implications for diabetic nephropathy. Kidney Int. 59, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 9. Jokela T. A., Lindgren A., Rilla K., Maytin E., Hascall V. C., Tammi R. H., Tammi M. I. (2008) Induction of hyaluronan cables and monocyte adherence in epidermal keratinocytes. Connect. Tissue Res. 49, 115–119 [DOI] [PubMed] [Google Scholar]
- 10. de la Motte C. A., Hascall V. C., Drazba J., Bandyopadhyay S. K., Strong S. A. (2003) Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-α-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 163, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lauer M. E., Erzurum S. C., Mukhopadhyay D., Vasanji A., Drazba J., Wang A., Fulop C., Hascall V. C. (2008) Differentiated murine airway epithelial cells synthesize a leukocyte-adhesive hyaluronan matrix in response to endoplasmic reticulum stress. J. Biol. Chem. 283, 26283–26296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Majors A. K., Austin R. C., de la Motte C. A., Pyeritz R. E., Hascall V. C., Kessler S. P., Sen G., Strong S. A. (2003) Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J. Biol. Chem. 278, 47223–47231 [DOI] [PubMed] [Google Scholar]
- 13. Bandyopadhyay S. K., de la Motte C. A., Kessler S. P., Hascall V. C., Hill D. R., Strong S. A. (2008) Hyaluronan-mediated leukocyte adhesion and dextran sulfate sodium-induced colitis are attenuated in the absence of signal transducer and activator of transcription 1. Am. J. Pathol. 173, 1361–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Potter-Perigo S., Johnson P. Y., Evanko S. P., Chan C. K., Braun K. R., Wilkinson T. S., Altman L. C., Wight T. N. (2010) Polyinosine-polycytidylic acid stimulates versican accumulation in the extracellular matrix promoting monocyte adhesion. Am. J. Respir. Cell Mol. Biol. 43, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evanko S. P., Potter-Perigo S., Johnson P. Y., Wight T. N. (2009) Organization of hyaluronan and versican in the extracellular matrix of human fibroblasts treated with the viral mimetic poly I:C. J. Histochem. Cytochem. 57, 1041–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clowes M. M., Lynch C. M., Miller A. D., Miller D. G., Osborne W. R., Clowes A. W. (1994) Long-term biological response of injured rat carotid artery seeded with smooth muscle cells expressing retrovirally introduced human genes. J. Clin. Invest. 93, 644–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hinek A., Braun K. R., Liu K., Wang Y., Wight T. N. (2004) Retrovirally mediated overexpression of versican v3 reverses impaired elastogenesis and heightened proliferation exhibited by fibroblasts from Costello syndrome and Hurler disease patients. Am. J. Pathol. 164, 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemire J. M., Merrilees M. J., Braun K. R., Wight T. N. (2002) Overexpression of the V3 variant of versican alters arterial smooth muscle cell adhesion, migration, and proliferation in vitro. J. Cell Physiol. 190, 38–45 [DOI] [PubMed] [Google Scholar]
- 19. Merrilees M. J., Lemire J. M., Fischer J. W., Kinsella M. G., Braun K. R., Clowes A. W., Wight T. N. (2002) Retrovirally mediated overexpression of versican v3 by arterial smooth muscle cells induces tropoelastin synthesis and elastic fiber formation in vitro and in neointima after vascular injury. Circ. Res. 90, 481–487 [DOI] [PubMed] [Google Scholar]
- 20. Evanko S. P., Potter-Perigo S., Bollyky P. L., Nepom G. T., Wight T. N. (2012) Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration. Matrix Biol. 31, 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evanko S. P., Angello J. C., Wight T. N. (1999) Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 19, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 22. Choudhury R., McGovern A., Ridley C., Cain S. A., Baldwin A., Wang M. C., Guo C., Mironov A., Jr., Drymoussi Z., Trump D., Shuttleworth A., Baldock C., Kielty C. M. (2009) Differential regulation of elastic fiber formation by fibulin-4 and -5. J. Biol. Chem. 284, 24553–24567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chapman S. L., Sicot F. X., Davis E. C., Huang J., Sasaki T., Chu M. L., Yanagisawa H. (2010) Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler. Thromb. Vasc. Biol. 30, 68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi J., Bergdahl A., Zheng Q., Starcher B., Yanagisawa H., Davis E. C. (2009) Analysis of dermal elastic fibers in the absence of fibulin-5 reveals potential roles for fibulin-5 in elastic fiber assembly. Matrix Biol. 28, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilkinson T. S., Potter-Perigo S., Tsoi C., Altman L. C., Wight T. N. (2004) Pro- and anti-inflammatory factors cooperate to control hyaluronan synthesis in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 31, 92–99 [DOI] [PubMed] [Google Scholar]
- 26. Béchard D., Scherpereel A., Hammad H., Gentina T., Tsicopoulos A., Aumercier M., Pestel J., Dessaint J. P., Tonnel A. B., Lassalle P. (2001) Human endothelial-cell specific molecule-1 binds directly to the integrin CD11a/CD18 (LFA-1) and blocks binding to intercellular adhesion molecule-1. J. Immunol. 167, 3099–3106 [DOI] [PubMed] [Google Scholar]
- 27. Needham L. A., Van Dijk S., Pigott R., Edwards R. M., Shepherd M., Hemingway I., Jack L., Clements J. M. (1994) Activation dependent and independent VLA-4 binding sites on vascular cell adhesion molecule-1. Cell Adhes. Commun. 2, 87–99 [DOI] [PubMed] [Google Scholar]
- 28. Keire P. A., L'Heureux N., Vernon R. B., Merrilees M. J., Starcher B., Okon E., Dusserre N., McAllister T. N., Wight T. N. (2010) Expression of versican isoform V3 in the absence of ascorbate improves elastogenesis in engineered vascular constructs. Tissue Eng. Part A 16, 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang M., Pierce R. A., Wachi H., Mecham R. P., Parks W. C. (1999) An open reading frame element mediates posttranscriptional regulation of tropoelastin and responsiveness to transforming growth factor β1. Mol. Cell Biol. 19, 7314–7326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parks W. C. (1997) Posttranscriptional regulation of lung elastin production. Am. J. Respir. Cell Mol. Biol. 17, 1–2 [DOI] [PubMed] [Google Scholar]
- 31. McGowan S. E., Jackson S. K., Olson P. J., Parekh T., Gold L. I. (1997) Exogenous and endogenous transforming growth factors-β influence elastin gene expression in cultured lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 17, 25–35 [DOI] [PubMed] [Google Scholar]
- 32. McGowan S. E., McNamer R. (1990) Transforming growth factor-β increases elastin production by neonatal rat lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 3, 369–376 [DOI] [PubMed] [Google Scholar]
- 33. Misra S., Toole B. P., Ghatak S. (2006) Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 281, 34936–34941 [DOI] [PubMed] [Google Scholar]
- 34. Misra S., Ghatak S., Toole B. P. (2005) Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 280, 20310–20315 [DOI] [PubMed] [Google Scholar]
- 35. Ghatak S., Misra S., Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 36. Ghatak S., Misra S., Toole B. P. (2002) Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J. Biol. Chem. 277, 38013–38020 [DOI] [PubMed] [Google Scholar]
- 37. Bourguignon L. Y., Gilad E., Peyrollier K. (2007) Heregulin-mediated ErbB2-ERK signaling activates hyaluronan synthases leading to CD44-dependent ovarian tumor cell growth and migration. J. Biol. Chem. 282, 19426–19441 [DOI] [PubMed] [Google Scholar]
- 38. Vigetti D., Genasetti A., Karousou E., Viola M., Moretto P., Clerici M., Deleonibus S., De Luca G., Hascall V. C., Passi A. (2010) Proinflammatory cytokines induce hyaluronan synthesis and monocyte adhesion in human endothelial cells through hyaluronan synthase 2 (HAS2) and the nuclear factor-κB (NF-κB) pathway. J. Biol. Chem. 285, 24639–24645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mohamadzadeh M., DeGrendele H., Arizpe H., Estess P., Siegelman M. (1998) Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44/HA-dependent primary adhesion. J. Clin. Invest. 101, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohkawa T., Ueki N., Taguchi T., Shindo Y., Adachi M., Amuro Y., Hada T., Higashino K. (1999) Stimulation of hyaluronan synthesis by tumor necrosis factor-α is mediated by the p50/p65 NF-κ B complex in MRC-5 myofibroblasts. Biochim. Biophys. Acta 1448, 416–424 [DOI] [PubMed] [Google Scholar]
- 41. Hernández D., Miquel-Serra L., Docampo M. J., Marco-Ramell A., Cabrera J., Fabra A., Bassols A. (2011) V3 versican isoform alters the behavior of human melanoma cells by interfering with CD44/ErbB-dependent signaling. J. Biol. Chem. 286, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miquel-Serra L., Serra M., Hernández D., Domenzain C., Docampo M. J., Rabanal R. M., de Torres I., Wight T. N., Fabra A., Bassols A. (2006) V3 versican isoform expression has a dual role in human melanoma tumor growth and metastasis. Lab. Invest. 86, 889–901 [DOI] [PubMed] [Google Scholar]
- 43. Vaday G. G., Franitza S., Schor H., Hecht I., Brill A., Cahalon L., Hershkoviz R., Lider O. (2001) Combinatorial signals by inflammatory cytokines and chemokines mediate leukocyte interactions with extracellular matrix. J. Leukoc. Biol. 69, 885–892 [PubMed] [Google Scholar]
- 44. Vaday G. G., Lider O. (2000) Extracellular matrix moieties, cytokines, and enzymes: dynamic effects on immune cell behavior and inflammation. J. Leukoc. Biol. 67, 149–159 [DOI] [PubMed] [Google Scholar]
- 45. Sorokin L. (2010) The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–723 [DOI] [PubMed] [Google Scholar]
- 46. Liu S. Q., Alkema P. K., Tieché C., Tefft B. J., Liu D. Z., Li Y. C., Sumpio B. E., Caprini J. A., Paniagua M. (2005) Negative regulation of monocyte adhesion to arterial elastic laminae by signal regulatory protein α and Src homology 2 domain-containing protein-tyrosine phosphatase-1. J. Biol. Chem. 280, 39294–39301 [DOI] [PubMed] [Google Scholar]
- 47. Liu S. Q., Tieche C., Alkema P. K. (2004) Neointima formation on vascular elastic laminae and collagen matrices scaffolds implanted in the rat aortae. Biomaterials 25, 1869–1882 [DOI] [PubMed] [Google Scholar]
- 48. Ito K., Shinomura T., Zako M., Ujita M., Kimata K. (1995) Multiple forms of mouse PG-M, a large chondroitin sulfate proteoglycan generated by alternative splicing. J. Biol. Chem. 270, 958–965 [DOI] [PubMed] [Google Scholar]
- 49. Hinek A. (1994) Nature and the multiple functions of the 67-kD elastin-/laminin binding protein. Cell Adhes. Commun. 2, 185–193 [DOI] [PubMed] [Google Scholar]
- 50. Hinek A., Wrenn D. S., Mecham R. P., Barondes S. H. (1988) The elastin receptor: a galactoside-binding protein. Science 239, 1539–1541 [DOI] [PubMed] [Google Scholar]
- 51. Hinek A., Mecham R. P., Keeley F., Rabinovitch M. (1991) Impaired elastin fiber assembly related to reduced 67-kD elastin-binding protein in fetal lamb ductus arteriosus and in cultured aortic smooth muscle cells treated with chondroitin sulfate. J. Clin. Invest. 88, 2083–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hinek A., Rabinovitch M. (1994) 67-kD elastin-binding protein is a protective “companion” of extracellular insoluble elastin and intracellular tropoelastin. J. Cell Biol. 126, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hinek A. (1996) Biological roles of the non-integrin elastin/laminin receptor. Biol. Chem. 377, 471–480 [PubMed] [Google Scholar]
- 54. Wu Y., Chen L., Zheng P. S., Yang B. B. (2002) β1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J. Biol. Chem. 277, 12294–12301 [DOI] [PubMed] [Google Scholar]
- 55. Wu Y., Sheng W., Chen L., Dong H., Lee V., Lu F., Wong C. S., Lu W. Y., Yang B. B. (2004) Versican V1 isoform induces neuronal differentiation and promotes neurite outgrowth. Mol. Biol. Cell 15, 2093–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu Y., Wu J., Lee D. Y., Yee A., Cao L., Zhang Y., Kiani C., Yang B. B. (2005) Versican protects cells from oxidative stress-induced apoptosis. Matrix Biol. 24, 3–13 [DOI] [PubMed] [Google Scholar]
- 57. Leask A., Hutchenreuther J. (2014) Activation of latent TGFβ by αβ integrin: of potential importance in myofibroblast activation in fibrosis. J. Cell Commun. Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Munger J. S., Harpel J. G., Giancotti F. G., Rifkin D. B. (1998) Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol. Biol. Cell 9, 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Asano Y., Ihn H., Yamane K., Jinnin M., Mimura Y., Tamaki K. (2005) Increased expression of integrin α(v) β3 contributes to the establishment of autocrine TGF-β signaling in scleroderma fibroblasts. J. Immunol. 175, 7708–7718 [DOI] [PubMed] [Google Scholar]
- 60. Asano Y., Ihn H., Yamane K., Jinnin M., Mimura Y., Tamaki K. (2005) Involvement of αvβ5 integrin-mediated activation of latent transforming growth factor β1 in autocrine transforming growth factor β signaling in systemic sclerosis fibroblasts. Arthritis Rheum. 52, 2897–2905 [DOI] [PubMed] [Google Scholar]
- 61. Mu D., Cambier S., Fjellbirkeland L., Baron J. L., Munger J. S., Kawakatsu H., Sheppard D., Broaddus V. C., Nishimura S. L. (2002) The integrin α(v)β8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. J. Cell Biol. 157, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., Sheppard D. (1999) The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 [DOI] [PubMed] [Google Scholar]
- 63. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tatler A. L., Jenkins G. (2012) TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 9, 130–136 [DOI] [PubMed] [Google Scholar]
- 65. Tatler A. L., John A. E., Jolly L., Habgood A., Porte J., Brightling C., Knox A. J., Pang L., Sheppard D., Huang X., Jenkins G. (2011) Integrin αvβ5-mediated TGF-β activation by airway smooth muscle cells in asthma. J. Immunol. 187, 6094–6107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang Z., Mu Z., Dabovic B., Jurukovski V., Yu D., Sung J., Xiong X., Munger J. S. (2007) Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 176, 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wipff P. J., Rifkin D. B., Meister J. J., Hinz B. (2007) Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Joddar B., Ramamurthi A. (2006) Elastogenic effects of exogenous hyaluronan oligosaccharides on vascular smooth muscle cells. Biomaterials 27, 5698–5707 [DOI] [PubMed] [Google Scholar]
- 69. Kothapalli C. R., Ramamurthi A. (2008) Benefits of concurrent delivery of hyaluronan and IGF-1 cues to regeneration of cross-linked elastin matrices by adult rat vascular cells. J. Tissue Eng. Regen. Med. 2, 106–116 [DOI] [PubMed] [Google Scholar]
- 70. Wilkinson T. S., Bressler S. L., Evanko S. P., Braun K. R., Wight T. N. (2006) Overexpression of hyaluronan synthases alters vascular smooth muscle cell phenotype and promotes monocyte adhesion. J. Cell Physiol. 206, 378–385 [DOI] [PubMed] [Google Scholar]
- 71. Tammi R. H., Passi A. G., Rilla K., Karousou E., Vigetti D., Makkonen K., Tammi M. I. (2011) Transcriptional and post-translational regulation of hyaluronan synthesis. FEBS J. 278, 1419–1428 [DOI] [PubMed] [Google Scholar]
- 72. Wu Y., Chen L., Cao L., Sheng W., Yang B. B. (2004) Overexpression of the C-terminal PG-M/versican domain impairs growth of tumor cells by intervening in the interaction between epidermal growth factor receptor and β1-integrin. J. Cell Sci. 117, 2227–2237 [DOI] [PubMed] [Google Scholar]
- 73. Xiang Y. Y., Dong H., Wan Y., Li J., Yee A., Yang B. B., Lu W. Y. (2006) Versican G3 domain regulates neurite growth and synaptic transmission of hippocampal neurons by activation of epidermal growth factor receptor. J. Biol. Chem. 281, 19358–19368 [DOI] [PubMed] [Google Scholar]
- 74. Zhang Y., Cao L., Yang B. L., Yang B. B. (1998) The G3 domain of versican enhances cell proliferation via epidermal growth factor-like motifs. J. Biol. Chem. 273, 21342–21351 [DOI] [PubMed] [Google Scholar]
- 75. Imhof B. A., Aurrand-Lions M. (2004) Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 4, 432–444 [DOI] [PubMed] [Google Scholar]
- 76. Pueyo M. E., Gonzalez W., Nicoletti A., Savoie F., Arnal J. F., Michel J. B. (2000) Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-κB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 20, 645–651 [DOI] [PubMed] [Google Scholar]
- 77. Landry D. B., Couper L. L., Bryant S. R., Lindner V. (1997) Activation of the NF-κB and IκB system in smooth muscle cells after rat arterial injury: induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am. J. Pathol. 151, 1085–1095 [PMC free article] [PubMed] [Google Scholar]
- 78. De Martin R., Hoeth M., Hofer-Warbinek R., Schmid J. A. (2000) The transcription factor NF-κB and the regulation of vascular cell function. Arterioscler. Thromb. Vasc. Biol. 20, E83–E88 [DOI] [PubMed] [Google Scholar]
- 79. Bourguignon L. Y., Wong G., Earle C. A., Xia W. (2011) Interaction of low molecular weight hyaluronan with CD44 and Toll-like receptors promotes the actin filament-associated protein 110-actin binding and MyD88-NFκB signaling leading to proinflammatory cytokine/chemokine production and breast tumor invasion. Cytoskeleton 68, 671–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim S., Takahashi H., Lin W. W., Descargues P., Grivennikov S., Kim Y., Luo J. L., Karin M. (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Oertli B., Beck-Schimmer B., Fan X., Wüthrich R. P. (1998) Mechanisms of hyaluronan-induced up-regulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-κB and activating protein-1. J. Immunol. 161, 3431–3437 [PubMed] [Google Scholar]
- 82. Schawalder A., Oertli B., Beck-Schimmer B., Wüthrich R. P. (1999) Regulation of hyaluronan-stimulated VCAM-1 expression in murine renal tubular epithelial cells. Nephrol. Dial. Transplant. 14, 2130–2136 [DOI] [PubMed] [Google Scholar]
- 83. McBeath R., Pirone D. M., Nelson C. M., Bhadriraju K., Chen C. S. (2004) Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–495 [DOI] [PubMed] [Google Scholar]
- 84. Spiegelman B. M., Ginty C. A. (1983) Fibronectin modulation of cell shape and lipogenic gene expression in 3T3-adipocytes. Cell 35, 657–666 [DOI] [PubMed] [Google Scholar]
- 85. Kim D. H., Lipke E. A., Kim P., Cheong R., Thompson S., Delannoy M., Suh K. Y., Tung L., Levchenko A. (2010) Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc. Natl. Acad. Sci. U.S.A. 107, 565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]