

Background: Orphan response transcription factor GlnR regulates nitrogen metabolism in important actinomycetes.
Results: GlnR has no typical “phosphorylation pocket,” where the only conserved Asp is unphosphorylated but is essential for functional homodimerization.
Conclusion: Actinomycete GlnR is an atypical response regulator functioning as a homodimer.
Significance: Conserved Asp-focused charge interactions of actinomycete GlnR are probably the mechanism that stabilizes the homodimer for physiological function.
Keywords: Actinobacteria, Crystal Structure, Gene Regulation, Phosphorylation, Protein Conformation, OmpR/PhoB Subfamily, Homodimer, Receiver Domain, Unphosphorylated Aspartate
Abstract
The OmpR/PhoB subfamily protein GlnR of actinomycetes is an orphan response regulator that globally coordinates the expression of genes related to nitrogen metabolism. Biochemical and genetic analyses reveal that the functional GlnR from Amycolatopsis mediterranei is unphosphorylated at the potential phosphorylation Asp50 residue in the N-terminal receiver domain. The crystal structure of this receiver domain demonstrates that it forms a homodimer through the α4-β5-α5 dimer interface highly similar to the phosphorylated typical response regulator, whereas the so-called “phosphorylation pocket” is not conserved, with its space being occupied by an Arg52 from the β3-α3 loop. Both in vitro and in vivo experiments confirm that GlnR forms a functional homodimer via its receiver domain and suggest that the charge interactions of Asp50 with the highly conserved Arg52 and Thr9 in the receiver domain may be crucial in maintaining the proper conformation for homodimerization, as also supported by molecular dynamics simulations of the wild type GlnR versus the deficient mutant GlnR(D50A). This model is backed by the distinct phenotypes of the total deficient GlnR(R52A/T9A) double mutant versus the single mutants of GlnR (i.e. D50N, D50E, R52A and T9A), which have only minor effects upon both dimerization and physiological function of GlnR in vivo, albeit their DNA binding ability is weakened compared with that of the wild type. By integrating the supportive data of GlnRs from the model Streptomyces coelicolor and the pathogenic Mycobacterium tuberculosis, we conclude that the actinomycete GlnR is atypical with respect to its unphosphorylated conserved Asp residue being involved in the critical Arg/Asp/Thr charge interactions, which is essential for maintaining the biologically active homodimer conformation.
Introduction
A two-component system, typically consisting of a membrane-associated sensor histidine kinase and a cognate intracellular response regulator (RR),3 is the predominant signal transduction system employed by bacteria, and also found in archaea and eukarya (1). Most typical RRs remain as monomers or “weak” dimers (2, 3) with their receiver domains unphosphorylated. Once the environmental stimulus triggers histidine kinase autophosphorylation, the phosphoryl group is transferred to a conserved Asp residue in the receiver domain of the cognate RR. The phosphorylated RR then undergoes a substantial conformational change for “tight” homodimerization that enables its binding to the target DNA sequences (cis-elements) and in turn affects the transcription (4–6). Based on the homology of their DNA-binding domains, most RRs can be categorized into four major subfamilies (i.e. OmpR/PhoB, NarL/FixJ, NtrC, and LytR), leaving the remaining RRs containing miscellaneous effector domains, such as RNA binding or enzymatic functions (2, 7).
In most bacteria, a two-component system is employed to sense and respond to the nitrogen status in the environment, and the NtrB/NtrC-mediated nitrogen assimilation regulation in enteric bacteria is one of the best studied (8, 9). However, in many actinomycetes, including the rifamycin-producing industrial actinomycete Amycolatopsis mediterranei, the model organism Streptomyces coelicolor, and the pathogenic Mycobacterium tuberculosis, nitrogen assimilation is globally regulated by an OmpR/PhoB subfamily protein, GlnR (10–13), which is considered an orphan RR because its cognate sensor histidine kinase has not been identified (10, 11, 14–16). Despite its great importance in global regulation of nitrogen metabolism, the understanding of the regulation of GlnR activity as well as its impact upon the GlnR-mediated global transcription regulation is limited, although much attention has been paid to the identification of the GlnR target genes and their corresponding cis-elements so far (10, 13, 17–19) .
The GlnR from S. coelicolor (ScoGlnR) was once predicted to be a typical RR subject to Asp phosphorylation as typical OmpR/PhoB subfamily members due to the presence of the conserved residues Asp50 and Thr83 of the active site quintet (14, 20, 21), essential in defining the so-called acidic “phosphorylation pocket” of the typical RRs (22). Recently, the atypical receiver domains of several orphan RRs (7, 22, 23) were shown to be generally similar to the typical receiver domains in their amino acid sequences and three-dimensional structures but lacked one or more residues of the highly conserved active site quintet (22, 24). Because the GlnR from actinomycetes other than streptomycetes (see Fig. 2) only has its putative phosphorylation site Asp residue found to be conserved in the active quintet (21, 25), we hypothesized that GlnR is an atypical RR, with its activity being independent of Asp phosphorylation. However, this hypothesis was mechanistically challenged by a mutational analysis, in which substitution of the potential phosphorylation site Asp residue with Ala abolished the GlnR function in Mycobacterium smegmatis (14).
FIGURE 2.

Structure and comparison of different GlnRRecs. A, structure-based sequence alignment of different GlnRRecs. The five residues constituting the phosphorylation pocket in typical RRs are highlighted in red, whereas the corresponding residues that differ from typical RRs in GlnR and atypical RRs are shown in yellow. The highly conserved residues among OmpR subfamily RRs are colored in blue, and the residues only showing conservation among GlnRs are colored in cyan. Red stars, conformation switch residues in typical RRs; red triangle, conserved residue Asp in the putative phosphorylation site; red circles, the two residues Thr and Arg forming interactions with the residue Asp in the putative phosphorylation site; red square, Arg residue essential for the dimerization. The secondary structure elements of AmeGlnR and PhoB (Protein Data Bank code 1ZES) are shown at the top and bottom, respectively. AmeGlnR, GlnR from A. mediterranei U32 (YP_003771100); MtbGlnR, GlnR from M. tuberculosis H37Rv (NP_215333); ScoGlnR, GlnR from S. coelicolor M145 (NP_628336); MseGlnR, GlnR from M. smegmatis strain MC2 155 (YP_890012); SveGlnR, GlnR from S. venezuelae ATCC 10712 (YP006879462). ChxR is from C. trachomatis 434/Bu (YP_001654963); NblR is from Synechococcus elongatus PCC 7942(AAC33849); HP1043 is from H. pylori (NP_207833); JadR1 is from S. venezuelae ATCC 10712(AAB36584); ArcA is from E. coli K12 (NP_418818); OmpR is from E. coli O157:H7 strain EDL933 (NP_289945); and PhoB is from E. coli K12 (NP_414933). B, ribbon diagram shows the structure of the AmeGlnRRec. The two molecules A and B constituting the homodimer are colored in yellow and cyan, and the dimer interface is shown in magenta. C, structure comparison of AmeGlnRRec (cyan) with phosphorylated PhoB (gray) (Protein Data Bank code 1ZES). β1-α1 and β3-α3 loops and residue Arg52 of AmeGlnRRec are colored in magenta. The residues important for phosphorylation in PhoB and corresponding ones in AmeGlnRRec are shown with side chains. A close-up view of the putative phosphorylation pocket and interactions is also shown.
In this study, the structure-function relationship of GlnR from A. mediterranei (AmeGlnR) is comprehensively analyzed. By integration of the knowledge learned from those of M. tuberculosis and S. coelicolor, the conserved Asp site of actinomycete GlnR is proved to be unphosphorylated but critical for homodimerization via its charge interactions with the surrounding residues, which, in turn, are essential for its physiological function in vivo and DNA binding ability in vitro.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
Escherichia coli strains were grown at 37 °C in lysogeny broth (LB) medium (26). A. mediterranei were grown at 30 °C in the nutrient-rich Bennett's medium (27). To examine the growth phenotypes of A. mediterranei U32 and its glnR mutants, strains were incubated at 30 °C in minimal medium (27) supplemented with 80 mm potassium nitrate or 60 mm ammonium sulfate as the sole nitrogen source, and the growth was observed after 7 days' cultivation. S. coelicolor M145 and its derivatives were generally cultured at 30 °C in the MS medium for spore suspension preparations (28), whereas phenotype analysis was conducted in nitrogen-limited N-Evans medium with 5 mm nitrate or 100 mm ammonium sulfate as the sole nitrogen source after 4 days' cultivation. If necessary, the media were supplemented with antibiotics (100 μg ml−1 for ampicillin, 50 μg ml−1 for kanamycin, 50 μg ml−1 for apramycin, and 50 μg ml−1 for thiostrepton).
Expression and Purification of GlnR Regulatory Domain (GlnRRec) Protein
DNA fragments encoding the receiver domains of GlnR proteins were PCR-amplified using the genomic DNA of A. mediterranei U32 (AmeglnRRec) and M. tuberculosis H37Rv (MtbglnRRec). The PCR products were digested and inserted into the pET28b expression vector, resulting in N-terminal His6-tagged pET-28bAmeglnRRec and pET-28bMtbglnRRec. The plasmid was transformed into E. coli BL21 (DE3) strain (Novagen), and the cells were cultured at 37 °C in LB medium containing 50 μg ml−1 kanamycin. Protein expression was induced by adding isopropyl β-d-thiogalactoside into the medium to a final concentration of 1 mm, when the A600 is 0.8. Then the cells were harvested by centrifugation at 5,000 × g for 10 min at 4 °C, resuspended in a lysis buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 1 mm PMSF), and disrupted using a French press. The recombinant protein was purified with affinity chromatography using a Ni2+-nitrilotriacetate Superflow column (Qiagen) pre-equilibrated with buffer A (50 mm Tris-HCl, pH 8.0, and 100 mm NaCl) and then washed with buffer B (buffer A supplemented with 50 mm imidazole) to remove nonspecific binding proteins. The target protein was eluted with buffer C (buffer A supplemented with 250 mm imidazole), and the eluted fractions were further purified using a gel filtration column (GE Healthcare). After the two-step purification, the target protein was of sufficient purity (above 95%) and was then concentrated to ∼10 mg ml−1 in buffer A by ultrafiltration for further structural and biochemical studies.
Selenomethionine-substituted AmeGlnRRec protein was prepared following a method described previously (29). Purification of the selenomethionine AmeGlnRRec protein was performed using the same methods as for the native protein. Gel filtration analysis of the purified protein was performed to measure the oligomeric state of AmeGlnRRec in solution. High and low molecular weight (mass) calibration kits (GE Healthcare) were used to calibrate the molecular mass of wild type and mutants of AmeGlnRRec. All of the above analysis was carried out on an FPLC system (GE Healthcare). Protein samples of 100 μl each were loaded into a 0.5-ml sample loop and injected into a Superdex 200 column. The apparent molecular mass of the protein sample was calculated according to the protocol provided in the kit.
Phos-tag Acrylamide Gel Analysis of GlnR Phosphorylation
For sample preparation for in vivo detection of phosphorylation, a standard protocol was used with minor modifications (30). A. mediterranei U32 or S. coelicolor M145 and related mutants were grown in Bennett's or MS medium (10) at 30 °C for 2 days and were then harvested by swabbing from the plate. Aliquots of the cells were washed and resuspended in minimal medium or N-Evans medium supplemented with 80 mm potassium nitrate or 60 mm ammonium sulfate as the sole nitrogen source for GlnR or with 4 or 0 mm potassium hydrogen phosphate as the sole phosphate source for AmePhoP. After 12 h of growth, cells were pelleted by centrifugation, immediately following harvest; cells were lysed with 3.3 ml of 1 m formic acid (0.55 m final concentration formic acid) per equivalent of pellet of 50 ml of 0.2 A600 of cells. A French press was used to lyse the frozen cell pellet. Each lysate was solubilized by the addition of 200 μl of 5 m NaOH (0.17 m final concentration) to neutralize the solution and 1.5 ml of 5× SDS loading solution. Resulting cell lysates (20 μl) were immediately loaded onto a Phos-tag gel for electrophoresis as described below; the whole lysis process should be kept at a low temperature to prevent the hydrolysis of phospho-Asp residues.
For in vitro phosphorylation experiments, solutions of 10 μm protein in phosphorylation buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 2 mm β-mercaptoethanol, 20 mm MgCl2) with or without incubation with 20 mm ammonium hydrogen phosphoramidate (synthesized as described previously (31)) for 30 min at 37 °C were prepared. The phosphorylation reactions were stopped by the addition of 5× SDS-loading buffer. Phos-tag acrylamide gels were prepared as described previously with minor modifications (30, 32, 33). Phos-tag acrylamide running gels contained 12% (w/v) 29:1 acrylamide/N,N-methylene-bisacrylamide, 375 mm Tris, pH 8.8, 0.1% (w/v) SDS. Gels were copolymerized with 25 μm Phos-tag acrylamide and 50 μm MnCl2 for analysis of purified GlnR and other positive control proteins. The stacking gels contained 5% (w/v) 29:1 acrylamide/N,N-methylene-bisacrylamide, 125 mm Tris, pH 6.8, 0.1% (w/v) SDS. All Phos-tag acrylamide-containing gels were run at 4 °C under constant voltage (120 V). Gels were fixed for 10 min in standard transfer buffer (20% (v/v) methanol, 50 mm Tris-HCl, 40 mm glycine, with 1 mm EDTA added to remove Mn2+) and then incubated for an additional 20 min in transfer buffer without EDTA to remove the chelated metal. Transfer to nitrocellulose membranes was performed using a Bio-Rad transfer apparatus under a constant 300 mA for 1 h. Western blotting assay was performed using standard protocols.
Crystallization, Data Collection, and Structure Determination
The AmeGlnRRec and MtbGlnRRec proteins were used in crystallization experiments at 4 °C using the sitting drop vapor diffusion method. Crystals were grown in the drop containing equal volumes (1 μl) of the protein solution (∼10 mg/ml AmeGlnRRec/MtbGlnRRec) and the reservoir solution (0.1 m Tris-HCl, pH 8.0, and 20% MPD (1.8 m sodium acetate trihydrate, pH 7.0, 0.1 m Bistris propane)), respectively. For diffraction data collection, the AmeGlnRRec and MtbGlnRRec crystals were first cryoprotected using paratone oil (Hampton Research) and then flash-cooled in liquid nitrogen. Selenium single-wavelength anomalous dispersion and native data of AmeGlnRRec were collected to a resolution of 3.0 and 2.8 Å, respectively, from flash-cooled crystals at 100 K at the Shanghai Synchrotron Radiation Facility, beamline BL17U. The native data of MtbGlnRRec were collected to 2.8 Å. The diffraction data were processed, integrated, and scaled together using the HKL2000 suite.
Structure of the AmeGlnRRec was solved using the Autosol implemented in Phenix (34). Over 60% of main chain residues were built, and the overall figure of merit was increased from 0.35 to 0.69 at 3.0 Å. The full structure model was built manually using the program Coot (35). Structure refinement was carried out using Phenix and refmac. Because the resolution and statistics of the single wavelength anomalous dispersion data set of AmeGlnRRec were better than the native data, the single wavelength anomalous dispersion data set was used in the final refinement of the structure of AmeGlnRRec. The structure of MtbGlnRRec was solved by molecular replacement using the structure of AmeGlnRRec as a starting model. The final model was refined to 2.8 Å. All of the statistics of data collection and structure refinement are summarized in Table 1.
TABLE 1.
Statistics of diffraction data collection and structure refinement
| MtbGlnRRec | AmeGlnRRec | AmeGlnRRec-Se | |
|---|---|---|---|
| Diffraction data | |||
| Wavelength (Å) | 0.9793 | 0.9793 | 0.9793 |
| Space group | I41 | P6522 | P6522 |
| Cell parameters | |||
| a (Å) | 144.2 | 93.1 | 93.1 |
| b (Å) | 144.2 | 93.1 | 93.1 |
| c (Å) | 92.2 | 308.4 | 310.4 |
| α (degrees) | 90.0 | 90.0 | 90 |
| β (degrees) | 90.0 | 90.0 | 90 |
| γ (degrees) | 90.0 | 120.0 | 120 |
| Resοlution (Å) | 50.0–2.80 | 50.0–2.80 | 50.0–3.00 |
| (2.90–2.80)a | (2.90–2.80) | (3.11–3.00) | |
| Observed reflections | 71,225 | 196,781 | 698891 |
| Unique reflections (I/σ(I) > 0) | 23,093 | 20,511 | 16,902 |
| Average redundancy | 3.1 (3.1) | 9.6 (9.9) | 41.3 (42.8) |
| Average I/σ (I) | 13.7 (4.4) | 19.1 (4.4) | 54.8 (10.9) |
| Completeness (%) | 99.0 (99.7) | 99.8 (100.0) | 100.0 (100.0) |
| Rmerge (%)b | 9.8 (31.9) | 11.7 (62.1) | 16.0 (63.0) |
| Refinement and structure model | |||
| Reflections (Fo ≥0σ(Fo)) | |||
| Working set | 21,918 | 19,390 | |
| Test set | 1,158 | 1,039 | |
| Rwork/Rfree (%)c | 18.0/22.5 | 22.1/26.9 | |
| No. of atoms | 4,283 | 3,391 | |
| Protein | 4,111 | 3,364 | |
| Water | 172 | 27 | |
| Average B factor (Å2) | |||
| All atoms | 38.8 | 54.5 | |
| Protein | 38.8 | 54.6 | |
| Water | 38.5 | 50.3 | |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.007 | 0.011 | |
| Bond angles (degrees) | 1.4 | 1.9 | |
| Ramachandran plot (%) | |||
| Most favored | 95.3 | 97.6 | |
| Allowed | 4.7 | 2.4 | |
a Numbers in parentheses represent the highest resolution shell.
b Rmerge = ΣhklΣi Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
c R = Σhkl‖Fo| − |Fc‖/Σhkl|Fo|.
Molecular Dynamics Simulations
The starting structure of AmeGlnR monomer was extracted from the crystal structures of AmeGlnR dimer. The D50A, D50E, and D50N mutants were built from the wild type by mutating the aspartic acid into glutamate or asparagine, respectively. After that, each system was solvated by TIP3P waters with 0.15 m NaCl. Finally, each simulation system includes about 17,000 atoms (55 × 55 × 55 Å).
MD simulations were carried out with the GROMACS version 4.6.1 package with the NPT ensemble and periodic boundary condition (36). The AMBER99SB-ILDN force field was applied for the simulations (37). Energy minimizations were first performed to relieve unfavorable contacts, followed with 2 ns in total to equilibrate the side chains of protein and solvent. The particle mesh Ewald method was used for long range electrostatic interactions with a short range cut-off of 1.2 nm. All simulations were run at 300 K using the v-rescale method with a coupling time of 0.1 ps (38). The pressure was kept at 1 bar using the Berendsen barostat τp = 1.0 ps and a compressibility of 4.5 × 10−5 bar−1. SETTLE constraints and LINCS constraints were applied on the hydrogen-involved covalent bonds in water molecules and in other molecules, respectively, and the time step was set to 2 fs. Each system was put into a 150-ns production run.
Electrophoretic Mobility Shift Assay (EMSA)
For expression of recombinant mutated AmeGlnR (i.e. D50A, D50N, T9A, R52A, T9A/R52A, R111A, and R111E mutants), the wild type AmeglnR gene on the expression plasmid pET28b was mutated using site-directed mutagenesis methods (18). The glnA promoter region of A. mediterranei U32 was generated by PCR and was then inserted into the HincII site of pUC18. The obtained plasmid was used as the template for preparation of the FAM-labeled probes using the universal primer pair of FAM-labeled M13F (−47) and M13R (−48). FAM-labeled probe (30 ng) was incubated with varying amounts of AmeGlnR or its mutants at 25 °C for 20 min in a buffer of 25 mm Tris-HCl (pH 8.0), 50 mm KCl, 2.5 mm MgCl2, 5% glycerol, 1 mm dithiothreitol (DTT), and 100 μg ml−1 sonicated salmon sperm DNA (Sangon) (total volume 20 μl). Because GlnR has been proved a specific regulator for binding of the glnA promoter region in A. mediterranei U32 (39) and the EMSA employed in this study is to measure the DNA binding affinities of GlnR as well as its mutants, the cold probe competition assay was unnecessary and was therefore omitted. The resulting DNA-protein complexes were subjected to electrophoresis on agarose gels with a running buffer containing 40 mm Tris-HCl (pH 7.8), 20 mm boric acid, and 1 mm EDTA at 150 V and 4 °C for 1 h. After electrophoresis, gels were directly scanned for fluorescent DNA using an ImageQuantTM LAS 4000 system (GE Healthcare).
Complementation Assay
The A. mediterranei glnR gene together with its native promoter region was amplified from A. mediterranei genome DNA and ligated with pRT803, which was excised by EcoRV, yielding plasmid pRT803AmeglnR, which was then used as the template for site-directed mutagenesis of AmeglnR. After verification by DNA sequencing, the complementation plasmids were transformed into A. mediterranei U32ΔglnR using a Bio-Rad Gene Pulser according to the methods described previously (40). Transformants were selected in Bennett's agar plates containing hygromycin. The S. coelicolor glnR gene together with its native promoter region was excised from pSETScoglnR with BamHI (18) and was subsequently cloned into the same site of pBluescript II SK (Stratagene), yielding plasmid pSKScoglnR, which was then used as the template for site-directed mutagenesis of scoglnR. The generated plasmids with various mutations of scoglnR were digested with BamHI and inserted into the same site of pSET1521 (41) to obtain the relevant plasmids for scoglnR complementation. After being verified by DNA sequencing, the complementation plasmids were conjugated into S. coelicolor M145ΔglnR (18). Exconjugants were selected by growth on MS agar flooded with nalidixic acid and thiostrepton.
In Vivo Chemical Cross-linking Experiment
A. mediterranei strains containing the mutated residues in glnR were made by complementing the A. mediterranei U32ΔglnR strain with mutated A. mediterranei glnR fused with a FLAG tag at the C terminus using the method described above. A. mediterranei was first cultured in liquid Bennett's medium at 30 °C for 48 h before being inoculated into fresh liquid minimal medium supplemented with either 80 mm KNO3 or 60 mm (NH4)2SO4 for another 24-h culture. Cells were then collected, and the cell pellets were resuspended in PBS and exposed to 5 mm DSS (Pierce). After a 20-min incubation at 25 °C, the reaction was quenched with the addition of 50 mm Tris-HCl (pH 8.0) (final concentration) for 15 min. The samples were subjected to SDS-PAGE and immunoblot assays were performed using the anti-FLAG antibody.
Reverse Transcription PCR (RT-PCR)
For RNA extraction, wild type A. mediterranei U32 and glnR mutants were grown in liquid Bennett's medium for 48 h before being inoculated into fresh liquid Bennett's medium supplemented with 80 mm KNO3 or 60 mm (NH4)2SO4 for further culture for 36 h. Total RNA was extracted using TRIzol reagent (Invitrogen). RNA was treated with RNase-free DNase I (Promega) to prevent contamination of trace genomic DNA. Reverse transcription was performed with a random hexamer primer using 3 μg of RNA in a total volume of 30 μl employing Super-Script III reverse transcriptase (Invitrogen). PCR was performed employing 20-ng reaction mixtures as the template to check the transcription of nasA and glnR genes and using the rpoB gene as the internal control. A negative control was made by following the same procedures except that the addition of reverse transcriptase was omitted. Two independent samples were used for analyses.
RESULTS
GlnR of Either A. mediterranei or S. coelicolor Is Unphosphorylated at the Potential Asp Phosphorylation Site
Because the Phos-tag methods (30, 32, 33) are able to characterize the phosphorylation state of the Asp residues in typical RRs, such as that shown for PhoB from E. coli (EcoPhoB) cell lysates (42), similar in vitro phosphorylation assays employing the high energy phospho-donor, ammonium hydrogen phosphoramidate, against the purified recombinant proteins of AmeGlnR and ScoGlnR proteins were conducted individually. Both of the annotated typical RRs (i.e. PhoP of A. mediterranei (AmePhoP) (13) and SCO5403 of S. coelicolor (21)), bearing the highly conserved acidic quintet in their amino acid sequences, as well as the purified well characterized EcoPhoB could be successfully phosphorylated in vitro by ammonium hydrogen phosphoramidate, probably at their corresponding Asp residues, whereas neither AmeGlnR nor ScoGlnR could (Fig. 1A). Therefore, it is unlikely that the conserved Asp residues of either of the two GlnRs can be phosphorylated by the high energy phosphodonor in vitro.
FIGURE 1.
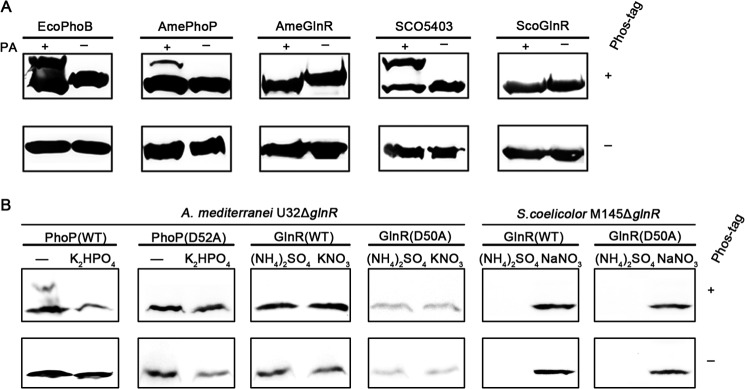
Biochemical analyses detecting the phosphorylation status of the conserved residue Asp of GlnR. A, Western blot monitoring the phosphorylation status of the His tag-fused E. coli PhoB (EcoPhoB), S. coelicolor GlnR/5403 (ScoGlnR/SCO5403), A. mediterranei U32 GlnR (AmeGlnR), and the A. mediterranei U32 PhoP (AmePhoP), purified by nickel affinity chromatography from the corresponding E. coli heterologous expression systems (see “Experimental Procedures”) treated with (+) or without (−) 20 mm ammonium hydrogen phosphoramidate (PA). B, Western blot monitoring the in vivo phosphorylation status of the FLAG tag-fused PhoP (WT, D52A) under phosphate-replete (K2HPO4) or -limited (−) conditions and the phosphorylation status of FLAG tag-fused GlnR (WT, D50A) under nitrogen-rich ((NH4)2SO4) or -limited (KNO3) conditions in the A. mediterranei U32ΔglnR and S. coelicolor M145ΔglnR strains. Cell lysates were treated as described under “Experimental Procedures.”
In order to test whether GlnR is phosphorylated or not in vivo, the phosphorylation status of AmeGlnR or ScoGlnR in vivo was tested via Western blot analysis against the cell lysates of A. mediterranei or S. coelicolor electrophoresed on Phos-tag gel, employing AmePhoP as a positive control and AmePhoP(D52A) as a negative control. In accord with the fact that the Asp52 of PhoP is phosphorylated under the phosphate limitation conditions (43), AmePhoP exhibited a single band on the gel when cells are grown in phosphate (K2HPO4)-rich medium, whereas double bands of AmePhoP were observed for cells grown in phosphate-limiting media. Obviously, the upper band corresponding to the phosphorylated AmePhoP disappeared in the D52A mutation, indicating that Asp52 was the phosphorylation site (Fig. 1B). In contrast, although GlnR is functional under nitrogen-limited conditions (39), no phosphorylation band of AmeGlnR or ScoGlnR could be observed in lysates of cells grown in either nitrogen-rich ((NH4)2SO4) or nitrogen-limited (KNO3) media. All of these results are in good agreement with the results of the in vitro experiments mentioned above (Fig. 1A), and therefore, we conclude that both AmeGlnR and ScoGlnR are unphosphorylated at their conserved Asp residues.
Crystal Structures of the GlnR Receiver Domains from A. mediterranei and M. tuberculosis Are Distinct from That of the Typical RRs Regarding the Potential “Phosphorylation Pocket” and Its Surrounding Amino Acid Residues
We determined the crystal structures of the receiver domains of GlnR from A. mediterranei U32 (AmeGlnRRec) and M. tuberculosis (MtbGlnRRec). The final structure models of both proteins were refined to 2.8 Å, with their statistics summarized in Table 1. Considering that AmeGlnRRec and MtbGlnRRec not only share 58% sequence identity (Fig. 2A) but also form similar homodimer structures (root mean square deviation = 1.0 Å), AmeGlnRRec alone is selected for further structural and functional analyses (Fig. 2B). The structures of AmeGlnRRec and MtbGlnRRec have been deposited to the Protein Data Bank with the codes 4O1H and 4O1I, respectively.
The structure of AmeGlnRRec consists of five alternating β-strands and α-helices folding into a five-stranded parallel β-sheet in the middle surrounded by two α-helices on one side and three on the other (Fig. 2B), which is similar to most of the known structures of the typical RR receiver domains (22) except that the helices α1, α2, and α4 of AmeGlnRRec are partially unwound (Fig. 2C). We notice that the so-called “phosphorylation pocket” defined in the typical RRs as well as its microenvironment are significantly altered in the structure of AmeGlnRRec (Fig. 2, A and C). All of the five residues essential for the phosphorylation of PhoB except Asp50 are neither conserved nor in proper position, which is also demonstrated by the structure-based sequence alignment among GlnR proteins from representative actinomycetes and the well characterized typical and atypical RRs from Gram-negative bacteria (Fig. 2A). The Glu9 and Asp10 residues known to bind with Mg2+ to promote phosphorylation and dephosphorylation in PhoB of E. coli are replaced by residues Thr9 and Ala10 in AmeGlnRRec, respectively. These changes are likely to exclude the binding of a divalent metal cation and hence reduce the possibility of phosphorylation at AmeGlnRRec residue Asp50. In addition, the Thr and Lys residues believed to communicate between the phosphorylation site and dimer interface in typical RRs are changed to Val81 and Leu100, respectively, and the critical conformation switch residue Tyr is also substituted with an Ile98 in AmeGlnRRec. These data suggest that the AmeGlnR is significantly diverged from the canonical RRs with respect to the potential “phosphorylation pocket” and its surrounding amino acid residues.
When the monomer structure of AmeGlnRRec is superimposed with that of the phosphorylated PhoB (root mean square deviation = 2.1 Å), we find that the β1-α1 and β3-α3 loops move toward the putative phosphorylation pocket, thus shrinking the size of the “pocket.” It is particularly significant that the Arg52 residue, conserved among all GlnR proteins but completely different in Gram-negative OmpR/PhoB subfamily proteins (Fig. 2A), protrudes from the β3-α3 loop into and occupied the “pocket” with its guanidinium side chain positioned at the close vicinity of the carboxyl side chain of the only conserved Asp50. Based on the measured distance of the interactions, an ionic bridge may form between the side chains of these two residues, and the side chain of Asp50 may further be stabilized by forming another hydrogen bond with Thr9. Thus, the configuration of Arg52 not only lessens the likelihood of residue Asp50 being phosphorylated but also introduces a hydrogen-bonding/ionic interaction network, which may provide an alternative mechanism for maintaining the homodimerization status of GlnR different from that of Asp phosphorylation in typical RRs (Fig. 2C).
Homodimerization of the Two GlnR Monomers through Their α4-β5-α5 Interface Is Essential for Their Physiological Function
The crystal structure data suggest that both AmeGlnRRec and MtbGlnRRec form homodimers. The interface involves the α4-β5-α5 secondary structure elements from both monomers and buries about 30% of the total surface area, which is similar to that of the phosphorylated typical RRs, such as PhoB and ArcA (Fig. 3, A and B), where homodimerization through such an interface is universal and essential for their physiological activities. On the other hand, in contrast to ArcA and PhoB, there is a lower percentage of hydrophilic residues but a higher percentage of hydrophobic residues within the interface of the two GlnR monomers, which presumably favors tighter protein-protein interactions (Fig. 3A). Two Arg111 residues in AmeGlnRRec from β5 of both monomers A and B, conserved among almost all OmpR/PhoB subfamily proteins except for the atypical ChxR from Chlamydia trachomatis (Fig. 2A), are found stacking against each other by π-π interactions. This Arg residue further stabilizes the interface by forming salt bridge and hydrogen bond networks with Asp97 and Glu107 from both monomers (Fig. 3C). The predominant role of this Arg residue in maintaining the homodimer is confirmed by site-directed mutagenesis analysis followed by gel filtration verification, in which an R111A single mutation results in a disruption of the homodimer of AmeGlnRRec (Fig. 3D). The hydrophilic interactions among Arg111, Asp97, and Glu107 are surrounded by hydrophobic residues Leu86, Val89, Ile98, Ala106, Ala110, and Leu114, which protrude from α4 and α5 helices of monomers A and B, strengthening the homodimer interaction (Fig. 3, A and C).
FIGURE 3.

Structure and in vitro characterization of the residues essential for AmeGlnRRec homodimer interaction. A, sequence alignment of the residues constituting the dimer interface of GlnR, ChxR, HP1043, ArcA, PhoB, and the hydrophobic and hydrophilic residues are colored in yellow and green, respectively. B, superimposition of the structural elements involved in the homodimer interface of AmeGlnRRec (magenta) and PhoB (gray). C, residues involved in the homodimer formation. The two molecules are colored in yellow and cyan. Hydrophobic interaction residues are shown with spheres, and hydrophilic residues forming hydrogen bonds and salt bridges (shown with red dashed lines and numbers indicating the distance) are shown with side chains. D, gel filtration analysis of wild type AmeGlnRRec and its mutants. The mobility profiles of wild type AmeGlnRRec, R111A, D50A, D50N, and D50E are shown, and the molecular masses were calculated based on the standard proteins indicated at the top. The 25- and 40-kDa peaks represent the monomer and dimer, respectively, whereas the 64- and 70-kDa peaks represent the oligomer.
The dimerization and extensive interactions of the receiver domains imply that GlnR may form a homodimer as its functional status under physiological conditions. However, we failed in crystallization and analysis of the oligomeric state of the full-length GlnR using the heterogeneously expressed protein, probably due to its aggregation in solution. Alternatively, as shown in Fig. 4, we demonstrate that the AmeGlnR protein exists mainly as dimers in vivo in the presence of cross-linking agent DSS under either nitrogen-rich or limited conditions. We further explore whether the α4-β5-α5 interface is essential for the homodimerization of full-length AmeGlnR. Indeed, AmeGlnR harboring an R111A mutation changes the oligomeric state from dimers to monomers, which suggests that the homodimerization of AmeGlnR is dependent on the α4-β5-α5 interface in vivo (Fig. 4).
FIGURE 4.

In vivo cross-linking analysis of AmeGlnR and its mutants. The oligomeric status of the wild type and mutated AmeGlnR (R111A, D50A, D50N, T9A, R52A, T9A/R52A, and D50E) in the presence (+) and absence (−) of cross-linking agent DSS are shown by Western blot analysis. The strains were cultured in the nitrogen-rich ((NH4)2SO4) or -limited (KNO3) conditions. Molecular markers are shown on the left. Di, dimer; Mo, monomer.
The glnR null mutants of both A. mediterranei U32 and S. coelicolor are proved unable to grow on minimal medium when nitrate is supplied as the sole nitrogen source (10, 39). Similar growth failure was observed when mutations of R111A/E (Fig. 5) or R108A/E (data not shown) were introduced individually into AmeglnR or ScoglnR, respectively. Consistent with these physiological phenotypes, the transcription of GlnR target genes, such as nasA and glnA, are not activated in A. mediterranei glnR(R111A/E) mutants either (Fig. 6). Further EMSA experiments using purified mutated proteins indicated that the DNA binding abilities of AmeGlnR(R111A) and AmeGlnR(R111E) proteins were all significantly reduced (Fig. 7). These data suggest that the homodimer formation through the α4-β5-α5 interface is indispensable for GlnR function in vivo, and the disruption of homodimer formation of GlnR may lead to the impairment of its DNA binding ability, therefore abolishing the regulatory function of GlnR.
FIGURE 5.
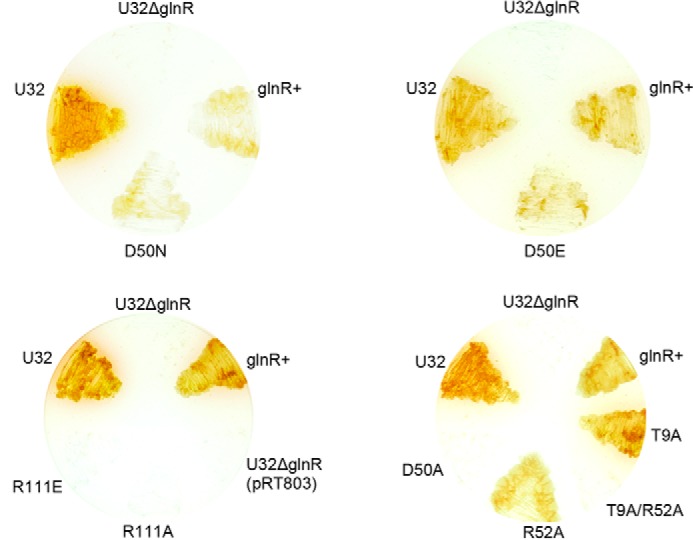
Growth phenotypes of A. mediterranei U32 mutants and complementation strains. Results of complementation by wild type AmeglnR or the glnR mutants (R111A/E, D50A, D50N, D50E, T9A, R52A, and T9A/R52A) grown on minimal medium supplemented with KNO3.
FIGURE 6.
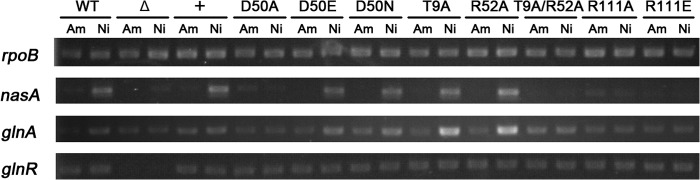
Transcriptional analysis of wild type U32 and glnR mutants for the transcription of GlnR target genes in A. mediterranei. The transcription of rpoB was used as the internal control. WT, wild type A. mediterranei U32; Δ, U32ΔglnR; +, AmeglnR+; D50A, AmeglnR(D50A); D50E, AmeglnR(D50E); D50N, AmeglnR(D50N); T9A, AmeglnR(T9A); R52A, AmeglnR(R52A); T9A/R52A, AmeglnR(T9A/R52A); R111A, AmeglnR(R111A); R111E, AmeglnR(R111E); Am, Bennett's medium with 60 mm ammonium; Ni, Bennett's medium with 80 mm KNO3.
FIGURE 7.
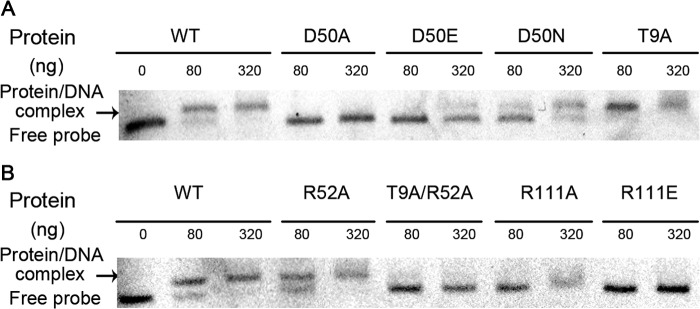
EMSA results of wild type AmeGlnR and its mutants. The FAM-labeled glnA promoter region was incubated with the indicated concentrations of AmeGlnR proteins (WT, D50A, D50E, D50N, T9A, R52A, T9A/R52A, R111A, and R111E), and salmon sperm DNA was added in each sample to mask the nonspecific binding effect. The signals of free DNA and protein-DNA complexes were scanned and shown.
The Conserved Asp50 of GlnR Is Critical for Maintaining the Receiver Domain-mediated Protein Dimerization and the Corresponding Physiological Function
The above biochemical and structural analyses clearly demonstrate that GlnR with the conserved Asp unphosphorylated is functional. However, because the D50A mutation supposed to mimic its unphosphorylated status of GlnR can neither complement for the growth defect of the glnR null mutants nor bind to its target promoter, such as that of glnA (Figs. 5 and 7; see Ref. 14 for the data of M. smegmatis), the underlying mechanism for the importance of Asp50 in maintaining the biological function of GlnR needs to be addressed.
The crystal structure of the wild type AmeGlnRRec protein indicates that Asp50 forms an ionic interaction with Arg52 and a hydrogen bond with Thr9 (Fig. 2C; see above). This is also true in the structure of MtbGlnRRec, in which Asp49 forms similar interactions with Arg51 and Thr8. Therefore, we propose that these charge interactions may stabilize the homodimeric conformation of GlnR (Fig. 2C), and D50A mutation in AmeGlnR may completely disrupt these interactions and thus result in dismantling the functional homodimerization.
The influence of this D50A mutation upon homodimer formation of either the AmeGlnRRec or the full-length AmeGlnR protein was verified using in vitro size exclusion chromatographic analysis or an in vivo chemical cross-linking assay, respectively. Both results suggest that whereas the wild type AmeGlnR forms homodimer, AmeGlnR(D50A) exists mainly as monomers (Figs. 3D and 4). To explore the possible mechanism underlying the effect of the D50A mutation upon dimerization, the structure of AmeGlnRRec(D50A) is modeled based on that of the wild type AmeGlnRRec through molecular dynamic (MD) simulations (Fig. 8). Comparing these two structural models, it is obvious that the Asp50-focused charge interaction network (with Arg52 and Thr9) is completely abolished in the D50A mutant, which may cause both the Arg52 residue and the β3-α3 loop (residues Ala50–Asp54) to move away from the so-called “phosphorylation pocket,” leaving a space to accommodate the β4-α4 loop (residues Val81–Val86) shifted away from the dimer interface (shown as arrows in Fig. 8). The conformational change in β4-α4 loop and the connecting α4 helix may greatly impair the α4-β5-α5 interface, through which the two monomers form a homodimer.
FIGURE 8.
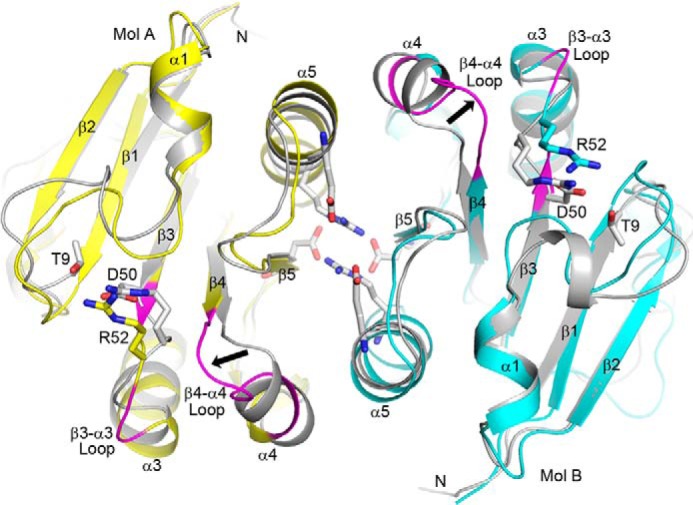
Structural differences between wild type AmeGlnRRec dimer and AmeGlnRRec(D50A) (MD simulations). The structure of wild type AmeGlnRRec dimer is colored gray, the MD simulated structure of AmeGlnRRec(D50A) is colored yellow (molecule A) and cyan (molecule B). The β3-α3 loop (residues Ala50–Asp54) and β4-α4 loop (residues Val81–Val86) are colored in magenta, and an arrow indicates the significant conformational change in β4-α4 loop. The residues constituting the putative phosphorylation pocket and the polar residues involved in the dimer interface are shown with sticks.
To further verify the above hypothesis, we generated two groups of mutants based on the structural information. The AmeglnR(D50N) and AmeglnR(D50E) mutants are designed to maintain the hydrogen bonds of either Asn50 or Glu50 with Arg52 and Thr9, although the interactions are weakened due to the loss of the proposed salt bridge or the varied bond lengths in between. The AmeGlnR mutants containing either R52A or T9A alone and the double mutant of R52A/T9A belong to the second group, which is designed to test the effect of charge interactions by altering the surrounding amino acid residues individually or together rather than simply mutating the Asp50 residue as in the first group.
All of these mutants were tested for their in vivo oligomeric status, growth properties, and in vitro DNA binding capabilities. The AmeGlnR containing the single mutation of D50N, D50E, R52A, or T9A alone still forms a homodimer, whereas the GlnRs with either a single mutation of D50A or double mutation of T9A/R52A exist as monomers in vivo (Fig. 4). Consistently, the AmeGlnR proteins containing the D50N, D50E, R52A, or T9A mutation individually can bind to the glnA promoter in vitro and activate its transcription in vivo, whereas the D50A or T9A/R52A mutated AmeGlnR cannot (Figs. 6 and 7). Physiologically, as expected, the mutants of D50N, D50E, R52A, or T9A alone can complement the growth defect of the ΔglnR host on minimal medium supplemented with nitrate as the sole nitrogen source (Fig. 5), but neither AmeGlnR(D50A) nor AmeGlnR(R52A/T9A) mutant can. In addition, the AmeGlnR(D50L) mutant, which diminishes the charge interaction completely as that of the D50A mutant but maintains the length of the side chain similar to that of Asp, is shown to completely impair the complementation function either (data not shown). It is also significant that similar results are obtained in S. coelicolor with corresponding mutations in ScoGlnR (data not shown). All of the above data suggest that the charge interactions among Asp50, Arg52, and Thr9 of AmeGlnR are critical for maintaining its receiver domain-mediated protein dimerization and the corresponding physiological function, which may be applied to the GlnRs of other actinomycetes.
DISCUSSION
The global transcription factor GlnR of the Gram-positive actinomycetes has been one of the major research focuses with respect to bacterial molecular physiology. It is due to not only its pivotal role in coordinating the expression of genes related to nitrogen metabolism of this industrially and medically important bacterial clade in response to the environmental nitrogen conditions but also its significantly different mode of action as an orphan RR in contrast to that of the well studied two-component system in Gram-negative enteric bacteria. However, the progress of the research has been largely hindered by the difficulties involved in biochemical determination and genetic characterization of GlnR phosphorylation status and its impact upon the protein's structure-function relationship under different physiological conditions. In this study, with a great deal of technological improvement, taking advantage of both the comprehensive research system developed in A. mediterranei and the highly conserved properties of GlnR from S. coelicolor and M. tuberculosis, multilevel evidence is gathered to support the conclusion that the actinomycete GlnR is an atypical OmpR/PhoB subfamily RR and functions as a homodimer stabilized by the critical charge interactions of the unphosphorylated conserved Asp residue with its spatially nearby polar amino acid residues.
Because of the universal presence of a so-called “phosphorylation pocket” within the N-terminal receiver domain of typical RRs, RRs without the pocket, usually determined by sequence alignment, are categorized as “atypical” and subject to various regulatory mechanisms different from that of phosphorylation at the conserved Asp residue in the “pocket” region (44). The crystal structures of AmeGlnRRec and MtbGlnRRec and the structure-based sequence alignment analysis presented in this study demonstrate that the actinomycete GlnR not only lacks the typical acidic pocket but also has the possibility of phosphorylation at the conserved Asp50 site being spatially excluded (Fig. 2). Therefore, in combination with the reproducible negative results in detecting the phosphorylated GlnR either in vitro or in vivo along with the clear positive controls, it is quite certain that the functional GlnR is not phosphorylated at its conserved aspartate residue (Fig. 1). However, this residue is still essential for the physiological function of AmeGlnR, as shown both in vitro and in vivo in this study (Fig. 5).
Usually the Glu/Ala mutation at the conserved phospho-accepting Asp residue is known to mimic the phosphorylation/unphosphorylation status of the RRs (45, 46), although exceptions do exist. For instance, the Asp → Glu mutant of VirG, an RR of the VirA/VirG two-component system in Agrobacterium tumefaciens, does not mimic the phenotype of the phosphorylated VirG. In fact, this study offers another case, where, in contrast to the completely deficient GlnR(D50A) mutant, the GlnR(D50N) is still functional, which is another frequently used mimetic model for the unphosphorylated status. Therefore, these mutations are useful models for mechanistic studies rather than for the proof of the presence of phosphorylation.
The current understanding of the mechanism of activation of the typical RRs is derived from comparisons of structures under the phosphorylated versus the unphosphorylated states (3, 8). For most of the typical RRs, the phosphorylation at their Asp residues induces conformational changes. Particularly, the reorientation of the β4-α4 loop and the two conserved switch residues, namely Ser/Thr from β4 and Tyr/Phe from β5, are changed to facilitate the transition of the RR from monomers (47–49) or weak dimers (2), both at their unphosphorylated states, to the “tight” functional homodimers through the formation of a common α4-β5-α5 ionic interface contributed by a set of highly conserved residues. This strong interaction further brings the DNA binding domains into close proximity, allowing them to bind to the direct repeat half-sites that comprise the recognition sequences for most OmpR/PhoB subfamily RRs (3, 50). On the other hand, the crystal structure of the orphan RR GlnR, unphosphorylated at the conserved Asp residue, forms a functional homodimer through the α4-β5-α5 interface, which is in accordance with the previous data regarding the GlnR binding consensus sequences, where two GlnR binding boxes are found in many cases (18, 51).
Various mechanisms are adopted for facilitating and stabilizing the functional dimerization in atypical RRs in addition to the universal α4-β5-α5 secondary structure element critical for a proper interface. First of all, both ionic and hydrophobic interactions within the dimer interface are employed as the main forces for stabilizing the functional dimer. In the cases of HP1043 from Helicobacter pylori and ChxR from C. trachomatis, no matter whether the conserved Arg residue (corresponding to Arg111 of AmeGlnR) is present in the former or absent in the latter, they all retain the conserved Tyr residues as in typical RRs as well as a few atypical RRs (Fig. 2A), and the side chains in both cases adopted a similar orientation toward the active site as that of the phosphorylated typical RRs in order to facilitate the formation of active dimers. In contrast, the α4 helix of AmeGlnRRec is partially unwound, and the residues corresponding to Ser/Thr and Tyr/Phe in typical RRs are replaced by Val81 and Ile98, the side chains of which adopt similar orientations as that of the unphosphorylated typical RRs (Fig. 9). However, the smaller side chain of Ile98, compared with that of Tyr/Phe, may allow a close interaction between the two monomers through the α4-β5-α5 interface, which is highly similar to that of the phosphorylated typical RRs. In fact, similar small side chain residues are found in the GlnR proteins from various actinomycetes (Fig. 2A), which suggests that a dimer-stabilizing mechanism distinct from that of HP1043 and ChxR is commonly adopted.
FIGURE 9.
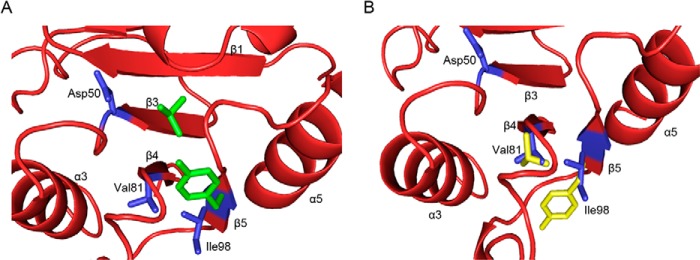
Different conformations of the “switch residues” in AmeGlnRRec and PhoB. The conformations of the “switch residues” in AmeGlnRRec (Val81 and Ile98, shown with blue sticks) differ from those in phosphorylated PhoB (green sticks) (A) but are similar to those in unphosphorylated PhoB (yellow stick) (B). The Protein Data Bank codes of phosphorylated and unphosphorylated PhoB are 1ZES and 1B00, respectively.
More significantly, along with resolving the controversial mutational analysis upon the conserved Asp residue within the missing “phosphorylation pocket” quintet of actinomycete GlnR, this study demonstrates that this Asp residue plays an important role in maintaining the functional conformation of the active homodimer via the formation of salt bridge/hydrogen bond interactions with two spatially close Arg and Thr residues (Thr9-Asp50-Arg52 interactions). This unique mechanism, novel among all of the atypical RRs studied so far, is comprehensively verified by mutational analysis, detecting their dimerization capabilities as well as related functional impacts both in vitro and in vivo. In addition, the MD simulation for wild type and all of the mutants indicates that a significant conformational difference that occurs in the β4-α4 loop is only observed in the GlnR(D50A) (Fig. 8) and GlnR(D50L) (data not shown) mutants, where the loop shifts toward the Asp50 position and may consequently influence or even disable the homodimerization of GlnR via weakening the α4-β5-α5 dimer interface. Interestingly, upon phosphorylation/dephosphorylation, the β4-α4 loop of the typical PhoB actually undergoes significant rearrangement, changing PhoB from homodimers to monomers, respectively. The spatial position of the loop in the unphosphorylated PhoB is the same as that in GlnR(D50A) (3), which may therefore explain the negative effect of D50A mutation upon the oligomerization of GlnR.
So far, this study has shown that the actinomycete GlnR is not phosphorylated at the conserved Asp residue in vitro or in vivo and thus is confirmed to be an atypical RR. Further RT-PCR analyses employing two GlnR target genes, nasA and glnA, show that the transcriptional activation ability of the AmeGlnR mutants is consistent with their corresponding growth phenotypes (Fig. 6); i.e. the active GlnR mutants, GlnR(D50E), GlnR(D50N), GlnR(T9A), and GlnR(R52A) are still able to respond to the extracellular nitrogen availabilities, whereas the inactive GlnR mutants, GlnR(D50A), GlnR(T9A/R52A), and GlnR(R111A/E), are not.
It is known that apart from the possible post-translational modification, transcription of the S. coelicolor glnR gene is stringently regulated by the environmental nitrogen availability (10), which may alter the quantity of functional GlnR available in vivo. However, the expression of glnR in A. mediterranei and M. smegmatis is not significantly affected by the extracellular nitrogen sources (15, 25). Considering the fact that the biological function of the above GlnRs is only found in nitrogen-limited conditions, at least the GlnRs of A. mediterranei and M. smegmatis are expected to be regulated by uncharacterized mechanisms, most likely post-translational modification, resulting in distinct activities in GlnR-mediated global transcriptional regulation. Although eukaryotic phosphorylation on Ser, Thr, or Tyr residues, often identified in prokaryotic proteins (52), is seemingly excluded by the Phos-tag assays under our tested conditions, in addition to that of the Asp residue, it might occur under other cultural conditions or detected by other assay methods. Meanwhile, other types of modifications besides phosphorylation have been reported to alter the activities of atypical RRs (e.g. posttranslational acetylation of RcsB (53, 54) and binding of the small ligand jadomycin B in the modulation of the JadR1 from Streptomyces venezuelae (44)). Interestingly, in the two cases mentioned above, although different mechanisms are adopted, they both inactivate rather than activate the atypical transcription factors under certain metabolic conditions. In the case of actinomycete GlnR, which is naturally active in its dimer status that is stabilized by a robust charge interaction network, “activation” of GlnR seems unnecessary, whereas a similar “inactivation” consequence conveyed by its special regulation mechanism(s) is expected under nitrogen-rich conditions. Therefore, efforts are currently under way in that direction.
Acknowledgment
We thank the staff at the Shanghai synchrotron facility beamline 17U1 for technical assistance with data collection.
This work is supported by National Basic Research Program of China Grant 2012CB721102; National Nature Science Foundation Grants 31322016, 31121001, and 30830002; and Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Grants 2012OHTP, 2009CSP001, and KSCX2-EW-J-12.
The atomic coordinates and structure factors (codes 4O1H and 4O1I) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RR
- response regulator
- GlnRRec
- GlnR regulatory domain
- DSS
- disuccinimidyl suberate
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- MD
- molecular dynamics
- FAM
- 6-carboxyfluorescein.
REFERENCES
- 1. Stock A. M., Robinson V. L., Goudreau P. N. (2000) Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 [DOI] [PubMed] [Google Scholar]
- 2. Gao R., Stock A. M. (2009) Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63, 133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachhawat P., Swapna G. V., Montelione G. T., Stock A. M. (2005) Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure 13, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jeon Y., Lee Y. S., Han J. S., Kim J. B., Hwang D. S. (2001) Multimerization of phosphorylated and non-phosphorylated ArcA is necessary for the response regulator function of the Arc two-component signal transduction system. J. Biol. Chem. 276, 40873–40879 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Birck C., Samama J. P., Hulett F. M. (2003) Residue R113 is essential for PhoP dimerization and function: a residue buried in the asymmetric PhoP dimer interface determined in the PhoPN three-dimensional crystal structure. J. Bacteriol. 185, 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mack T. R., Gao R., Stock A. M. (2009) Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J. Mol. Biol. 389, 349–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hong E., Lee H. M., Ko H., Kim D. U., Jeon B. Y., Jung J., Shin J., Lee S. A., Kim Y., Jeon Y. H., Cheong C., Cho H. S., Lee W. (2007) Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282, 20667–20675 [DOI] [PubMed] [Google Scholar]
- 8. Kern D., Volkman B. F., Luginbühl P., Nohaile M. J., Kustu S., Wemmer D. E. (1999) Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature 402, 894–898 [DOI] [PubMed] [Google Scholar]
- 9. Amon J., Titgemeyer F., Burkovski A. (2010) Common patterns, unique features: nitrogen metabolism and regulation in Gram-positive bacteria. FEMS Microbiol. Rev. 34, 588–605 [DOI] [PubMed] [Google Scholar]
- 10. Tiffert Y., Supra P., Wurm R., Wohlleben W., Wagner R., Reuther J. (2008) The Streptomyces coelicolor GlnR regulon: identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol. Microbiol. 67, 861–880 [DOI] [PubMed] [Google Scholar]
- 11. Malm S., Tiffert Y., Micklinghoff J., Schultze S., Joost I., Weber I., Horst S., Ackermann B., Schmidt M., Wohlleben W., Ehlers S., Geffers R., Reuther J., Bange F. C. (2009) The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology 155, 1332–1339 [DOI] [PubMed] [Google Scholar]
- 12. Tullius M. V., Harth G., Horwitz M. A. (2003) Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect. Immun. 71, 3927–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao W., Zhong Y., Yuan H., Wang J., Zheng H., Wang Y., Cen X., Xu F., Bai J., Han X., Lu G., Zhu Y., Shao Z., Yan H., Li C., Peng N., Zhang Z., Zhang Y., Lin W., Fan Y., Qin Z., Hu Y., Zhu B., Wang S., Ding X., Zhao G. P. (2010) Complete genome sequence of the rifamycin SV-producing Amycolatopsis mediterranei U32 revealed its genetic characteristics in phylogeny and metabolism. Cell Res. 20, 1096–1108 [DOI] [PubMed] [Google Scholar]
- 14. Jenkins V. A., Robertson B. D., Williams K. J. (2012) Aspartate D48 is essential for the GlnR-mediated transcriptional response to nitrogen limitation in Mycobacterium smegmatis. FEMS Microbiol. Lett. 330, 38–45 [DOI] [PubMed] [Google Scholar]
- 15. Yu H., Yao Y., Liu Y., Jiao R., Jiang W., Zhao G. P. (2007) A complex role of Amycolatopsis mediterranei GlnR in nitrogen metabolism and related antibiotics production. Arch. Microbiol. 188, 89–96 [DOI] [PubMed] [Google Scholar]
- 16. Tiffert Y., Franz-Wachtel M., Fladerer C., Nordheim A., Reuther J., Wohlleben W., Mast Y. (2011) Proteomic analysis of the GlnR-mediated response to nitrogen limitation in Streptomyces coelicolor M145. Appl. Microbiol. Biotechnol. 89, 1149–1159 [DOI] [PubMed] [Google Scholar]
- 17. Wang Y., Cen X. F., Zhao G. P., Wang J. (2012) Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J. Bacteriol. 194, 5237–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J., Zhao G. P. (2009) GlnR positively regulates nasA transcription in Streptomyces coelicolor. Biochem. Biophys. Res. Commun. 386, 77–81 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y., Wang J. Z., Shao Z. H., Yuan H., Lu Y. H., Jiang W. H., Zhao G. P., Wang J. (2013) Three of four GlnR binding sites are essential for GlnR-mediated activation of transcription of the Amycolatopsis mediterranei nas operon. J. Bacteriol. 195, 2595–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fink D., Weissschuh N., Reuther J., Wohlleben W., Engels A. (2002) Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol. Microbiol. 46, 331–347 [DOI] [PubMed] [Google Scholar]
- 21. Hutchings M. I., Hoskisson P. A., Chandra G., Buttner M. J. (2004) Sensing and responding to diverse extracellular signals? Analysis of the sensor kinases and response regulators of Streptomyces coelicolor A3(2). Microbiology 150, 2795–2806 [DOI] [PubMed] [Google Scholar]
- 22. Bourret R. B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hickey J. M., Lovell S., Battaile K. P., Hu L., Middaugh C. R., Hefty P. S. (2011) The atypical response regulator protein ChxR has structural characteristics and dimer interface interactions that are unique within the OmpR/PhoB subfamily. J. Biol. Chem. 286, 32606–32616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Connor T. J., Nodwell J. R. (2005) Pivotal roles for the receiver domain in the mechanism of action of the response regulator RamR of Streptomyces coelicolor. J. Mol. Biol. 351, 1030–1047 [DOI] [PubMed] [Google Scholar]
- 25. Amon J., Bräu T., Grimrath A., Hänssler E., Hasselt K., Höller M., Jessberger N., Ott L., Szököl J., Titgemeyer F., Burkovski A. (2008) Nitrogen control in Mycobacterium smegmatis: nitrogen-dependent expression of ammonium transport and assimilation proteins depends on the OmpR-type regulator GlnR. J. Bacteriol. 190, 7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertani G. (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mejía A., Barrios-González J., Viniegra-González G. (1998) Overproduction of rifamycin B by Amycolatopsis mediterranei and its relationship with the toxic effect of barbital on growth. J. Antibiot. 51, 58–63 [DOI] [PubMed] [Google Scholar]
- 28. Kieser T., Hopwood D. A. (1991) Genetic manipulation of Streptomyces: integrating vectors and gene replacement. Methods Enzymol. 204, 430–458 [DOI] [PubMed] [Google Scholar]
- 29. Ma J., Zhang P., Zhang Z., Zha M., Xu H., Zhao G., Ding J. (2008) Molecular basis of the substrate specificity and the catalytic mechanism of citramalate synthase from Leptospira interrogans. Biochem. J. 415, 45–56 [DOI] [PubMed] [Google Scholar]
- 30. Boulanger A., Chen Q., Hinton D. M., Stibitz S. (2013) In vivo phosphorylation dynamics of the Bordetella pertussis virulence-controlling response regulator BvgA. Mol. Microbiol. 88, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheridan R. C., McCullough J. F., Wakefield Z. T. (1971) Phosphoramidic acid and its salts. Inorg. Synth. 13, 23–26 [Google Scholar]
- 32. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 33. Barbieri C. M., Stock A. M. (2008) Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal. Biochem. 376, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 35. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 36. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory. Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 37. Piana S., Lindorff-Larsen K., Shaw D. E. (2011) How robust are protein folding simulations with respect to force field parameterization. Biophys. J. 100, L47–L49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 39. Yu H., Peng W. T., Liu Y., Wu T., Yao Y. F., Cui M. X., Jiang W. H., Zhao G. P. (2006) Identification and characterization of glnA promoter and its corresponding trans-regulatory protein GlnR in the rifamycin SV producing actinomycete, Amycolatopsis mediterranei U32. Acta Biochim. Biophys. Sin. 38, 831–843 [DOI] [PubMed] [Google Scholar]
- 40. Ding X. M., Zhang N., Tian Y. Q., Jiang W. H., Zhao G. P., Jiao R. S. (2002) Establishment of gene replacement/disruption system through homologous recombination in Amycolatopsis mediterranei U32. Sheng Wu Gong Cheng Xue Bao 18, 431–437 [PubMed] [Google Scholar]
- 41. Shu D., Chen L., Wang W., Yu Z., Ren C., Zhang W., Yang S., Lu Y., Jiang W. (2009) afsQ1-Q2-sigQ is a pleiotropic but conditionally required signal transduction system for both secondary metabolism and morphological development in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 81, 1149–1160 [DOI] [PubMed] [Google Scholar]
- 42. McCleary W. R. (1996) The activation of PhoB by acetylphosphate. Mol. Microbiol. 20, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 43. Darbon E., Martel C., Nowacka A., Pegot S., Moreau P. L., Virolle M. J. (2012) Transcriptional and preliminary functional analysis of the six genes located in divergence of phoR/phoP in Streptomyces lividans. Appl. Microbiol. Biotechnol. 95, 1553–1566 [DOI] [PubMed] [Google Scholar]
- 44. Wang L., Tian X., Wang J., Yang H., Fan K., Xu G., Yang K., Tan H. (2009) Autoregulation of antibiotic biosynthesis by binding of the end product to an atypical response regulator. Proc. Natl. Acad. Sci. U.S.A. 106, 8617–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao R., Mukhopadhyay A., Fang F., Lynn D. G. (2006) Constitutive activation of two-component response regulators: characterization of VirG activation in Agrobacterium tumefaciens. J. Bacteriol. 188, 5204–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arribas-Bosacoma R., Kim S. K., Ferrer-Orta C., Blanco A. G., Pereira P. J., Gomis-Rüth F. X., Wanner B. L., Coll M., Solà M. (2007) The X-ray crystal structures of two constitutively active mutants of the Escherichia coli PhoB receiver domain give insights into activation. J. Mol. Biol. 366, 626–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Friedland N., Mack T. R., Yu M., Hung L. W., Terwilliger T. C., Waldo G. S., Stock A. M. (2007) Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry 46, 6733–6743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nowak E., Panjikar S., Konarev P., Svergun D. I., Tucker P. A. (2006) The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J. Biol. Chem. 281, 9659–9666 [DOI] [PubMed] [Google Scholar]
- 49. Robinson V. L., Wu T., Stock A. M. (2003) Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 185, 4186–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. West A. H., Stock A. M. (2001) Histidine kinases and response regulator proteins in two-component signaling systems. Trends. Biochem. Sci. 26, 369–376 [DOI] [PubMed] [Google Scholar]
- 51. Lewis R. A., Shahi S. K., Laing E., Bucca G., Efthimiou G., Bushell M., Smith C. P. (2011) Genome-wide transcriptomic analysis of the response to nitrogen limitation in Streptomyces coelicolor A3(2). BMC Res. Notes 4, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Appleby J. L., Bourret R. B. (1999) Activation of CheY mutant D57N by phosphorylation at an alternative site, Ser-56. Mol. Microbiol. 34, 915–925 [DOI] [PubMed] [Google Scholar]
- 53. Thao S., Chen C. S., Zhu H., Escalante-Semerena J. C. (2010) Nepsilon-lysine acetylation of a bacterial transcription factor inhibits Its DNA-binding activity. PLoS. One 5, e15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hu L. I., Chi B. K., Kuhn M. L., Filippova E. V., Walker-Peddakotla A. J., Bäsell K., Becher D., Anderson W. F., Antelmann H., Wolfe A. J. (2013) Acetylation of the response regulator RcsB controls transcription from a small RNA promoter. J. Bacteriol. 195, 4174–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]