

Background: DNA damage-induced activation of the Rac1/JNK cascade is required for apoptosis.
Results: Upon chemotherapeutic drug treatment, Tiam1, a Rac1-specific GEF, is accumulated through inhibition of CK1/β-TrCP-mediated degradation.
Conclusion: DNA damage induces up-regulation of Tiam1, which contributes to Rac1/JNK activation.
Significance: This work uncovers how the Rac1/JNK cascade is activated upon DNA damage signaling and subsequent apoptosis.
Keywords: Apoptosis, DNA Damage, E3 Ubiquitin Ligase, c-Jun N-terminal Kinase (JNK), Rac1, Tiam1, Chemotherapy
Abstract
The Rac1/JNK cascade plays important roles in DNA damage-induced apoptosis. However, how this cascade is activated upon DNA damage remains to be fully understood. We show here that, in untreated cells, Tiam1, a Rac1-specific guanine nucleotide exchange factor, is phosphorylated by casein kinase 1 (CK1) at its C terminus, leading to Skp, Cullin, F-box-containingβ-TrCP recognition, ubiquitination, and proteasome-mediated degradation. Upon DNA-damaging anticancer drug treatment, CK1/β-TrCP-mediated Tiam1 degradation is abolished, and the accumulated Tiam1 contributes to downstream activation of Rac1/JNK. Consistently, tumor cells overexpressing Tiam1 are hypersensitive to DNA-damaging drug treatment. In xenograft mice, Tiam1-high cells are more susceptible to doxorubicin treatment. Thus, our results uncover that inhibition of proteasome-mediated Tiam1 degradation is an upstream event leading to Rac1/JNK activation and cell apoptosis in response to DNA-damaging drug treatment.
Introduction
During treatment with chemotherapeutic DNA damage drugs, eukaryotic cells initiate an interlaced and complicated signaling network that results in cell cycle arrest or apoptosis (1, 2). The DNA damage response triggers a series of protein modifications, like phosphorylation and ubiquitination, to execute DNA repair, apoptosis, senescence, and cell cycle control (3). During the DNA damage response-induced apoptotic process, the phosphorylation and activation of JNK is required (4–6). As reported, the expression of a constitutively active MEKK1 in PC-12 cells induced apoptosis by activating JNK (7). Also, MAP3Ks could induce JNK activation by stimulating MKK4 or MKK7 in the apoptotic pathway (5). Importantly, MKK4/7-JNK activation was Rac1- or CDC42-dependent (8, 9), suggesting a positive role of Rac1 in promoting apoptotic cell death. Actually, accumulating evidence has indicated that activated Rac1 was able to induce apoptosis. For example, acute activation of JNK through rapid expression of constitutive mutant Rac1 V12 led to apoptosis in thyroid cells (10). Plitidepsin, a potent inducer of apoptosis, was dependent on the Rac1/JNK pathway to function (11). All of these results strongly suggest that the Rac1/JNK cascade is important in DNA damage response-induced cell apoptosis. However, how Rac1 is activated in response to stress stimuli has yet to be fully investigated.
Ubiquitination and degradation of Ras GTPase and Rho GTPase are vital for cell signaling (12). Rac1 belongs to the Rho GTPase family (13). The HECT domain- and ankyrin repeat-containing E3 ubiquitin protein ligase 1 (HACE1) and inhibitors of apoptosis proteins are both E3 ligases for Rac1 (14, 15). However, the ubiquitination control of guanine nucleotide exchange factor (GEF)3 is not well studied. T cell lymphoma invasion and metastasis 1 (Tiam1) serves as a specific GEF for Rho GTPase Rac1 (16). Although it has been well established that Tiam1 plays important roles in tumor progression and metastasis (17–20), the specific mechanisms regulating its activity and protein stability are far from clear (21, 22). It has been reported that Src-induced adherens junction disassembly and cell migration involve phosphorylation and degradation of Tiam1, likely through calpain proteases (20). However, no specific E3 ubiquitin ligase has been identified for Tiam1 to control Rac1 activation.
Loss of Tiam1 sensitizes keratinocytes to apoptosis induced by growth factor starvation, indicating a role of Tiam1 in cell survival (23). Moreover, Tiam1 negatively regulated anoikis-induced apoptosis in a Rac1-independent manner, suggesting that other effectors are probably involved in this form of cell death (18). It has also been reported that Tiam1 is required for bufalin-induced apoptosis (24). Therefore, the function and molecular mechanism of Tiam1 in apoptotic signaling remain contradictory. It is plausible that cell type and DNA damage drugs are key determinants for the function of Tiam1 in apoptosis. Camptothecin (CPT) and doxorubicin (DOX), which target DNA topoisomerase I and II, respectively, are both anticancer drugs (25, 26). Understanding the mechanisms that determine the cell response to these drugs is of high value for cancer treatment in the clinic.
In the ubiquitin proteasome pathway, β-TrCP (β-transducin repeat-containing protein) is a substrate recognition subunit of the SCFβ-TrCP E3 ubiquitin ligase (27, 28). By regulating substrate degradation, β-TrCP is involved in multiple cellular processes, including cell division and various signal transduction pathways (28–30). Among many, IκB, β-catenin, and Mdm2 have been demonstrated to be important substrates (31, 32). In general, β-TrCP recognizes a conserved degron motif, DSGXXS, in a phosphorylation-dependent manner (33). Kinases, including GSK3 and casein kinase family members, have been identified as essential upstream players for β-TrCP to recognize its substrates (27, 30). In this study, initially aimed to search upstream GEFs for Rac1 activation, we found incidentally that β-TrCP-dependent Tiam1 polyubiquitination/degradation is inhibited in response to CPT- or DOX-induced DNA damage signaling and that subsequent Tiam1 accumulation contributes to Rac1/JNK activation and apoptosis.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, and Reagents
HEK293T and HeLa cells were cultured in Dulbecco's modified Eagle's medium. Except where noted, all culture media were supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Anti-c-Myc (catalog no. sc-40), anti-Tiam1 (catalog no. sc-872), anti-casein kinase1ϵ (catalog no. sc-81446), and protein A/G Plus-agarose beads were purchased from Santa Cruz Biotechnology. Anti-FLAG M2, anti-FLAG M2-agarose, anti c-myc (rabbit)-agarose, propidium iodide, MG132, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, citric acid, thymidine, and CPT were purchased from Sigma-Aldrich. D4476 and doxorubicin were purchased from Tocris Bioscience. Anti-Rac1 (catalog no. 610651) was purchased from BD Transduction Laboratories. Anti c-myc (rabbit polyclonal) was from MBL. Anti-GFP was from Proteintech. Polyclonal anti-ubiquitin was purchased from Enzo Life Sciences. Anti-SAPK/JNK (catalog no. 9252), anti-phospho-SAPK/JNK (Thr-183/Tyr-185) (catalog no. 4668), anti-β-TrCP (catalog no. 4394), anti-cleaved caspase 3 (catalog no. 9664), and rabbit IgG (catalog no. 2729) were purchased from Cell Signaling Technology. Anti c-myc (mouse)-agarose was from Abmart. Protein A-Sepharose 4B (catalog no. 10-1041), donkey-anti-mouse Alexa Fluor 568, and goat-anti-rabbit Alexa Fluor 488 were purchased from Invitrogen. Anti-casein kinase1δ (catalog no. A302-136A) was purchased from Bethyl.
Plasmids
The Tiam1-HA construct was provided by Dr. John G. Collard, and MT5-Tiam1 was constructed by cloning Tiam1-HA into the pCS2+vector fused with an N-terminal Myc5 tag. DN-CK1δ was a gift from Dr. Xi He. Tiam1-C1199 was subcloned into the pEGFP-N1 vector to obtain Tiam1-C1199-GFP. CK1 constructs have been described previously (34). Five fragments of Tiam, 1named Tiam1-A (aa 1–392), Tiam1-B (aa 393–750), Tiam1-C (aa 751–1000), Tiam1-D (aa 1001–1250), and Tiam1-E (aa 1251–1591) were subcloned into the pcDNA 3.1 vector fused with an N-terminal Myc tag. Tiam1 (aa 1001–1084) and Tiam1 (aa 1400–1483) were subcloned into the pPGH vector containing an N-terminal GST tag. Mutations in full-length Tiam1, Tiam1-D, and Tiam1-E were generated by site-directed mutagenesis and confirmed by DNA sequencing. Other plasmids have been described previously (35).
Caspase 3 Activity Assay
Caspase 3 activity was detected according to the instructions of the manufacturer of the caspase 3 activity kit (Beyotime), which is on the basis of the hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide into the fluorescent p-nitroanilide. Stable HeLa cells were placed in 6-well plates at 1 × 106 cells/well and treated with DNA-damaging drugs for 10 h. Cells were lysed with 50 μl of lysis buffer for 15 min on ice. Cell lysates were obtained by centrifugation at 14,000 × g for 15 min, and protein concentration was determined by Bradford protein assay (Beyotime). 20 μl of cell lysates (about 30 μg of protein) were firstly mixed with 70 μl of reaction buffer and then incubated with 10 μl of acetyl-Asp-Glu-Val-Asp p-nitroanilide (2 mm) in a 96-well plate for 2 h at 37 °C. Caspase 3 activity was measured through the substrate p-nitroanilide at an absorbance at 405 nm in a microplate spectrophotometer (Thermo). Caspase 3 activity was presented as a ratio of enzyme activity by comparing it to that of untreated cells.
Transfection and RNA Interference
Plasmid DNA and siRNA transfections were performed using VigoFect transfection reagent (Vigorous Biotech, Beijing, China) or Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturers. Human β-TrCP1/2 siRNA (sense, 5′-AAGUGGAAUUUGUGGAACAUC-3′) has been validated extensively previously (32, 36, 37). shRNA oligonucleotides that target luciferase (sense, 5′-CGTACGCGGAATACTTCGA-3′) and Tiam1 (sense, 5′-GCGAAGGAGCAGGTTTTCT-3′) have also been validated previously (38). siRNA sequences for CK1δ and CK1ϵ were as follows: CK1δ oligo 1, 5′-GCAACCTGGTGTACATCAT-3′; CK1δ oligo 2, 5′-GCACCTTGGAATTGAACAA-3′; CK1ϵ oligo 1, 5′-CCCGCAAATTCAGCCTCAA-3′; and CK1ϵ oligo 2, 5′-CCCTCCGAATTCTCAACAT-3′.
Real-time PCR
Real-time PCR (quantitative PCR) was performed according to the instructions of Fast-Plus EvaGreen® Master Mix with High ROX (catalog no. 31015, Biotium, Hayward, CA). Primers for real-time PCR were as follows: Tiam1, 5′-GAGCCTTCCCTCATCCCAGCAAT-3′ (sense) and 5′-CACGGACTCACAGAACTCATCATCG-3′ antisense; RPLP0, 5′-ACCCAGCTCTGGAGAAACTGC-3′ (sense) and 5′-TGAGGTCCTCCTTGGTGAACA-3′ (antisense).
Western Blot Analysis and Coimmunoprecipitation Assay
For Western blot analysis, cells were lysed with radioimmune precipitation assay buffer (50 mm Tris-Cl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 0.2% SDS, 1 mm DTT supplemented with protease inhibitor mixture, 2 mm Na3VO4, and 25 mm NaF). The protein concentration was measured by BCA protein assay (Pierce), and then equal amount of total proteins were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. For coimmunoprecipitation, the appropriate antibodies (1–2 μg) were incubated with protein A-Sepharose beads for 2 h at 4°C, followed by another incubation with cell lysates for 4 h or overnight. Immunocomplexes were washed five times with wash buffer (25 mm Tris-Cl (pH 7.5), 100 mm NaCl, 1% Nonidet P-40, 30 mm MgCl2) and resolved by Western blot analysis.
Rac1 Activation Assay and Immunofluorescence
Rac1 activation assays (39) and immunofluorescence (35) were performed as described previously.
Immunohistochemistry
Dissected tumors were fixed in 10% formaldehyde for 24 h, embedded in paraffin, and sectioned in 4- to 6-μm-thick slices. Paraffin sections were deparaffinized with xylene and rehydrated in a graded series of ethanol. Antigen was retrieved in 0.01 m citrate buffer by boiling for 15 min. Sections washed with PBS were blocked in peroxidase quenching solution for 10 min and then immersed in 10% goat serum for 10 min. The following procedures were performed according to the instructions for the immunohistochemistry kit (catalog no. 879263, Invitrogen). Tiam1 (dilution: 1:50) and cleaved caspase 3 (dilution: 1:200) antibodies were applied. Sections counterstained with hematoxylin were dehydrated, cleared, and mounted.
Protein Extraction from Tumors
Dissected tumors were ground in mortars with liquid nitrogen. The tumor powder was incubated with radioimmune precipitation assay buffer and sonicated with high-intensity sound waves at 4 °C. Cell lysates were then centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatant was resolved by SDS-PAGE after measuring protein concentration by BCA protein assay (Pierce).
In Vitro Binding Assay
CK1 protein (catalog no. P6030S) was purchased from New England Biolabs. Recombinant GST-Tiam1-D′ (aa 1001–1084) or GST-Tiam1-E′ (aa 1400–1483) was incubated with CK1 in the presence of 1 mm ATP and 1× CK1 kinase buffer for 30 min. The reactions were ready for immobilized GST fusion proteins binding assays. For the in vitro binding assay, HEK293T cells transfected with FLAG-β-TrCP were lysed with lysis buffer and then immunoprecipitated with anti-FLAG M2 beads. The precipitates were washed three times with radioimmune precipitation assay buffer and twice with lysis buffer and finally eluted with FLAG peptides (Sigma-Aldrich, St. Louis, MO) in lysis buffer. Purified FLAG-β-TrCP was incubated at 4°C with phosphorylated or unphosphorylated GST fusion proteins bound to glutathione-Sepharose beads (GE Healthcare) for 1 h. After incubation, the mixture was washed three times with lysis buffer plus 200 mm NaCl, and the precipitates were subjected to SDS-PAGE for Western blot analyses.
In Vivo Ubiquitination
Cells were resuspended with 100 μl of denaturing lysis buffer (50 mm Tris-Cl (pH 7.5), 150 mm NaCl, 0.5% Triton, 1% SDS, and 1 mm DTT) and boiled for 10 min, followed by addition of 1 ml of lysis buffer (50 mm Tris-Cl (pH 7.5), 150 mm NaCl, and 0.5% Triton, supplemented with protease inhibitor mixture). Cell lysates were incubated with the indicated antibodies for 4 h at 4 °C, followed by another incubation with protein A for 1 h. Immunocomplexes were washed four times with lysis buffer and resolved by immunoblotting with the indicated antibodies.
Sub-G1 Analysis
Stable HeLa cells were seeded into 12-well plates at a density of 2 × 105 cells/well. Cells were serum-starved for 24 h and then treated with DNA-damaging drugs for the indicated times. Trypsin-treated cells were washed once with cold PBS and then resuspended in 1 ml of cold 80% ethanol to be fixed in −20 °C overnight or for several days. For sub-G1 analysis, cells were pelleted and washed twice in phosphate-citrate buffer (200 mm Na2HPO4 and 4 mm citric acid (pH 7.8)) at room temperature. Then, 500 μl of propidium iodide solution (50 μg/ml ribonuclease A and 50 μg/ml propidium iodide) was added, and the cells were incubated for 30 min before being analyzed by flow cytometry (FACSAria II, BD Biosciences).
Cell Viability Assay
Cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Stable HeLa cells were seeded into a 96-well plate in triplicate at a density of 2 × 103 cells/well. On the second day, cells were treated with medium with or without DNA-damaging drugs for 0, 48, and 72 h. At each time point, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (5 mg/ml) was added into each well for incubation for another 4 h at 37°C, and then culture media were discarded. Dimethyl sulfoxide (150 μl) was then added to the wells, and the plate was subjected to vibration for 5–10 min until the purple precipitate dissolved. Finally, the absorbance was measured at 490 nm. Results in figures were presented as the A490 relative to that of untreated cells.
In Vivo Chemotherapy
Mouse experiments were approved by the Experimental Animal Management and Ethics Committee of Tsinghua University. All nude female mice 6 weeks of age were used for the tumor xenograft experiment. HeLa cells (2 × 106 cells) expressing GFP or Tiam1-C1199-GFP in 100 μl of DMEM/Matrigel (BD Biosciences) (1:1) solution were implanted subcutaneously into the right flank of each mouse. Treatment began when the diameter of the tumor reached 0.5–1 cm. Mice (GFP, n = 12; Tiam1-C1199-GFP, n = 12) were separated randomly into four groups (GFP + saline, GFP + DOX, Tiam1-C1199-GFP + saline, and Tiam1-C1199-GFP + DOX). Mice were administered 2.5 mg/kg doxorubicin every 3 days by intraperitoneal injection. The tumor size was measured every 3 days by digital caliper, and tumor volume was calculated by the formula volume = length × width2 / 2. To detect up-regulation of Tiam1 in solid tumors after doxorubicin treatment, HeLa cells (5 × 105 cells) expressing GFP were injected subcutaneously into the right flanks of four mice. When the tumor was visible, mice were separated into two groups that were treated with 2.5 mg/kg doxorubicin or saline by intraperitoneal injection three times a week. After 2 weeks, tumors were dissected for immunohistochemistry and protein extraction.
Statistical Analysis
Data are presented as mean ± S.D. Two-tailed Student's t test was used to evaluate the statistical significance in the mean value between two populations (**, p < 0.01).
RESULTS
β-TrCP Interacts with Tiam1 and Controls Its Stability
Tiam1 is an important GEF for Rac1 activation, but its stability control is largely unknown, and no E3 ligase has been identified that specifically targets Tiam1. Here we found that β-TrCP (SCFβ-TrCP) specifically bound toTiam1 (Fig. 1A). The association between endogenous Tiam1 and β-TrCP was confirmed further (Fig. 1B), suggesting that β-TrCP might be an E3 ubiquitin ligase for Tiam1. Next, we observed that, upon MG132 treatment, endogenous Tiam1 was up-regulated (Fig. 1C). Moreover, MG132 treatment could rescue the reduction of exogenous Tiam1 caused by coexpression of β-TrCP, whereas cotransfected GFP was not affected (Fig. 1D), suggesting that Tiam1 was degraded by the proteasome pathway. Consistent with β-TrCP being an E3 ligase, β-TrCP coexpression markedly induced Tiam1 polyubiquitination (Fig. 1E), and knockdown of β-TrCP efficiently diminished endogenous Tiam1 polyubiquitination (Fig. 1F). Moreover, β-TrCP controlled Tiam1 via posttranscriptional regulation because depletion of β-TrCP by siRNA resulted in Tiam1 protein accumulation with its mRNA level unaffected (Fig. 1G). β-TrCP normally recognizes substrates in a phosphorylation-dependent manner (40). Therefore, we performed a coimmunoprecipitation assay after λ-phosphatase treatment and found that the interaction between endogenous as well as overexpressed β-TrCP and Tiam1 were abolished (Fig. 1, H and I). Taken together, our results suggest that SCFβ-TrCP, an E3 ubiquitin ligase for multiple proteins, binds Tiam1 and regulates its stability in a phosphorylation-dependent manner.
FIGURE 1.

β-TrCP interacts with Tiam1 and controls its stability in a phosphorylation-dependent manner. A, cell lysates from HEK293T cells transfected with Myc-Tiam1 and FLAG-β-TrCP plasmids were subjected to IP with anti-FLAG M2 beads. WCL and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Cells were treated with 10 μm MG132 overnight before harvesting. HC, heavy chain. B, cell lysates from HeLa cells were subjected to IP with anti-β-TrCP. IgG was used as a negative control. Cells were treated with 10 μm MG132 overnight before harvesting. C, Western blot analysis of total cell lysates derived from HEK293T cells treated with 10 μm MG132 for 6 h. DMSO, dimethyl sulfoxide. D, Western blot analysis of total cell lysates derived from HEK293T cells transfected with Myc-Tiam1 and FLAG-β-TrCP. Where noted, cells were treated with 10 μm MG132 for 8 h. pEGFP-N1 was used to monitor transfection efficiency. E, cell lysates from HEK293T cells transfected with Myc-Tiam1, FLAG-β-TrCP, and HA-ubiquitin (Ub) plasmids were subjected to IP with anti-Myc beads. Whole cell lysates and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Where noted, cells were treated with 10 μm MG132 for 6 h before harvesting. F, cell lysates from HeLa cells transfected with non-targeting siRNA (Ctrl) and β-TrCP siRNA for 48 h were subjected to IP with anti-Tiam1. Where noted, cells were treated with 10 μm MG132 for 6 h before harvesting. G, Western blot analysis of total cell lysates (top panel) and quantitative PCR analysis of the Tiam1 mRNA level (bottom panel) derived from HeLa cells transfected with non-targeting siRNA (Ctrl) and β-TrCP siRNA for 48 h. H, cell lysates from HEK293T cells transfected with Myc-Tiam1 and FLAG-β-TrCP plasmids were subjected to IP with anti-FLAG M2 beads. Where noted, whole cell lysates were treated with λ-phosphatase before IP. Cells were treated with 10 μm MG132 overnight before harvesting. I, cell lysates from HeLa cells were subjected to IP with anti-β-TrCP. IgG was used as a negative control. Cells were treated with 10 μm MG132 overnight before harvesting. Where noted, whole cell lysates were treated with λ-phosphatase before IP.
Casein Kinase 1 Regulates Tiam1 Stability Dependent on the Ubiquitin Pathway
It is generally accepted that β-TrCP recognizes a phospho-degron motif in target protein phosphorylated by one or several kinases (28, 41). Hence, we decided to identify the specific kinases responsible for Tiam1 phosphorylation that led to β-TrCP recognition. Analysis of the amino acid sequence in Tiam1 revealed many serine and threonine residues coincident with CK1 consensus phosphorylation motifs, D/EXXS and S/T-PO4XXS/T (42). Therefore, we performed a coimmunoprecipitation assay and found that Tiam1 interacted predominantly with CK1δ and CK1ϵ, whereas its interaction with CK1α was much weaker and, with CK1γ, undetectable (Fig. 2A). Endogenous association between Tiam1 and CK1δ was also detected (Fig. 2B). Interestingly, depletion of either CK1δ or CK1ϵ alone resulted in Tiam1 accumulation in HeLa cells (Fig. 2C), suggesting that CK1 family members are likely involved in regulating the Tiam1 protein level through the ubiquitin pathway. Supporting this hypothesis, depletion of either CK1δ or CK1ϵ by siRNA reduced endogenous Tiam1 polyubiquitination (Fig. 2D).
FIGURE 2.

CK1 is involved in regulating Tiam1 stability. A, cell lysates from HEK293T cells transfected with Myc-Tiam1 and various FLAG-CK1 plasmids were subjected to IP with anti-FLAG M2 beads. WCL and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Cells were treated with 10 μm MG132 overnight before harvesting. B, cell lysates from HeLa cells were subjected to IP with anti-CK1δ. IgG was used as a negative control. Cells were treated with 10 μm MG132 for 6 h before harvesting. C, Western blot analysis of total cell lysates derived from HeLa cells transfected with non-targeting siRNA (Ctrl), CK1δ, or CK1ϵ siRNA for 48 h. D, cell lysates from HeLa cells transfected with non-targeting siRNA (Ctrl), CK1δ, or CK1ϵ siRNA were subjected to IP with anti-Tiam1. WCL and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Where noted, cells were treated with 10 μm MG132 for 6 h before harvesting. Ub, ubiquitin. E, Western blot analysis of total cell lysates derived from HeLa cells treated with the CK1 inhibitor D4476 for 24 h at the indicated concentrations. F, cell lysates from HeLa cells were subjected to IP with anti-Tiam1 for ubiquitination analysis. Where noted, cells were treated with D4476 for 24 h and 10 μm MG132 for 6 h before harvesting.
To further test the importance of CK1 in controlling Tiam1 stabilization, a small molecule inhibitor, D4476, was utilized to inhibit the activity of endogenous CK1 (43). As expected, the treatment caused significant Tiam1 accumulation in a dose-dependent manner (Fig. 2E). Furthermore, D4476 treatment evidently reduced the polyubiquitination of endogenous Tiam1 proteins (Fig. 2F), indicating that D4476 stabilized Tiam1 through inhibiting its polyubiquitination and degradation. Overall, these results suggest that CK1 controls Tiam1 stability by mediating its polyubiquitination.
CK1 Is Required for Tiam1 Recognition and Destruction by β-TrCP
Next, we verified whether CK1 was involved in regulating Tiam1/β-TrCP interaction. A dominant negative CK1δ (DN-CK1δ) was applied to inhibit endogenous CK1 activity, and it ultimately abolished the interaction between Tiam1 and β-TrCP (Fig. 3A). In addition, D4476 could also dramatically inhibit Tiam1/β-TrCP association (Fig. 3B). Consistently, depletion of CK1δ and CK1ϵ led to a much weaker interaction between Tiam1 and β-TrCP in comparison with control (Ctrl) siRNA treatment (Fig. 3C). Endogenous association between Tiam1 and β-TrCP also relied on CK1δ/ϵ because either CK1δ or CK1ϵ knockdown abolished Tiam1/β-TrCP interaction (Fig. 3D). To further demonstrate a role of CK1 in β-TrCP mediated Tiam1 destruction, CK1δ was coexpressed with Tiam1 and β-TrCP. As expected, CK1δ drastically enhanced Tiam1 degradation, and MG132 treatment clearly rescued Tiam1 degradation, implicating the proteasome-dependent pathway (Fig. 3E). Therefore, Tiam1 is likely phosphorylated by CK1, subsequently recognized and ubiquitinated by β-TrCP, and degraded via the proteasome.
FIGURE 3.

CK1 is required for β-TrCP/Tiam1 interaction. A, cell lysates from HEK293T cells transfected with Myc-Tiam1, FLAG-β-TrCP and Myc-dominant negative (DN) CK1δ plasmids were subjected to IP with anti-FLAG M2 beads. WCL and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Cells were treated with 10 μm MG132 overnight before harvesting. HC, heavy chain. B, cell lysates from HEK293T cells transfected with Myc-Tiam1 and FLAG-β-TrCP plasmids were subjected to IP with anti-FLAG M2 beads. Where noted, cells were treated with the CK1 inhibitor D4476 for 24 h. Cells were treated with 10 μm MG132 overnight before harvesting. C, HEK293T cells were transfected with non-targeting siRNA (Ctrl), CK1δ, or CK1ϵ siRNA for 24 h, followed by transfection with Myc-Tiam1 and FLAG-β-TrCP plasmids. 36 h post-transfection, cell lysates were immunoprecipitated with anti-FLAG beads. Cells were treated with 10 μm MG132 overnight before harvesting. D, HeLa cells were transfected with non-targeting siRNA (Ctrl), CK1δ, or CK1ϵ siRNA. 48 h post-transfection, cell lysates were immunoprecipitated with anti-β-TrCP. IgG was used as a negative control. Cells were treated with 10 μm MG132 for 6 h before harvesting. E, Western blot analysis of total cell lysates derived from HEK293T cells transfecting with FLAG-β-TrCP and Myc-Tiam1 in the presence or absence of Myc-CK1δ. Where noted, 10 μm MG132 was added for 6 h. pEGFP was used to monitor transfection efficiency.
CK1 Phosphorylates Tiam1 at Its C terminus to Trigger Its Interaction with β-TrCP
Because most β-TrCP substrates have a conserved destruction motif, DSGXXS (29), we analyzed the amino acid sequence of Tiam1 proteins and recognized a DSGXXS motif conserved in distinct species (Fig. 4A). To investigate the importance of this motif, we generated three mutant Tiam1 proteins, including two single point mutations (S370A or S374A) and a double point mutation (S370A/S374A) and then verified their interaction with β-TrCP. Surprisingly, none of the mutations influenced Tiam1/β-TrCP association (Fig. 4B), indicating that the DSGXXS motif in Tiam1 was likely not required for its binding to β-TrCP.
FIGURE 4.

CK1 phosphorylates Tiam1 at its C terminus to trigger β-TrCP recognition. A, alignment of DSGXXS motifs in Tiam1 of different species. The asterisks indicate the DSGXXS motif. B, cell lysates from HEK293T cells transfected with FLAG-β-TrCP, Myc-Tiam1 WT, and its point mutant plasmids were subjected to IP with anti-FLAG M2 beads. WCL and immunoprecipitates were subjected to Western blot analysis with the indicated antibodies. Cells were treated with 10 μm MG132 overnight before harvesting. HC, heavy chain. C, schematic of Tiam1 and five truncated Tiam1 fragments containing two PEST sequences (P), an N-terminal pleckstrin homology (PHn) domain, a Ras-binding domain (RBD), a PDZ domain, a Discs-large homology (DH) region, and a C-terminal PH (PHc) domain. Tiam1 C1199 is an N-terminal truncated form. D, cell lysates from HEK293T cells transfected with β-TrCP and Myc-Tiam1 fragments were subjected to IP with anti-FLAG M2 beads. Cells were treated with 10 μm MG132 overnight before harvesting. E, cell lysates from HEK293T cells transfected with FLAG-CK1δ and Myc-Tiam1 fragments were subjected to IP with anti-FLAG M2 beads. Cells were treated with 10 μm MG132 overnight before harvesting. F, cell lysates from HEK293T cells transfected with FLAG-β-TrCP, Myc-Tiam1-A, or C1199 were subjected to IP with anti-FLAG M2 beads. Cells were treated with 10 μm MG132 overnight before harvesting. G and H, cell lysates from HEK293T cells transfected with FLAG-β-TrCP and Myc-Tiam1-D (G) or Myc-Tiam1-E (H) were subjected to IP with anti-FLAG M2 beads. Cells were treated with 10 μm MG132 overnight before harvesting. Where noted, whole cell lysates were treated with λ-phosphatase before IP. I, in vitro binding assay using GST-Tiam1-D′ (Tiam1-D′, aa 1001–1084) or GST-Tiam1-E′ (Tiam1-E′, aa 1400–1483) purified from Escherichia coli and FLAG-β-TrCP purified from transfected HEK293T cells. CK1 and ATP were added as indicated. GST was used as a negative control. GST proteins were shown by Coomassie Blue staining, and β-TrCP was detected by Western blotting.
To elucidate the phospho-degron sequences in Tiam1, we constructed five truncated fragments of Tiam1 (Tiam1-A, Tiam1-B, Tiam1-C, Tiam1-D, and Tiam1-E) (Fig. 4C), according to its distinct functional domains (16), and tested their interaction with β-TrCP. Coimmunoprecipitation results indicated that Tiam1-C, Tiam1-D, and Tiam1-E interacted preferentially with β-TrCP, although other two fragments also associated with β-TrCP to a lesser extent (Fig. 4D), indicating that the phospho-degron likely resides in the carboxyl terminus of Tiam1. Coincidently, we found that CK1δ specifically bound to Tiam1-D and Tiam1-E but not to the other fragments (Fig. 4E), suggesting that CK1δ may phosphorylate Ser/Thr residues in these two fragments and trigger β-TrCP recognition. Tiam1-C1199, a well characterized, constitutively active form lacking an N terminus (22, 44), was also recognized by β-TrCP (Fig. 4F). The Tiam1-D and Tiam1-E fragments were recognized by β-TrCP in a phosphorylation-dependent manner because λ-phosphatase treatment largely reduced their association (Fig. 4, G and H). To further confirm that Ser/Thr residues within fragments D and E were indeed phosphorylated by CK1 and functioned as β-TrCP recognition sequences, we purified GST-Tiam1-D′ and GST-Tiam1-E′ (two shorter fragments of Tiam1-D and Tiam1-E containing most potential phosphorylation sites) and performed an in vitro phosphorylation and binding assay. The results showed that GST-Tiam1-D′ or Tiam1-E′ was recognized by β-TrCP in a phosphorylation-dependent manner (Fig. 4I). This result confirmed that CK1 could indeed phosphorylate Tiam1 and that the phosphorylation ensured the recognition by β-TrCP in vitro. Taken together, CK1 mainly phosphorylates the C terminus of Tiam1 and then triggers β-TrCP recognition, subsequent ubiquitination, and degradation.
Tiam1 Is up-regulated in Response to DNA Damage by Disturbing CK1/β-TrCP-dependent Degradation
It has been shown that Tiam1 is involved in apoptotic signaling in response to genotoxic stress (24, 45). This prompted us to examine whether the level of Tiam1 ubiquitination was changed in response to genotoxic stress. As shown in Fig. 5A, CPT or DOX treatment significantly reduced Tiam1 polyubiquitination, suggesting that a DNA damage agent might cause Tiam1 accumulation through blocking its ubiquitination/degradation. Consistent with this hypothesis, treatment of CPT or DOX reduced the interaction between Tiam1 and CK1δ (Fig. 5B). Consequently, the interaction between β-TrCP and Tiam1 was largely abolished upon CPT or DOX treatment (Fig. 5C). These results indicate that chemotherapeutic drugs might regulate the Tiam1 protein level by controlling the CK1/β-TrCP-dependent degradation pathway. Therefore, we examined whether the Tiam1 protein level was increased in response to CPT or DOX treatment. As shown in Fig. 5D, CPT treatment in HeLa cells led to gradual Tiam1 accumulation in a time-dependent manner with a concomitant increase of phospho-JNK. Similarly, DOX treatment also led to Tiam1 accumulation, accompanied by increased Rac1-GTP and phosphorylated JNK (Fig. 5E), implying that up-regulated Tiam1 was likely responsible for activation of the Rac1/JNK cascade.
FIGURE 5.

Tiam1 is up-regulated by disrupting β-TrCP-dependent degradation in response to DNA-damaging drug treatment. A, cell lysates from HeLa cells treated with 100 μm CPT or 5 μm DOX for 4 h were subjected to IP with anti-Tiam1. WCL and immunoprecipitates were immunoblotted with the indicated antibodies. Where noted, cells were treated with 10 μm MG132 for 6 h before harvesting. Ub, ubiquitin. B, cell lysates from HeLa cells treated with 100 μm CPT or 5 μm DOX for 6 h were subjected to IP with anti-CK1δ. WCL and immunoprecipitates were immunoblotted with the indicated antibodies. Cells were treated with 10 μm MG132 for 6 h before harvesting. DMSO, dimethyl sulfoxide. HC, heavy chain. C, cell lysates from HeLa cells treated with 100 μm CPT or 5 μm DOX for 4 h were subjected to IP with anti-β-TrCP. WCL and immunoprecipitates were immunoblotted with the indicated antibodies. Cells were treated with 10 μm MG132 overnight before harvesting. exp., exposure. D, Western blot analysis of total cell lysates derived from HeLa cells treated with 100 μm CPT at the indicated time points. Cells were pretreated with 2.5 μm thymidine for 24 h before CPT treatment to enrich the S phase population. E, Western blot analysis of total cell lysates derived from HeLa cells treated with 5 μm DOX for 4 h (left panel) or 100 μm CPT for 6 h (right panel). Cells were pretreated with 2.5 μm thymidine for 24 h before DNA-damaging drug treatment. Dimethyl sulfoxide was used as a control because CPT was dissolved in dimethyl sulfoxide. The right and left panels are from two independent experiments. F, protein samples from dissected tumors of xenograft mice treated with saline or DOX were immunoblotted with anti-phospho-JNK (p-JNK) and anti-JNK antibodies. G, immunohistochemistry analysis of Tiam1 protein level and cleaved caspase 3 in dissected tumors. Scale bar = 500 μm.
Next, to test whether Tiam1 was accumulated after treatment of chemotherapeutic drugs in vivo, we applied a xenograft model using HeLa cells expressing GFP (also see below) implanted subcutaneously into nude mice. One week later, the mice were treated with DOX or saline alone. After 2 weeks, tumors were dissected for immunohistochemistry assay and protein extraction. As shown in Fig. 5F, phospho-JNK was induced in mice receiving DOX treatment. Consistent with JNK activation, cleaved caspase 3 was detected in DOX-treated samples (Fig. 5G). Accordingly, the protein level of Tiam1 was up-regulated markedly in mice treated with doxorubicin in comparison with saline-treated ones (Fig. 5G). Therefore, Tiam1 is stabilized in response to a chemotherapeutic drug-induced DNA damage signal, likely because of inhibition of CK1/β-TrCP-dependent degradation.
Tiam1 Overexpression Renders HeLa Cells Hypersensitive to Chemotherapeutic Drugs
We next generated stable HeLa cells expressing GFP or Tiam1-C1199-GFP to mimic chemotherapeutic drug-induced Tiam1 up-regulation (Fig. 6A). It is known that Tiam1-C1199 is mainly localized at the cell membrane (22, 44) and is a potent activator of Rac1 (46). Our results confirmed these observations (Fig. 6, A and B). Subsequently, we treated these two stable cell lines with CPT or DOX, and found that Tiam1-C1199-overexpressing cells presented higher levels of activated Rac1 and its downstream target phospho-JNK than GFP-expressing cells (Fig. 6C), indicating that Tiam1-C1199 overexpression rendered HeLa cells more sensitive to DNA damage signaling. We further measured cell viability under DOX or CPT treatment in the two stable cell lines. As expected, Tiam1-C1199 overexpression reduced cell viability in comparison with control cells under DOX treatment (Fig. 6D). Moreover, flow cytometry analysis revealed that Tiam1-C1199-expressing cells displayed a higher level of sub-G1 population upon treatment (Fig. 6E). Because JNK activates caspase 3 (47), we monitored caspase 3 activity and found a 2-fold higher activation in Tiam1-C1199-overexpressing cells compared with control cells after DOX treatment (Fig. 6F). All of these results suggest that Tiam1-C1199-expressing cells are more sensitive to chemotherapeutic drug-induced DNA damage, at least in part, through a concomitant activation of the Rac1/JNK cascade.
FIGURE 6.

Tiam1 overexpression enhanced cellular sensitivity to DNA damage. A, Western blot analysis of HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP. B, immunofluorescence confocal images of HeLa cells stably expressing pEGFP-N1 and Tiam1-C1199-GFP. Nuclei were counterstained with DAPI (blue). Scale bar = 20 μm. C, HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP were pretreated with 2.5 mm thymidine for 24 h and then exposed to 100 μm CPT or 5 μm DOX for 5 h. Total cell lysates were immunoblotted with the indicated antibodies. p-JNK, phospho-JNK; exp., exposure. D, cell viability assay of HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP treated with 2 μm DOX at the indicated time points. Cell viability assay was analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. E, flow cytometry analysis of HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP exposed to 40 μm CPT and 5 μm DOX for 24 h. Cells were serum-starved for 24 h before DNA-damaging drug treatment. **, p < 0.01. F, relative caspase 3 activity of HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP incubated with 10 μm DOX for 10 h compared with untreated GFP-expressing cells. Cells were serum-starved for 24 h before DOX treatment. **, p < 0.01. G, xenograft mice with HeLa cells stably expressing pEGFP-N1 or Tiam1-C1199-GFP were subjected to saline or DOX treatment. The tumor volume was measured at the indicated time points. Data are presented as mean ± S.D. (n = 6). H, mice as shown in F were sacrificed 18 days after treatment to measure the tumor weight. Data were presented as mean ± S.D. (n = 6). I, top panels, representative images of dissected tumors. Bottom panels, cleaved caspase 3 (apoptosis marker) staining. Scale bar = 500 μm.
To further confirm these phenomena, HeLa cells expressing GFP or Tiam1-C1199-GFP were injected into nude mice. Consistent with a previous study, Tiam1-C1199 accelerated tumor development (48) (Fig. 6G). Upon treatment, DOX very mildly reduced tumor growth from GFP/HeLa cells in comparison with saline treatment. In contrast, tumor growth from Tiam1-C1199-GFP/HeLa cells was reduced significantly with DOX treatment in comparison with saline-treated ones (Fig. 6G). Tumor weight was also obviously reduced in Tiam1-C1199-GFP/HeLa tumor-bearing mice treated with DOX, whereas there was no significant change in GFP/HeLa tumor-bearing mice treated with saline or DOX (Fig. 6H). Consistently, the staining of cleaved caspase 3, a marker for apoptotic cells, was clearly higher in Tiam1-C1199-GFP/HeLa tumors treated with DOX (Fig. 6I). These results indicated that Tiam1-C1199 overexpression rendered tumors more sensitive to the treatment of chemotherapeutic drugs. All of these results suggest that chemotherapeutic drug-induced Tiam1 up-regulation might contribute to apoptotic signaling.
DISCUSSION
Tiam1, a specific GEF for Rac1 activation, is important for Rac1 signaling during tumorigenesis (17). It has also been well demonstrated that Rac1 activation in response to DNA damage stimuli is required for efficient apoptosis (49–51). We show here that CK1 phosphorylates Tiam1 at its C terminus, leading to β-TrCP recognition and subsequent degradation. In response to DNA damage, CK1/β-TrCP-dependent Tiam1 destabilization is abolished and, as a result, Tiam1 becomes stabilized and accumulates in the cytoplasm. Accumulated Tiam1 stimulates the Rac1/JNK cascade, subsequent caspase 3 activation, and cell apoptosis (Fig. 7).
FIGURE 7.
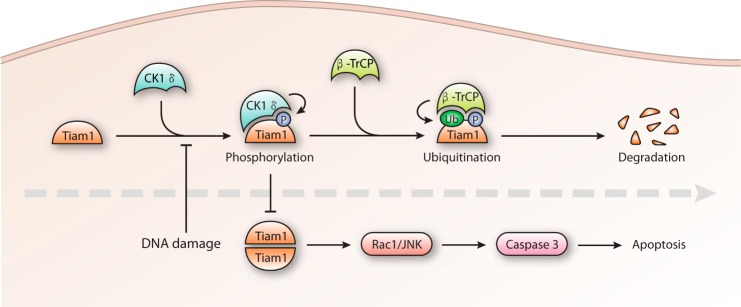
A model. CK1/β-TrCP-mediated Tiam1 degradation is released by DNA damage signaling, and accumulated Tiam1 contributes to Rac1/JNK activation and apoptotic cell death. Ub, ubiquitin; P, phosphorylation.
How could DNA damage signaling release Tiam1 from CK1/β-TrCP regulation? Previous studies have reported that CK1δ is recruited into the nucleus in response to doxorubicin treatment (32, 52). Drosophila CK1α can also translocate into the nucleus, accompanied by high kinase activity after irradiation (53). More recently, it has been shown that DNA damage-induced activation of ATM (ataxia-telangiectasia mutated) directly phosphorylated CK1δ to promote its nuclear localization, thereby facilitating nuclear Mdm2 degradation (54). In this study, we observed that Tiam1/CK1 interaction and subsequent Tiam1 ubiquitination and degradation were abolished in response to DNA damage (Fig. 5). We thus propose that CK1 nuclear localization caused by DNA damaging signal not only promotes nuclear Mdm2 ubiquitination and degradation (32, 54), but also diminishes cytoplasmic Tiam1 ubiquitination and destabilization, both working synergistically to activate apoptotic cascade. Our results thus provided an explanation on how chemotherapy drug-induced DNA damage signaling led to Rac1/JNK activation. Moreover, our observation that DNA damage signaling releases CK1 from Tiam1 establishes a link between the nuclear events after DNA damage-induced ATM activation and the cytoplasmic activation of Rac1/JNK cascade.
Generally, the protein substrates of SCFβ-TrCP contain a destruction motif DSGΦXS (Φ indicating a hydrophobic and X indicating any amino acid) (41). Although there is a conserved DSGXXS in Tiam1 from different species, it lacks the hydrophobic residue at Φ position (serine or tyrosine in human and mouse Tiam1) (Fig. 4A), providing a possible explanation why the DSGXXS in Tiam1 is not recognized by β-TrCP. β-TrCP is capable of recognizing different types of phospho-degron besides the canonical DSGXXS motif (29). Even a non-phospho-motif in Cdc25A is recognized by β-TrCP (55), indicating the complexity of β-TrCP recognition. Here we show that motifs within fragments D and E of Tiam1 are phosphorylated by CK1 and are sufficient and necessary for β-TrCP binding, at least in vitro. Similar to Tiam1, Mdm2 and Per1 are also phosphorylated by CK1 to ensure β-TrCP recognition (32, 56). There is a strong evolutionary relationship between CK1δ and CK1ϵ (56), which also share an autoinhibitory mechanism in controlling their catalytic activity (57, 58). Hence, CK1δ and CK1ϵ often have similar functions in regulating β-TrCP-dependent protein degradation (32, 59). Our data indicate that CK1δ and CK1ϵ are both involved, but not redundant, in mediating Tiam1 degradation because knockdown of either of them induced Tiam1 up-regulation. A similar phenomenon has been reported with Mdm2 stability control (32). Notably, CK1 sometimes needs another kinase to provide a priming phosphorylation (59). Hence, we could not exclude the possibility that other kinases cooperate with CK1 to control Tiam1 stability.
Cancer chemotherapy using DNA-damaging drugs is still a widely applied strategy for cancer patients, although adverse effects are relatively common. On one hand, it is of great importance to maximize the therapeutic efficacy of DNA-damaging agents and minimize the side effects (25). On the other hand, it is of equal importance to predict the sensitivity of each patient toward chemotherapeutic agents. In this sense, specific biomarkers are crucial indicators. Thus, Tiam1, as a positive regulator of DNA damage-induced apoptosis, might be a good biomarker for chemotherapy with at least the treatment of CPT or DOX.
Acknowledgments
We thank Drs. Ye-Guang Chen, John G. Collard, and Xi He for reagents.
This work was supported by National Natural Science Foundation of China Grants 31221064 and 81272235 (to W. W) .
- GEF
- guanine nucleotide exchange factor
- CPT
- camptothecin
- aa
- amino acids
- Ctrl
- control
- IP
- immunoprecipitation
- WCL
- whole cell lysate(s).
REFERENCES
- 1. Polo S. E., Jackson S. P. (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sperka T., Wang J., Rudolph K. L. (2012) DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 13, 579–590 [DOI] [PubMed] [Google Scholar]
- 3. Harper J. W., Elledge S. J. (2007) The DNA damage response: ten years after. Mol. Cell 28, 739–745 [DOI] [PubMed] [Google Scholar]
- 4. Deng Y., Ren X., Yang L., Lin Y., Wu X. (2003) A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115, 61–70 [DOI] [PubMed] [Google Scholar]
- 5. Dhanasekaran D. N., Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J., Lin A. (2005) Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 15, 36–42 [DOI] [PubMed] [Google Scholar]
- 7. Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331 [DOI] [PubMed] [Google Scholar]
- 8. Xu Z., Maroney A. C., Dobrzanski P., Kukekov N. V., Greene L. A. (2001) The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell Biol. 21, 4713–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu Z., Kukekov N. V., Greene L. A. (2003) POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 22, 252–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shirokawa J. M., Elisei R., Knauf J. A., Hara T., Wang J., Saavedra H. I., Fagin J. A. (2000) Conditional apoptosis induced by oncogenic Ras in thyroid cells. Mol. Endocrinol. 14, 1725–1738 [DOI] [PubMed] [Google Scholar]
- 11. Suárez Y., González-Santiago L., Zarich N., Dávalos A., Aranda J. F., Alonso M. A., Lasunción M. A., Rojas J. M., Muñoz A. (2006) Plitidepsin cellular binding and Rac1/JNK pathway activation depend on membrane cholesterol content. Mol. Pharmacol. 70, 1654–1663 [DOI] [PubMed] [Google Scholar]
- 12. Nethe M., Hordijk P. L. (2010) The role of ubiquitylation and degradation in RhoGTPase signalling. J. Cell Sci. 123, 4011–4018 [DOI] [PubMed] [Google Scholar]
- 13. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 14. Oberoi-Khanuja T. K., Rajalingam K. (2012) IAPs as E3 ligases of Rac1: shaping the move. Small GTPases 3, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torrino S., Visvikis O., Doye A., Boyer L., Stefani C., Munro P., Bertoglio J., Gacon G., Mettouchi A., Lemichez E. (2011) The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 21, 959–965 [DOI] [PubMed] [Google Scholar]
- 16. Habets G. G., Scholtes E. H., Zuydgeest D., van der Kammen R. A., Stam J. C., Berns A., Collard J. G. (1994) Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell 77, 537–549 [DOI] [PubMed] [Google Scholar]
- 17. Malliri A., van der Kammen R. A., Clark K., van der Valk M., Michiels F., Collard J. G. (2002) Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 417, 867–871 [DOI] [PubMed] [Google Scholar]
- 18. Minard M. E., Ellis L. M., Gallick G. E. (2006) Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin. Exp. Metastasis 23, 301–313 [DOI] [PubMed] [Google Scholar]
- 19. Minard M. E., Kim L. S., Price J. E., Gallick G. E. (2004) The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res. Treat. 84, 21–32 [DOI] [PubMed] [Google Scholar]
- 20. Woodcock S. A., Rooney C., Liontos M., Connolly Y., Zoumpourlis V., Whetton A. D., Gorgoulis V. G., Malliri A. (2009) SRC-induced disassembly of adherens junctions requires localized phosphorylation and degradation of the Rac activator Tiam1. Mol. Cell 33, 639–653 [DOI] [PubMed] [Google Scholar]
- 21. Schmidt A., Hall A. (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 22. Mertens A. E., Roovers R. C., Collard J. G. (2003) Regulation of Tiam1-Rac signalling. FEBS Lett. 546, 11–16 [DOI] [PubMed] [Google Scholar]
- 23. Rygiel T. P., Mertens A. E., Strumane K., van der Kammen R., Collard J. G. (2008) The Rac activator Tiam1 prevents keratinocyte apoptosis by controlling ROS-mediated ERK phosphorylation. J. Cell Sci. 121, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 24. Cao-Hong, Shibayama-Imazu T., Masuda Y., Shinki T., Nakajo S., Nakaya K. (2007) Involvement of Tiam1 in apoptosis induced by bufalin in HeLa cells. Anticancer Res. 27, 245–249 [PubMed] [Google Scholar]
- 25. Nitiss J. L. (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hertzberg R. P., Caranfa M. J., Hecht S. M. (1989) On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme-DNA complex. Biochemistry 28, 4629–4638 [DOI] [PubMed] [Google Scholar]
- 27. Fuchs S. Y., Spiegelman V. S., Kumar K. G. (2004) The many faces of β-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene 23, 2028–2036 [DOI] [PubMed] [Google Scholar]
- 28. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 29. Frescas D., Pagano M. (2008) Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nat. Rev. Cancer 8, 438–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lau A. W., Fukushima H., Wei W. (2012) The Fbw7 and βTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci. 17, 2197–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winston J. T., Strack P., Beer-Romero P., Chu C. Y., Elledge S. J., Harper J. W. (1999) The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 13, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inuzuka H., Tseng A., Gao D., Zhai B., Zhang Q., Shaik S., Wan L., Ang X. L., Mock C., Yin H., Stommel J. M., Gygi S., Lahav G., Asara J., Xiao Z. X., Kaelin W. G., Jr., Harper J. W., Wei W. (2010) Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(β-TRCP) ubiquitin ligase. Cancer Cell 18, 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Margottin F., Bour S. P., Durand H., Selig L., Benichou S., Richard V., Thomas D., Strebel K., Benarous R. (1998) A novel human WD protein, h-β TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1, 565–574 [DOI] [PubMed] [Google Scholar]
- 34. Liang J., Fu Y., Cruciat C. M., Jia S., Wang Y., Tong Z., Tao Q., Ingelfinger D., Boutros M., Meng A., Niehrs C., Wu W. (2011) Transmembrane protein 198 promotes LRP6 phosphorylation and Wnt signaling activation. Mol. Cell Biol. 31, 2577–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu G., Wang Y., Huang B., Liang J., Ding Y., Xu A., Wu W. (2012) A Rac1/PAK1 cascade controls β-catenin activation in colon cancer cells. Oncogene 31, 1001–1012 [DOI] [PubMed] [Google Scholar]
- 36. Seki A., Coppinger J. A., Du H., Jang C. Y., Yates J. R., 3rd, Fang G. (2008) Plk1- and β-TrCP-dependent degradation of Bora controls mitotic progression. J. Cell Biol. 181, 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mailand N., Bekker-Jensen S., Bartek J., Lukas J. (2006) Destruction of Claspin by SCFβTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 23, 307–318 [DOI] [PubMed] [Google Scholar]
- 38. Malliri A., van Es S., Huveneers S., Collard J. G. (2004) The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J. Biol. Chem. 279, 30092–30098 [DOI] [PubMed] [Google Scholar]
- 39. Habas R., He X. (2006) Activation of Rho and Rac by Wnt/frizzled signaling. Methods Enzymol. 406, 500–511 [DOI] [PubMed] [Google Scholar]
- 40. Petroski M. D., Deshaies R. J. (2005) Function and regulation of Cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 41. Wu G., Xu G., Schulman B. A., Jeffrey P. D., Harper J. W., Pavletich N. P. (2003) Structure of a β-TrCP1-Skp1-β-catenin complex: destruction motif binding and lysine specificity of the SCF(β-TrCP1) ubiquitin ligase. Mol. Cell 11, 1445–1456 [DOI] [PubMed] [Google Scholar]
- 42. Marin O., Bustos V. H., Cesaro L., Meggio F., Pagano M. A., Antonelli M., Allende C. C., Pinna L. A., Allende J. E. (2003) A noncanonical sequence phosphorylated by casein kinase 1 in β-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 100, 10193–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rena G., Bain J., Elliott M., Cohen P. (2004) D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 5, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hordijk P. L., ten Klooster J. P., van der Kammen R. A., Michiels F., Oomen L. C., Collard J. G. (1997) Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science 278, 1464–1466 [DOI] [PubMed] [Google Scholar]
- 45. Kawazoe N., Watabe M., Masuda Y., Nakajo S., Nakaya K. (1999) Tiam1 is involved in the regulation of bufalin-induced apoptosis in human leukemia cells. Oncogene 18, 2413–2421 [DOI] [PubMed] [Google Scholar]
- 46. Michiels F., Stam J. C., Hordijk P. L., van der Kammen R. A., Ruuls-Van Stalle L., Feltkamp C. A., Collard J. G. (1997) Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J. Cell Biol. 137, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tournier C., Hess P., Yang D. D., Xu J., Turner T. K., Nimnual A., Bar-Sagi D., Jones S. N., Flavell R. A., Davis R. J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 48. van Leeuwen F. N., van der Kammen R. A., Habets G. G., Collard J. G. (1995) Oncogenic activity of Tiam1 and Rac1 in NIH3T3 cells. Oncogene 11, 2215–2221 [PubMed] [Google Scholar]
- 49. Kadara H., Tahara E., Kim H. J., Lotan D., Myers J., Lotan R. (2008) Involvement of Rac in fenretinide-induced apoptosis. Cancer Res. 68, 4416–4423 [DOI] [PubMed] [Google Scholar]
- 50. Kang M. A., So E. Y., Simons A. L., Spitz D. R., Ouchi T. (2012) DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 3, e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mayr M., Hu Y., Hainaut H., Xu Q. (2002) Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J. 16, 1423–1425 [DOI] [PubMed] [Google Scholar]
- 52. Alsheich-Bartok O., Haupt S., Alkalay-Snir I., Saito S., Appella E., Haupt Y. (2008) PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 27, 3653–3661 [DOI] [PubMed] [Google Scholar]
- 53. Santos J. A., Logarinho E., Tapia C., Allende C. C., Allende J. E., Sunkel C. E. (1996) The casein kinase 1 α gene of Drosophila melanogaster is developmentally regulated and the kinase activity of the protein induced by DNA damage. J. Cell Sci. 109, 1847–1856 [DOI] [PubMed] [Google Scholar]
- 54. Wang Z., Inuzuka H., Zhong J., Fukushima H., Wan L., Liu P., Wei W. (2012) DNA damage-induced activation of ATM promotes β-TRCP-mediated Mdm2 ubiquitination and destruction. Oncotarget 3, 1026–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanemori Y., Uto K., Sagata N. (2005) β-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc. Natl. Acad. Sci. U.S.A. 102, 6279–6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shirogane T., Jin J., Ang X. L., Harper J. W. (2005) SCFβ-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 280, 26863–26872 [DOI] [PubMed] [Google Scholar]
- 57. Cegielska A., Gietzen K. F., Rivers A., Virshup D. M. (1998) Autoinhibition of casein kinase I ϵ (CKI ϵ) is relieved by protein phosphatases and limited proteolysis. J. Biol. Chem. 273, 1357–1364 [DOI] [PubMed] [Google Scholar]
- 58. Graves P. R., Roach P. J. (1995) Role of COOH-terminal phosphorylation in the regulation of casein kinase I δ. J. Biol. Chem. 270, 21689–21694 [DOI] [PubMed] [Google Scholar]
- 59. Zhao B., Li L., Tumaneng K., Wang C. Y., Guan K. L. (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev. 24, 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]