

Background: The molecular mechanisms mediating the oncogenic activity of the transcription factor GLI1 remain elusive.
Results: GLI1 interacts with SMAD factors and PCAF to regulate TGFβ-induced gene expression.
Conclusion: These results define a novel epigenetic mechanism underlying the role of GLI1 as an oncogene.
Significance: This study increases our understanding of gene expression regulation in cancer cells and its potential impact in tumor development.
Keywords: Cancer, Gene Expression, SMAD Transcription Factor, Transcription Factor, Transforming Growth Factor β (TGFβ), GLI1, PCAF
Abstract
The biological role of the transcription factor GLI1 in the regulation of tumor growth is well established; however, the molecular events modulating this phenomenon remain elusive. Here, we demonstrate a novel mechanism underlying the role of GLI1 as an effector of TGFβ signaling in the regulation of gene expression in cancer cells. TGFβ stimulates GLI1 activity in cancer cells and requires its transcriptional activity to induce BCL2 expression. Analysis of the mechanism regulating this interplay identified a new transcriptional complex including GLI1 and the TGFβ-regulated transcription factor, SMAD4. We demonstrate that SMAD4 physically interacts with GLI1 for concerted regulation of gene expression and cellular survival. Activation of the TGFβ pathway induces GLI1-SMAD4 complex binding to the BCL2 promoter whereas disruption of the complex through SMAD4 RNAi depletion impairs GLI1-mediated transcription of BCL2 and cellular survival. Further characterization demonstrated that SMAD2 and the histone acetyltransferase, PCAF, participate in this regulatory mechanism. Both proteins bind to the BCL2 promoter and are required for TGFβ- and GLI1-stimulated gene expression. Moreover, SMAD2/4 RNAi experiments showed that these factors are required for the recruitment of GLI1 to the BCL2 promoter. Finally, we determined whether this novel GLI1 transcriptional pathway could regulate other TGFβ targets. We found that two additional TGFβ-stimulated genes, INTERLEUKIN-7 and CYCLIN D1, are dependent upon the intact GLI1-SMAD-PCAF complex for transcriptional activation. Collectively, these results define a novel epigenetic mechanism that uses the transcription factor GLI1 and its associated complex as a central effector to regulate gene expression in cancer cells.
Introduction
GLI1 is a C2-H2-type zinc finger transcription factor that belongs to the GLI family of transcription factors (1). This family consists of three different members (GLI1, 2, and 3), which are involved in the regulation of tissue patterning, size, and shape by integrating multiple signaling inputs. GLI1, a potent inducer of cellular transformation (1, 2), was originally identified by Kinzler and colleagues in 1987 as an amplified gene in malignant glioma (3). GLI1 was found to be overexpressed in these tumors and its derived cell lines. In addition to gliomas, GLI1 is highly expressed in multiple neoplasms such as small cell lung, pancreatic, stomach, and prostate cancer where its expression is in general associated with unfavorable overall patient survival (4–8). Numerous reports have demonstrated that the sole overexpression of GLI1 is sufficient to induce transformation in vitro and in vivo (9, 10). For instance, transgenic overexpression of GLI1 in epithelial cells generates hyperplastic lesions and tumor development (9, 10). These findings support the notion that increased expression of GLI1 is sufficient for the development of a subset of tumors. GLI1 activity is regulated by different oncogenic cascades, including the HEDGEHOG, EGFR, RAS, and TGFβ pathways (1, 11–20). It has been demonstrated that malignant transformation induced by some of these cascades requires an intact GLI1 transcriptional activity (17, 20, 21). Knowledge of the molecular mechanisms underlying the biological role of GLI1 during carcinogenesis will be important for the understanding of gene regulation as well as development of future therapeutic approaches for tumors with an active GLI1 pathway.
Here, we provide evidence of a novel mechanism regulating the activity of GLI1 in cancer cells. We demonstrate a physical and functional interaction between GLI1 and SMAD4, a downstream effector of the TGFβ pathway. In addition, we show that this novel complex requires the histone acetyltransferase, PCAF,3 to modulate gene expression. Finally, we provide evidence that GLI1 and its cofactors are recruited to the BCL2 promoter upon TGFβ signaling and are required by the TGFβ signaling pathway to control gene expression in tumoral cells. These findings define a new mechanism underlying the role of GLI1 during tumorigenesis and expand the repertoire of epigenetic pathways controlling gene expression in cancer cells.
EXPERIMENTAL PROCEDURES
Cell Lines and TGFβ1 Treatment
PANC1 cells were obtained from American Type Culture Collection (ATCC) and cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS; SAFC BioScience). RMS13 cells were from ATCC and cultured in RPMI 1640 medium with 10% FBS. Smad2−/− and Smad3−/− mouse embryonic fibroblast (MEF) cell lines were a gift from Dr. Hawse (Mayo Clinic, Rochester, MN) and cultured in DMEM supplemented with 10% FBS. For TGFβ treatment, 5 ng/ml TGFβ1 recombinant ligand (R&D Systems) was added to cells in serum-free medium. Samples were collected 24 h after treatment.
Plasmids
Full-length human GLI1 cDNA was cloned in pCMV-3×FLAG vector (Sigma-Aldrich) to make FLAG-GLI1 and into pcDNA 3.1/HisC vector (Invitrogen) to make His-GLI1 using standard recombinant DNA methodology (22). The HA-GLI1 plasmid was a gift from Dr. Cheng (Igen International, Gaithersburg, MD). The FLAG-SMAD2 and FLAG-SMAD4 constructs were generous gifts from Dr. Leof (Mayo Clinic, Rochester, MN). GFP-SMAD4 (epB-Bsd-CAG-EGFP-hSMAD4) and untagged SMAD2 (epB-Puro-CAG-mSMAD2) constructs were from Dr. Brivanlou (The Rockefeller University, New York, NY) (23). The FLAG-PCAF expression construct was kindly provided by Dr. Faubion (Mayo Clinic, Rochester, MN). The BCL2 promoter-luciferase construct was a gift from Dr. Boxer (Center of Molecular Biology in Medicine, Veterans Affairs Palo Alto Health Care System, Palo Alto, CA) (24). The GLI-luciferase reporter (GLI-Luc) was provided by Dr. Hui (University of Toronto, Toronto, ON, Canada) (25). The FLAG-BCL2 construct was purchased from Addgene (Cambridge, MA). The shRNAs targeting human GLI1 (NM_005269.1–2761s1c1, and NM_005269.1–3414s1c1), PCAF (NM_020005.1–950s1c1 and NM_020005.1–962s1c1) and the nontargeting control (NT-SHC016) were obtained from Sigma. The shRNAs targeting SMAD4 (TG309253-5 and TG309253-7), SMAD2 (TG309255C and TG309255D), and Scramble control (TR30013) were from Origene (Rockville, MD). siRNA for GLI1 and SMAD2, and nontargeting control (NT) was purchased from Qiagen (Valencia, CA).
Transfections
Cells were transfected with Lipofectamine (Invitrogen) as described previously (26), with X-tremeGENE HP (Roche Applied Sciences) or Oligofectamine (Invitrogen) according to manufacturers' instructions. For luciferase reporter assays, 1 × 105 cells were plated into 6-well plates and transfected 24 h later. For each condition 0.3 μg of BCL2 promoter-luciferase reporter or GLI-luciferase reporter was used, along with 3 μg of GLI1, SMAD4, or empty vectors. For transfections of multiple constructs, equal amounts of the expression vectors were used. For expression experiments, 1 × 106 cells were plated in 10-cm dishes and transfected 24 h later with 10 μg of DNA. In each set of experiments, equal amounts of plasmid were used by adding empty vectors. Cells were harvested 24–48 h after transfection for overexpression studies or siRNA treatments and 72 h after transfection when performing shRNA knockdown assays. For chromatin immunoprecipitation (ChIP) experiments, 1.8 × 106 cells were plated in 150-mm dishes and transfected 24 h later with 30 μg of DNA.
Luciferase Reporter Assay
Cells were grown and transfected as indicated above. For luciferase reporter assays, cells were plated in triplicate into 6-well plates in medium containing 10% FBS. Samples were harvested and prepared in accordance with the manufacturer's protocol (Promega). To control for intersample variations in transfection efficiency, the total protein of samples in each well was quantitated (Bio-Rad), and luciferase readouts were normalized to protein content. Relative luciferase activity represents luciferase readouts/protein concentrations normalized to control cells within each experiment.
Reverse Transcription and Real-time PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen) following the manufacture's protocol. Five μg of total RNA was reverse-transcribed using a high capacity cDNA synthesis kit (Applied Biosystems). A portion of the total cDNA was amplified by real-time PCR. Samples were prepared with 1×IQ SYBR Green Supermix (Bio-Rad) and the following primers: GLI1, 5′-tgccttgtaccctcctcccgaa-3′ (forward) and 5′-gcgatctgtgatggatgagattccc-3′ (reverse); BCL2, 5′-gagccacgacccttcttaagacat-3′ (forward) and 5′-caggggtcaattaatccatgacac-3′ (reverse); SMAD4, 5′-ttactgttgatggatacgtggacc-3′ (forward) and 5′-agtatgcataagcgacgaaggtca-3′ (reverse); PCAF, 5′-ctgtcagaggaagagatggacaga-3′ (forward) and 5′-tggacgcaggtgaagaggtact-3′ (reverse); SMAD2, 5′-atgtcgtccatcttgccattc-3′ (forward) and 5′-aaccgtcctgttttcttt-3′ (reverse); NMYC, 5′-ctcagtacctccggagag-3′ (forward) and 5′-ggcatcgtttgaggatc-3′ (reverse); IL6, 5′-ccacacagacagccactcacc-3′ (forward) and 5′-ctacatttgccgaagagccctc-3′ (reverse); IL7, 5′-cgcaagttgaggcaatttct-3′ (forward) and 5′-ctctttgttggttgggcttc-3′ (reverse); CYCLIN D1, 5′-gaagatcgtcgccacctg-3′ (forward) and 5′-gacctcctcctcgcacttct-3′ (reverse); and GAPDH, 5′-gacctgacctgccgtctagaaaaa-3′ (forward) and 5′-accaccctgttgctgtagccaaat-3′ (reverse). Amplification was performed using the C1000 Thermal Cycler (Bio-Rad) under the following reaction conditions: 95 °C for 3 min followed by 40 cycles of 30 s at 95 °C, 30 s at 60 °C, and 20 s at 72 °C. Each mRNA level was normalized by comparison with GAPDH RNA levels in the same sample. The results were calculated following the 2ΔCp method.
ChIP Assay
ChIP was conducted using a modification of the Magna ChIP kit protocol (EMD Millipore) as described previously (26). Chromatin was digested with micrococcal nuclease (2.5 units/ml; New England Biolabs) for 20 min at 37 °C and then sonicated for 15 min. Aliquots of the sheared chromatin were subjected to immunoprecipitation using the following antibodies: GLI1 (Novus Biologicals, Littleton, CO), SMAD4 (Cell Signaling Technology, Danvers, MA), PCAF (Abcam, Cambridge, MA), and SMAD2 (Gene Tex, Irvine, CA). Normal rabbit IgG (EMD Millipore) was used as control. Real-time PCR of the ChIP products and genomic input DNA were performed using primers that amplify two areas of the BCL2 promoter containing consensus GLI binding sites. The sequences of the primers are the following: area 1 (−481/−200), 5′-tccgcactccgtcgtccgcccggc-3′ (forward) and 5′-tggcgcgtcccgccgggggcacat-3′ (reverse); and area 2 (−1282/−979), 5′-gctaggggctattcatgctgatta-3′ (forward) and 5′-gggaaggggtttatcaagggcttt-3′ (reverse). Quantitative SYBR PCR was performed in triplicate for each sample or control using the C1000 Thermal Cycler. Results are represented as -fold enrichment, where each antibody signal was relative to its respective input and then normalized to the nonimmune IgG control signal.
Western Blotting
Cells were grown and transfected as described previously. Whole cell extracts were prepared using lysis buffer (150 mm NaCl, 0.5% Nonidet P-40, 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2) supplemented with Complete inhibitor tablets (Roche Applied Science) and incubated for 1 h on ice. To shear the DNA, samples were passed through a 27 ½-gauge needle with a 1-ml syringe. After the lysates were cleared at 15,000 ×g for 30 min, supernatants were collected. Equal amounts of protein (20–50 μg/lane) were separated by electrophoresis and then transferred to PVDF membrane. Antibodies against GLI1, SMAD4, phosphorylated and total SMAD3 were purchased from Cell Signaling; anti-HA antibody was from Roche Applied Science; anti-PCAF was from Abcam; the tubulin antibody and anti-FLAG were obtained from Sigma. Anti-Peroxidase-conjugated secondary antibodies were used, and immunoreactive proteins were detected by chemiluminescence (GE Healthcare).
Immunoprecipitation
PANC1 cells transfected for 48 h or untransfected RMS13 cells (2–3 confluent 10-cm culture plates for each immunoprecipitation) were lysed in 50 mm Tris, pH 7.5, 300 mm NaCl, 0.5% Nonidet P-40 with Complete inhibitor. Lysates were passed through a 27 ½-gauge needle five times and then diluted with 50 mm Tris, pH 7.5, 0.5% Nonidet P-40 to 150 mm NaCl. Samples were centrifuged at 17,000 × g for 10 min. Supernatants were subjected to immunoprecipitation following the Dynabeads Protein G immunoprecipitation kit protocol (Invitrogen). The following antibodies were cross-linked to Dynabeads Protein G for 1 h at room temperature: anti-GLI1 (R&D Systems), anti-PCAF, and normal goat and rabbit IgGs (Santa Cruz Biotechnology). Dynabeads-antibody complexes and lysates were incubated overnight at 4 °C with rotation. Proteins were eluted by the addition of SDS sample buffer and incubation at 95 °C for 5 min. These eluates were subjected to Western blot analysis using primary antibodies as mentioned above.
Immunofluorescence
PANC1 cells were fixed in 4% formaldehyde for 40 min and permeabilized in 0.1% Triton-X-100 at 4 °C for 5 min. The following primary antibodies were used at the indicated dilutions: anti-HA (Roche, 1:300), anti-GFP (Invitrogen; 1:200), and anti-PCAF (1:200). Fluorescent secondary antibodies were from Invitrogen and used at 1:200. After staining, cells were mounted in Prolong with DAPI (Invitrogen). Microscopy was performed using an Olympus AX70 equipped with a Hamamatsu C4742-95 camera. Images were captured using MetaMorph (Universal Imaging Corp.).
MTS Colorimetric Assay
Cells were grown and transfected as indicated above. For MTS assays, 24 h after transfection 5000 cells were plated in 96-well plates in medium containing 10% FBS. Twenty-four h after plating the cells, a first measurement was done and this considered as time = 0. Cells were then cultured under starvation conditions, and the final measurement was performed after 24 h. Samples were harvested and prepared in accordance with the manufacturer's protocol (Promega). Each value was normalized to its time = 0 within each condition. In some experiments cells were treated with 20 μm BCL2 inhibitor, ABT737 (Selleck Biochemical, Houston, TX), for 24 h.
Annexin V Binding Assay
Binding of allophycocyanin-conjugated annexin V (BD Biosciences) to cells was assessed as originally described (27, 28). The cells were transfected with plasmids for GLI1 or BCL2 expression plus GFP and then serum-starved (0.5% FBS) for 48 h. Cells were then stained with annexin V (at the concentration suggested by the supplier) and 100 μl of annexin V buffer consisting of 140 mm NaCl, 2.5 mm CaCl2, and 10 mm HEPES (pH 7.4). After a 15-min incubation, samples were diluted with 400 μl of annexin V buffer and immediately subjected to flow cytometry. In all, 20,000 events were collected from appropriate channels of a FACSCanto flow cytometer (BD Biosciences). Analysis was performed in double positive cells (GFP and allophycocyanin annexin V).
RESULTS
GLI1 Mediates TGFβ-induced Gene Expression in Cancer Cells
Previous studies have demonstrated that TGFβ can induce GLI1 transcription activity (18, 29). Here, we investigated whether the GLI1 and TGFβ-SMAD pathways interact and can act as transcriptional effector of the TGFβ cascade. We focused on the regulation of BCL2, a known transcriptional target of GLI1 (30, 31). GLI1 overexpression increased both mRNA levels (Fig. 1A, left) and promoter activity (Fig. 1A, right) of BCL2 in the TGFβ-responsive cell line, PANC1 (30, 31). Conversely, knockdown of GLI1 using two independent shRNA constructs decreased BCL2 expression in these cells (Fig. 1B) and as well as RMS13 (data not shown). We evaluated whether BCL2 plays a role in the known abilities of GLI1 to promote cell survival (32–34). We found that BCL2 overexpression was able to reduce the levels of apoptosis induced by the knockdown of GLI1 as detected by changes in annexin V staining (Fig. 1C, left). GLI1 depletion also lowered cell growth, and this effect was rescued by the overexpression of BCL2 (Fig. 1C, right). However, pharmacological inhibition of BCL2 blocks GLI1-mediated survival in PANC1 cells cultured under starvation conditions (Fig. 1D).
FIGURE 1.

BCL2 up-regulation underlies the mechanism of GLI1-mediated survival. A, left, increased BCL2 mRNA expression in PANC1 cells transfected with a FLAG-tagged GLI1 expression vector compared with vector control as analyzed by real-time PCR. Right, luciferase assay of cells cotransfected with BCL2 promoter reporter vector and either control vector or GLI1, showing that GLI1 increases BCL2 promoter activity. B, decreased BCL2 mRNA expression in PANC1 cells cotransfected with two independent GLI1 shRNA (#1 and #2) constructs compared with nontargeting (NT) shRNA control measured by real-time PCR. The inset includes Western blot analysis to demonstrate the knockdown efficacy. TUBULIN was used as housekeeping control. C, FACS-based detection of annexin V (left) and MTS assay (right) showing that effects of the knockdown of GLI1 on apoptosis and cell viability can be rescued by the overexpression of BCL2. D, MTS assay of cells overexpressing GLI1 or control vector and treated with the BCL2 inhibitor (ABT737). The data show that BCL2 inhibitor antagonized the effect of GLI1 on cell survival. Bar graphs represent average levels in each group ± S.E. (error bars) from two or more replicates.
Significantly, TGFβ was also able to induce the expression of BCL2. As shown in Fig. 2A, treatment with TGFβ1 ligand (5 ng/ml) increased BCL2 mRNA compared with vehicle controls. Immunoblotting confirmed activation of the TGFβ pathway, as indicated by an increase in SMAD3 phosphorylation in TGFβ-treated cells (Fig. 2A, inset). Using an artificial GLI1 luciferase reporter (GLI-Luc) (25) we showed that TGFβ1 treatment promoted GLI1 activity (Fig. 2B) although no significant change in GLI1 protein expression was observed by immunoblotting (Fig. 2B, inset). TGFβ activation also increased the binding of endogenous GLI1 to a region in the BCL2 promoter spanning from −481 to −200 bp upstream of the first exon containing canonical GLI1 binding sites (Fig. 2C). No increase in GLI1 binding induced by TGFβ1 was detected in the other area analyzed (−1282 to −979 bp upstream of the first exon) (Fig. 2C). Next, we investigated the possibility that TGFβ requires GLI1 to activate the BCL2 promoter. Knockdown of GLI1 completely abrogated the induction of BCL2 mRNA expression by TGFβ1 (Fig. 2D). Noteworthy, this effect was not due to decreased TGFβ-mediated activation of the pathway in GLI1-depleted cells, as shown by p-SMAD3 levels in TGFβ1-treated cells (Fig. 2D, inset). Collectively, these data identified GLI1 as a novel effector molecule for the TGFβ pathway by increasing BCL2 transcription in response to TGFβ signaling.
FIGURE 2.
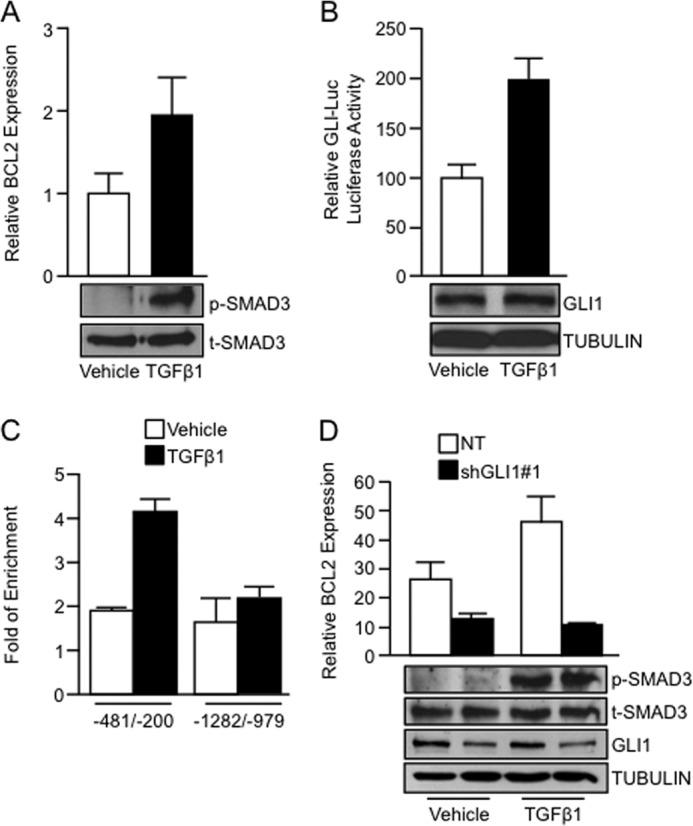
GLI1 is required by TGFβ to regulate BCL2 gene expression. A, real-time PCR showing increased BCL2 mRNA expression in PANC1 cells treated with TGFβ1 ligand. The inset shows increased levels of phospho-SMAD3 (p-SMAD3) in TGFβ1-treated cells, indicative of TGFβ pathway activation. Total SMAD3 was used as loading control. B, PANC1 cells treated with TGFβ1 exhibiting increased GLI-Luc reporter activity compared with vehicle-treated cells. Western blot showed no significant increase in endogenous GLI1 after treatment with TGFβ1 (inset). TUBULIN was used as a housekeeping control. C, ChIP assay was performed using an anti-GLI1 antibody. The data show that upon TGFβ1 stimulation, endogenous GLI1 binding is increased in a region of the BCL2 promoter spanning from −481 to −200 bp upstream the first exon (−481/−200). There was no binding to an upstream sequence in this promoter (−1282/−979). D, real-time PCR showing BCL2 mRNA expression in PANC1 cells transfected with NT vector or GLI1 shRNA targeting construct and treated with vehicle or TGFβ1. The inset shows the levels of the GLI1, phospho-SMAD3 (p-SMAD3), total levels of SMAD3 (t-SMAD3), and TUBULIN. Bar graphs represent average levels in each group ± S.E. (error bars) from three or more replicates.
SMAD4 Cooperates with GLI1 to Regulate Gene Expression
This interplay between TGFβ and GLI1 suggests the possibility of an interaction between GLI1 and members of the SMAD family of transcription factors, known transcriptional effectors of TGFβ signaling (35). Similar to GLI1, SMAD4 binding to the −481/−200 area of the BCL2 promoter was induced by TGFβ (Fig. 3A). Of note, no enrichment of SMAD4 upon TGFβ stimulation was detected in the upstream −1282/−979 area of the BCL2 promoter (Fig. 3A). Using the GLI-Luc reporter we showed a promoting effect of SMAD4 on GLI1-dependent transcription (Fig. 3B). The combined overexpression of SMAD4 and GLI1 resulted in a significant increase of the reporter activity compared with the control vector or the GLI1-transfected cells (Fig. 3B, left). Conversely, there was a lack of effect of SMAD4 overexpression on the GLI-Luc reporter activity in cells transfected with a shRNA construct targeting GLI1 (Fig. 3B, right). Thus, SMAD4 has no direct effect on this reporter, but only shows activation activity when GLI1 is present.
FIGURE 3.

GLI1 functionally interacts with SMAD4 to regulate gene expression. A, data from a ChIP assay in PANC1 cells using a SMAD4 antibody showing enrichment of the binding of endogenous SMAD4 to the −481/−200 BCL2 promoter area after treatment with TGFβ1. Similar to GLI1, SMAD4 does not increase the binding to an upstream sequence in this promoter (−1282/−979) upon TGFβ1 stimulation. B, luciferase assay using PANC1 cells cotransfected with the indicated expression vectors along with GLI-Luc reporter construct (left). The right panel includes reporter data showing the lack of activation of the GLI-Luc reporter by SMAD4 overexpression in the absence of GLI1. Verification of SMAD4 overexpression and GLI1 knockdown is included in the inset. C, real-time PCR for BCL2 mRNA expression in PANC1 cells transfected with the indicated expression vectors. D, decreased BCL2 mRNA expression in PANC1 cells co-transfected with two independent SMAD4 shRNA (#1 and #2) constructs compared with the NT shRNA control measured by real-time PCR. Inset shows the levels of SMAD4 knockdown by Western blotting. E, real-time PCR showing BCL2 mRNA expression in PANC1 cells transfected with the NT or SMAD4 shRNA along with vector control or GLI1 expression constructs. Western blot analysis (inset) shows that the GLI1 was not affected by the SMAD4 knockdown. TUBULIN was used as housekeeping control. F, real-time PCR showing BCL2 mRNA expression in PANC1 cells transfected with NT or SMAD4 shRNA targeting construct and treated with vehicle or TGFβ1. The inset shows the levels of phospho-SMAD3 (p-SMAD3) and total levels of SMAD3 (t-SMAD3). G, cell viability in PANC1 cells transfected with control vector or GLI1 expression construct along with NT vector or two shRNA targeting SMAD4. MTS assay was performed at 72 h post-transfection. Western blot analysis (inset) shows the expression of GLI1 and SMAD4. TUBULIN was used as housekeeping control. Bar graphs represent average levels in each group ± S.E. (error bars) from two or more replicates.
Similar to effects seen with the GLI-Luc artificial reporter, SMAD4 overexpression increases GLI1 activation of BCL2 expression (Fig. 3C). Finally, we demonstrate the requirement of SMAD4 to regulate BCL2 expression downstream of the TGFβ-GLI1 axis. SMAD4 shRNA knockdown impairs basal (Fig. 3D), GLI1-induced (Fig. 3E), as well as TGFβ-stimulated expression of BCL2 (Fig. 3F). Of note, the RNAi knockdown of SMAD4 impairs the induction of the mRNA levels of BCL2 without affecting GLI1 expression or the activation of the pathway by TGFβ1 ligand (Fig. 3, E and F, insets). Finally, we demonstrate that the GLI1-mediated increase in cell viability requires SMAD4 because knockdown of this SMAD4 impaired GLI1-induced survival in PANC1 cells (Fig. 3G). Of note, knockdown of SMAD4 did not affect survival under basal conditions (Control group). Western blot expression controls for the transfected GLI1 construct as well as the efficiency of knockdown of SMAD4 are shown in Fig. 3G (inset). Together, these findings functionally connect SMAD4 with the TGFβ-GLI1 axis in the regulation of gene expression.
GLI1 Physically Interacts with SMAD4
To further define the mechanism underlying this functional interaction between GLI1 and SMAD4, we determined whether these transcription factors function in a complex. Immunoprecipitation of GLI1 from transiently transfected PANC1 cells overexpressing FLAG-SMAD4 and HA-GLI1 demonstrated that both proteins occurred in the same protein complex and this interaction is enhanced by TGFβ (Fig. 4A). Next, we examined the colocalization of GLI1 and SMAD4 in PANC1 cells. Immunofluorescence of cells transfected with GFP-SMAD4 and HA-GLI1 demonstrated that SMAD4 was mainly cytosolic in the absence of TGFβ1, whereas GLI1 localization was primarily nuclear. TGFβ1 treatment caused a dramatic increased nuclear presence of SMAD4 and a smaller but significant shift of GLI1 to increased nuclear localization (Fig. 4B). Thus, the colocalization of these two transcription factors increases upon stimulation with TGFβ1, supporting the premise that GLI1-SMAD4 are within the same complex and functioning as coeffectors of the TGFβ pathway.
FIGURE 4.
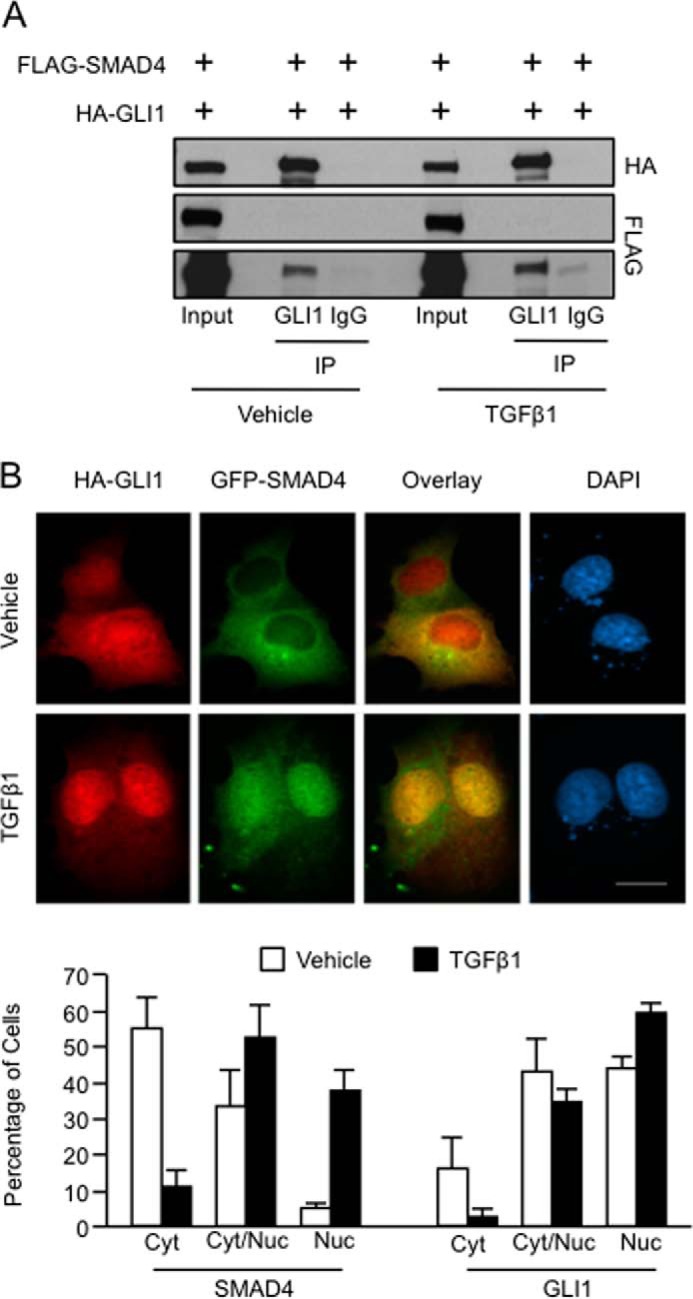
GLI1 and SMAD4 complex in TGFβ-responsive cells. A, PANC1 cells co-transfected with FLAG-SMAD4 and HA-GLI1, treated with TGFβ1, and then immunoprecipitated (IP) using a GLI1 antibody or nonimmune rabbit IgG. Western blotting demonstrated coimmunoprecipitation between GLI1 and SMAD4. B, immunofluorescence of PANC1 cells transfected with HA-GLI1, GFP-SMAD4, and unlabeled SMAD2, and treated with TGFβ1 or control vehicle. SMAD2 was coexpressed in these studies to increase the responsiveness of GFP-SMAD4 to TGFβ1 (23). Lower panel shows the quantification of cytosolic versus nuclear localization of GFP-SMAD4 and HA-GLI1 in cells treated ± TGFβ1. Double-stained cells were graded as primarily cytosolic (Cyt), similar intensity for cytosol and nucleus (Cyt/Nuc), or primarily nuclear (Nuc) for green (GFP-SMAD4) and red (HA-GLI1) signals. Results are expressed as percentage of cells in each category of total cells counted within that experiment. Values are means ± S.E. (error bars) of results from five independent experiments with ≥20 cells/condition for each experiment.
Because SMAD4 requires the interaction with one of the R-SMADs (SMAD2 and SMAD3) to modulate gene expression, we sought to define the role of these R-SMADs in the SMAD4-dependent regulation of GLI-mediated transcription. We used Smad2 and Smad3 knock-out MEFs to address this question. In wild type MEFS (data not shown) and smad3-null MEFS (Fig. 5A, left), there is a robust activation of the GLI-Luc reporter by SMAD4. However, in Smad2-null cells, SMAD4 is not able to increase the activity of GLI-Luc reporter (Fig. 5A, right).
FIGURE 5.

SMAD2 and SMAD4 are required for the recruitment of GLI1 to the BCL2 promoter. A, Smad3−/− and Smad2−/− MEFs were cotransfected with GLI-Luc reporter and either control or SMAD4 expression constructs. Samples were collected for luciferase assay 48 h post-transfection. Basal GLI-Luc activity due to endogenous GLI1 was set at a value of 100 for each control fibroblast line. B, real-time PCR shows BCL2 mRNA expression levels in PANC1 cells transfected with NT or two independent shRNA constructs targeting SMAD2 along with vector control or GLI1 expression constructs. The inset shows levels of expression of GLI1 and SMAD2. TUBULIN was use as housekeeping. Of note is that SMAD2 knockdown does not affect GLI1 expression. C, immunofluorescence in PANC1 cells transfected with HA-GLI1 and GFP-SMAD2 treated with TGFβ1 or control vehicle is shown. D, ChIP assay done in PANC1 cells treated with TGFβ1 shows binding of SMAD2 to the GLI1 binding region of the BCL2 promoter. E, endogenous immunoprecipitation (IP) of GLI1 shows binding of GLI1 with SMAD2 and SMAD4 in RMS13 cells treated with TGFβ1. F, GLI1 ChIP assays in PANC1 cells transfected with the NT, SMAD2, or SMAD4 shRNA and treated with TGFβ1 show that the knockdown of the SMAD2 and SMAD4 factors diminished the binding of GLI1 to the BCL2 promoter. G, similarly, depletion of SMAD2 using two independent siRNAs impairs the binding of SMAD4 to this promoter upon treatment with TGFβ1. Bar graphs represent average levels in each group ± S.E. (error bars) from three or more replicates.
Furthermore, knockdown of SMAD2 in PANC1 cells impairs GLI1-induced expression of BCL2 without affecting GLI1 expression (Fig. 5B). Similar to SMAD4, immunolocalization studies (Fig. 5C) show that SMAD2 and GLI1 colocalize in the nucleus upon TGFβ stimulation. Further, ChIP assays show that the treatment with the TGFβ1 only promotes the binding of SMAD2 to the BCL2 promoter at the same GLI1-SMAD4 binding region (−481/−200) (Fig. 5D).
Finally, we demonstrate that endogenous SMAD2 and SMAD4 can interact in TGFβ-stimulated cells with GLI1 as shown by coimmunoprecipitation of these proteins by an antibody against GLI1 (Fig. 5E). In addition, ChIP assays in cells depleted of SMAD4 and SMAD2 demonstrated a requirement for these transcription factors in the maximal binding of GLI1 to the BCL2 promoter induced by TGFβ1 ligand (Fig. 5F). The knockdown of SMAD2 impairs the binding of SMAD4 to this regulatory sequence (Fig. 5G). Taken together, these data suggest that GLI1 and SMAD4 form a functional complex to regulate transcription and support a functional role for SMAD2 in this complex.
TGFβ-GLI1 Axis Requires the Histone Acetyltransferase PCAF to Modulate Gene Expression
Transcription factors regulate gene expression in part through the interaction of coregulator molecules that modulate histone post-translational modifications. These modifications, known as the histone code, are interpreted by protein complexes (code readers) and translated into transcriptional activation or repression signals (36). In the search of potential coregulators cooperating with GLI1 to regulate gene expression we identified the acetyltransferase PCAF (37) as a mediator of this phenomenon. Knockdown experiments using two independent targeting shRNA vectors demonstrated that PCAF is required for both GLI1-induced (Fig. 6A) and TGFβ-induced BCL2 expression (Fig. 6B). Similar to SMAD proteins, this effect was not due to changes in the expression of GLI1 or activation of the TGFβ pathway (Fig. 6, A and B, insets). Immunoprecipitation of PCAF in cells transfected with FLAG-PCAF and HA-GLI1 demonstrated that these two proteins form a complex (Fig. 6C). The endogenous interaction between PCAF and GLI1 was confirmed by immunoprecipitation of GLI1 using an antibody for PCAF (Fig. 6D). Immunofluorescence studies showed the presence of GLI1 and PCAF in the nucleus of transfected cells and their colocalization upon TGFβ1 stimulation (Fig. 6E). Next, we assessed by ChIP assay the requirement of GLI1 for the binding of PCAF to the BCL2 promoter. RNAi knockdown of GLI1 impaired binding of PCAF to this regulatory sequence in cells treated with TGFβ1 (Fig. 6F). Finally, ChIP assay demonstrated that TGFβ1 induced PCAF binding to the BCL2 promoter (Fig. 6G, left) and increased acetylation of lysine 14 of the histone 3, a histone mark known to be regulated by PCAF (37), in the same region occupied by GLI1 and SMAD4 (Fig. 6G, right). In summary, these results demonstrate the involvement of the histone acetyltransferase PCAF in GLI1-dependent transcriptional activation and provide novel insight into the transcriptional mechanisms underlying GLI1 regulation of gene expression downstream of TGFβ signaling.
FIGURE 6.

The histone acetyltransferase PCAF is required for activation of gene expression regulated by the TGFβ-GLI1 axis. A, real-time PCR shows mRNA levels of BCL2 in PANC1 cells cotransfected with control or GLI1 expression constructs along with NT or PCAF shRNA vectors. GLI1 expression control by Western blotting showed that the knockdown of PCAF does not affect GLI1 expression. TUBULIN was used as housekeeping control. B, BCL2 mRNA expression is shown in PANC1 cells transfected with NT or PCAF shRNA targeting construct and treated with vehicle or TGFβ1. The inset shows the levels of PCAF knockdown, phospho-SMAD3 (p-SMAD3), and total levels of SMAD3 (t-SMAD3). TUBULIN was use as housekeeping. C, PANC1 cells were cotransfected with FLAG-PCAF and HA-GLI1 and then immunoprecipitated (IP) using an anti-PCAF antibody. Western blot analysis was performed to demonstrate coimmunoprecipitation between GLI1 and PCAF. D, an antibody specific for PCAF was used to immunoprecipitate endogenous GLI1 with PCAF in cells treated with TGFβ1. The immunoprecipitated complex was analyzed by Western blotting. E, PANC1 cells transfected with HA-GLI1 and FLAG-PCAF were treated with TGFβ1 ligand. Immunofluorescence was performed to detect the localization of GLI1 and PCAF using anti-HA and anti-PCAF antibodies, respectively. F, PCAF ChIP assay in PANC1 cells transfected with the NT, or GLI1 siRNA and treated with TGFβ1 showed that GLI1 is required for the recruitment of PCAF to BCL2 promoter. G, ChIP assay performed on PANC1 cells treated with the TGFβ1 ligand or vehicle control shows increase binding of PCAF (left) and an enrichment of histone 3 acetylated lysine 14 (H3K14Ac) in the GLI1 binding region of the BCL2 promoter (right) in cells treated with TGFβ1. Total histone 3 (H3) was used to normalize the levels of H3K14Ac. Bar graphs represent average levels in each group ± S.E. (error bars) from three or more replicates.
Finally, we examined whether the TGFβ-GLI1 axis targets additional cancer-related genes besides BCL2. Importantly, expression studies of several known GLI1 and/or TGFβ target genes (1, 2, 10, 11, 17, 38, 39) demonstrated a role for GLI1, SMAD4, and PCAF in the TGFβ-stimulated induction of INTERLEUKIN-7 (IL7) and CYCLIN D1 (Fig. 7, A, C, and D). In contrast, GLI1 depletion by shRNA treatment had no effect on the TGFβ-mediated induction of NMYC and INTERLEUKIN-6 (IL6) (Fig. 7A). Interestingly, overexpression of SMAD4 is able to increase the expression of the target genes in the presence of an active TGFβ pathway (Fig. 7B). Further, Real-time PCR assays show that GLI1 requires an intact PCAF (left) and SMAD4 (right) to modulate the expression of IL7 and CYCLIN D1 (Fig. 7C), thus, further supporting the role of this newly identified histone-modifying complex in the regulation of gene expression downstream of the TGFβ signaling.
FIGURE 7.

GLI1 serves as an effector of a subset of TGFβ-inducible genes. A, PANC1 cells transfected with NT or GLI1-targeted shRNA and treated with vehicle or TGFβ1. Real-time PCR analysis was done to analyze the mRNA expression levels of NMYC, IL6, IL7, and CYCLIN D1. B, mRNA expression of NMYC, IL6, IL7, and CYCLIN D1 in PANC1 cells transfected with SMAD4 expression or control vectors and treated with TGFβ1. C, real-time PCR showing IL7 (upper panels) and CYCLIN D1 (lower panels) mRNA expression in PANC1 cells transfected with NT or PCAF (left panels) or SMAD4 (right panel) shRNA targeting constructs and vector control or GLI1 expression constructs. D, real-time PCR showing CYCLIN D1 (lower panel) and IL7 (upper panel) mRNA expression in PANC1 cells transfected with NT or PCAF shRNA-targeting construct and treated with vehicle or TGFβ1. Bar graphs represent average levels in each group ± S.E. (error bars) from three or more replicates.
DISCUSSION
Several groups have shown a role for GLI1 as an effector of multiple signaling pathways in different cancer types including HEDGEHOG, KRAS, MEK, PI3K, and AKT (21, 40, 41). These pathways modulate GLI1 activity mainly via regulation of the expression of this transcription factor (1, 11–20). Here, we provide evidence of a novel regulatory mechanism involving the interaction of components of the TGFβ pathway (SMAD proteins) modulating GLI1 activity in cancer cells. Specifically, we demonstrate that GLI1 can complex with SMAD4, and this interaction regulates the activation of a subset of TGFβ-inducible target genes, BCL2, IL7, and CYCLIN D1 (Fig. 7). In addition, we show that the histone acetyltransferase, PCAF, and SMAD2 are required for this TGFβ/GLI1-mediated gene activation (Fig. 8).
FIGURE 8.
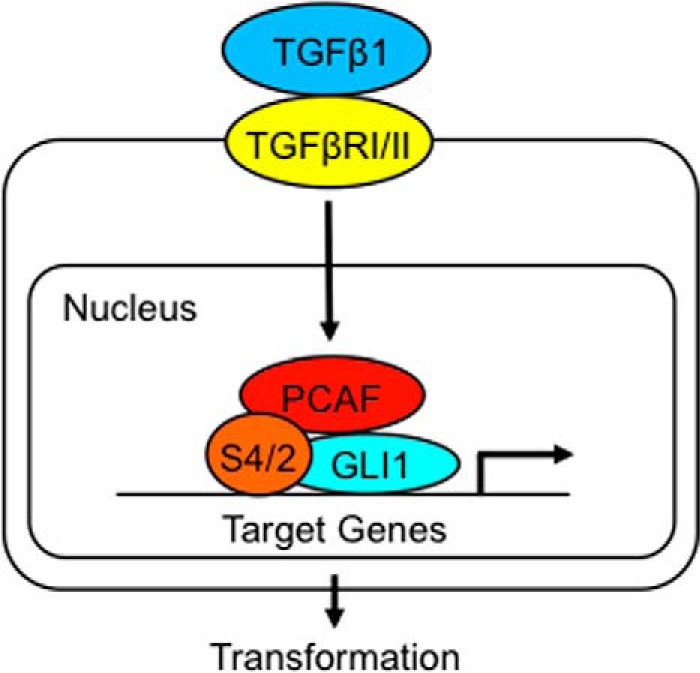
Schematic representation of the proposed mechanism underlying GLI1 action as a transcriptional effector of the TGFβ pathway. This mechanism includes the TGFβ-simulated interplay between GLI1 and SMAD proteins (S4/2) and the involvement of PCAF at the promoters of a subset of TGFβ-inducible genes.
Canonical TGFβ signaling is initiated by the ligand binding to paired type II receptors which recruit type I receptor dimers, forming a heterotetrameric complex and phosphorylating the type I receptor. The activated type I receptor then phosphorylates receptor-regulated SMADs (R-SMADs), such as SMAD2 and SMAD3. The phosphorylated R-SMADs then associate with the Co-SMAD, SMAD4, to form activated SMAD complexes, which are able to translocate to the nucleus. Once in the nucleus they regulate transcription in cooperation with other transcription factors and coactivators/corepressors (42, 43). In our studies, we identified GLI1 as an effector of the canonical TGFβ pathway acting as a transcriptional partner of the SMADs. We have found that SMAD2 and SMAD4 are required for the GLI1-mediated transcription of BCL2, but SMAD3 is dispensable for this interplay. In addition to identifying genes that are regulated by GLI1 and TGFβ, presumably via GLI1/SMAD interactions, we identified genes (NMYC and IL6) that are transcriptionally activated by TGFβ without apparent participation of GLI1. These data suggest that different categories of TGFβ-responsive genes are regulated with or without the involvement of GLI1. We previously showed that TGFβ could stimulate epithelial-to-mesenchymal transition via changes in gene expression in cells depleted of GLI1, consistent with the ability of TGFβ to alter the expression of some genes via GLI1-independent mechanisms (44). Thus, our studies increase the repertoire of mechanisms utilized by the TGFβ pathway in cancer cells by showing that GLI1 can act as a downstream mediator of TGFβ signaling via cooperation with the SMADs for the regulation of certain genes.
Our findings also suggest that in cancer cells in which GLI1 is activated (e.g. by HEDGEHOG or KRAS) and TGFβ signaling is active, these pathways may interact synergistically by GLI1-SMAD complex formation to stimulate transcription of key gene targets. Our studies also have potential translational significance for the development of treatments against cancer. Many cancer cell types exhibit activation of multiple signal cascades that can interact at the level of GLI1. Thus, treatments that inhibit a single pathway such as inhibitors of HEDGEHOG signaling may be insufficient to shut down GLI1 activation when a second pathway (e.g. TGFβ) is active. Such targeted therapies may require multiple drugs directed against different pathways to inhibit GLI1 activity successfully.
A defining feature of transcription factor complexes is the requirement for coregulators that control gene expression, often via modifications in histone amino acids (36, 45). However, no such histone-modifying partners have been previously identified for GLI1. We provide evidence that the histone acetyltransferase PCAF is a partner of GLI1, and we define their interaction as a key determinant for the regulation of gene expression downstream of the TGFβ signaling. PCAF has been implicated in global and locus-specific histone acetylation and plays a key role in oncogene-mediated gene transcription, and it is crucial for cell cycle progression in different cell types (37, 46–48). This histone acetyltransferase has been associated with chemoresistance, promotion of cell growth, and invasiveness in cancer cells. PCAF has been shown to acetylate nucleosomal histone 3, lysine 14 residues, a chromatin mark associated with gene activation (49, 50). In addition, PCAF is associated with large, multiprotein complexes, which possess further histone-modifying capabilities (49). Our findings suggest that GLI1 binding to gene promoters may recruit PCAF, which then modifies histones, thereby contributing to alterations in chromatin leading to a more active state. During the revision of our manuscript, two groups independently demonstrated an interaction between GLI1 and PCAF. One group supported our findings, showing that GLI1 and PCAF can function together to positively regulate GLI1 gene target transcription in cancer cells (51). A second group found that PCAF can acetylate GLI1 under conditions of genotoxic stress, leading to increased GLI1 proteosomal degradation (52). Thus, our work confirms and expands these findings by identifying a novel mechanism in which GLI1 regulates signaling-induced gene transcription via chromatin-based epigenetic modifications.
In summary, our data demonstrate a novel positive transcriptional cooperativity between GLI1 and the TGFβ signaling pathway, which also involves the histone acetyltransferase PCAF. These novel interactions provide insight into molecular mechanisms that may lead to tumor progression in cancer types that have a dependence on GLI1 signaling and an intact TGFβ signaling cascade. The interaction between GLI1 and SMADs has great potential to be further investigated as a molecular target for cancer patients.
Acknowledgment
We thank Emily Porcher for secretarial assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant GM55252 and by National Institutes of Health Grant CA136526 (to M. E. F.-Z.). This work was also supported by the Mayo Clinic College of Medicine Graduate School, Division of Oncology Research (Mayo Clinic), and Mayo Clinic Pancreatic SPORE P50 Grant CA102701 (to M. E. F.-Z.), Mayo Clinic Center for Cell Signaling in Gastroenterology Grant P30 DK84567 (to M. E. F.-Z.), Deutsche Forschungsgemeinschaft Grant KFO210 (to V. E.), SFB-TR17, and the German Cancer Research Foundation 109423 “Inflammation and Cancer” (to V. E.).
- PCAF
- p300/CREB-binding protein-associated factor
- Luc
- luciferase
- MEF
- mouse embryonic fibroblast
- NT
- nontargeting.
REFERENCES
- 1. Hui C. C., Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 [DOI] [PubMed] [Google Scholar]
- 2. Robbins D. J., Fei D. L., Riobo N. A. (2012) The Hedgehog signal transduction network. Sci. Signal. 5, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kinzler K. W., Bigner S. H., Bigner D. D., Trent J. M., Law M. L., O'Brien S. J., Wong A. J., Vogelstein B. (1987) Identification of an amplified, highly expressed gene in a human glioma. Science 236, 70–73 [DOI] [PubMed] [Google Scholar]
- 4. Bermudez O., Hennen E., Koch I., Lindner M., Eickelberg O. (2013) Gli1 mediates lung cancer cell proliferation and sonic hedgehog-dependent mesenchymal cell activation. PLoS One 8, e63226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen M., Carkner R., Buttyan R. (2011) The hedgehog/Gli signaling paradigm in prostate cancer. Expert Rev. Endocrinol. Metab. 6, 453–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ciucci A., De Stefano I., Vellone V. G., Lisi L., Bottoni C., Scambia G., Zannoni G. F., Gallo D. (2013) Expression of the glioma-associated oncogene homolog 1 (gli1) in advanced serous ovarian cancer is associated with unfavorable overall survival. PLoS One 8, e60145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savani M., Guo Y., Carbone D. P., Csiki I. (2012) Sonic hedgehog pathway expression in non-small cell lung cancer. Ther. Adv. Med. Oncol. 4, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saze Z., Terashima M., Kogure M., Ohsuka F., Suzuki H., Gotoh M. (2012) Activation of the sonic hedgehog pathway and its prognostic impact in patients with gastric cancer. Dig. Surg. 29, 115–123 [DOI] [PubMed] [Google Scholar]
- 9. Fiaschi M., Kolterud A., Nilsson M., Toftgård R., Rozell B. (2011) Targeted expression of GLI1 in the salivary glands results in an altered differentiation program and hyperplasia. Am. J. Pathol. 179, 2569–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fiaschi M., Rozell B., Bergström A., Toftgård R. (2009) Development of mammary tumors by conditional expression of GLI1. Cancer Res. 69, 4810–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eberl M., Klingler S., Mangelberger D., Loipetzberger A., Damhofer H., Zoidl K., Schnidar H., Hache H., Bauer H. C., Solca F., Hauser-Kronberger C., Ermilov A. N., Verhaegen M. E., Bichakjian C. K., Dlugosz A. A., Nietfeld W., Sibilia M., Lehrach H., Wierling C., Aberger F. (2012) Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 4, 218–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goel H. L., Pursell B., Chang C., Shaw L. M., Mao J., Simin K., Kumar P., Vander Kooi C. W., Shultz L. D., Greiner D. L., Norum J. H., Toftgard R., Kuperwasser C., Mercurio A. M. (2013) GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 5, 488–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Javelaud D., Alexaki V. I., Dennler S., Mohammad K. S., Guise T. A., Mauviel A. (2011) TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 71, 5606–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Javelaud D., Pierrat M. J., Mauviel A. (2012) Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 586, 2016–2025 [DOI] [PubMed] [Google Scholar]
- 15. Keysar S. B., Le P. N., Anderson R. T., Morton J. J., Bowles D. W., Paylor J. J., Vogler B. W., Thorburn J., Fernandez P., Glogowska M. J., Takimoto S. M., Sehrt D. B., Gan G. N., Eagles-Soukup J., Serracino H., Hirsch F. R., Lucia M. S., Thorburn A., Song J. I., Wang X. J., Jimeno A. (2013) Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 73, 3381–3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lauth M., Bergström A., Shimokawa T., Tostar U., Jin Q., Fendrich V., Guerra C., Barbacid M., Toftgård R. (2010) DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 17, 718–725 [DOI] [PubMed] [Google Scholar]
- 17. Mills L. D., Zhang Y., Marler R. J., Herreros-Villanueva M., Zhang L., Almada L. L., Couch F., Wetmore C., Pasca di Magliano M., Fernandez-Zapico M. E. (2013) Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating K-Ras oncogene-induced transformation. J. Biol. Chem. 288, 11786–11794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nolan-Stevaux O., Lau J., Truitt M. L., Chu G. C., Hebrok M., Fernández-Zapico M. E., Hanahan D. (2009) GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasca di Magliano M., Hebrok M. (2003) Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 3, 903–911 [DOI] [PubMed] [Google Scholar]
- 20. Rajurkar M., De Jesus-Monge W. E., Driscoll D. R., Appleman V. A., Huang H., Cotton J. L., Klimstra D. S., Zhu L. J., Simin K., Xu L., McMahon A. P., Lewis B. C., Mao J. (2012) The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, E1038–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ji Z., Mei F. C., Xie J., Cheng X. (2007) Oncogenic Kras activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 282, 14048–14055 [DOI] [PubMed] [Google Scholar]
- 22. Elsawa S. F., Almada L. L., Ziesmer S. C., Novak A. J., Witzig T. E., Ansell S. M., Fernandez-Zapico M. E. (2011) GLI2 transcription factor mediates cytokine cross-talk in the tumor microenvironment. J. Biol. Chem. 286, 21524–21534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warmflash A., Zhang Q., Sorre B., Vonica A., Siggia E. D., Brivanlou A. H. (2012) Dynamics of TGF-β signaling reveal adaptive and pulsatile behaviors reflected in the nuclear localization of transcription factor Smad4. Proc. Natl. Acad. Sci. U.S.A. 109, E1947–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duan H., Heckman C. A., Boxer L. M. (2007) The immunoglobulin heavy-chain gene 3′ enhancers deregulate bcl-2 promoter usage in t(14;18) lymphoma cells. Oncogene 26, 2635–2641 [DOI] [PubMed] [Google Scholar]
- 25. Sasaki H., Hui C., Nakafuku M., Kondoh H. (1997) A binding site for Gli proteins is essential for HNF-3β floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 26. Lo Re A. E., Fernandez-Barrena M. G., Almada L. L., Mills L., Elsawa S. F., Lund G., Ropolo A., Molejon M. I., Vaccaro M. I., Fernandez-Zapico M. E. (2012) A novel AKT1-GLI3-VMP1 pathway mediates Kras-induced autophagy in cancer cells. J. Biol. Chem. 287, 25325–25334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koopman G., Reutelingsperger C. P., Kuijten G. A., Keehnen R. M., Pals S. T., van Oers M. H. (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415–1420 [PubMed] [Google Scholar]
- 28. Martin S. J., Reutelingsperger C. P., McGahon A. J., Rader J. A., van Schie R. C., LaFace D. M., Green D. R. (1995) Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dennler S., André J., Alexaki I., Li A., Magnaldo T., ten Dijke P., Wang X. J., Verrecchia F., Mauviel A. (2007) Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 67, 6981–6986 [DOI] [PubMed] [Google Scholar]
- 30. Bigelow R. L., Chari N. S., Unden A. B., Spurgers K. B., Lee S., Roop D. R., Toftgard R., McDonnell T. J. (2004) Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through Gli-1. J. Biol. Chem. 279, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 31. Wu J. Y., Xu X. F., Xu L., Niu P. Q., Wang F., Hu G. Y., Wang X. P., Guo C. Y. (2011) Cyclopamine blocked the growth of colorectal cancer SW116 cells by modulating some target genes of Gli1 in vitro. Hepatogastroenterology 58, 1511–1518 [DOI] [PubMed] [Google Scholar]
- 32. Guo J., Gao J., Li Z., Gong Y., Man X., Jin J., Wu H. (2013) Adenovirus vector-mediated Gli1 siRNA induces growth inhibition and apoptosis in human pancreatic cancer with Smo-dependent or Smo-independent Hh pathway activation in vitro and in vivo. Cancer Lett. 339, 185–194 [DOI] [PubMed] [Google Scholar]
- 33. Thomas Z. I., Gibson W., Sexton J. Z., Aird K. M., Ingram S. M., Aldrich A., Lyerly H. K., Devi G. R., Williams K. P. (2011) Targeting GLI1 expression in human inflammatory breast cancer cells enhances apoptosis and attenuates migration. Br. J. Cancer 104, 1575–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng X., Vittar N. B., Gai X., Fernandez-Barrena M. G., Moser C. D., Hu C., Almada L. L., McCleary-Wheeler A. L., Elsawa S. F., Vrabel A. M., Shire A. M., Comba A., Thorgeirsson S. S., Kim Y., Liu Q., Fernandez-Zapico M. E., Roberts L. R. (2012) The transcription factor GLI1 mediates TGFβ1-driven EMT in hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One 7, e49581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morikawa M., Koinuma D., Miyazono K., Heldin C. H. (2013) Genome-wide mechanisms of Smad binding. Oncogene 32, 1609–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roeder R. G. (2005) Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 579, 909–915 [DOI] [PubMed] [Google Scholar]
- 37. Nagy Z., Tora L. (2007) Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26, 5341–5357 [DOI] [PubMed] [Google Scholar]
- 38. Colvin Wanshura L. E., Galvin K. E., Ye H., Fernandez-Zapico M. E., Wetmore C. (2011) Sequential activation of Snail1 and N-Myc modulates sonic hedgehog-induced transformation of neural cells. Cancer Res. 71, 5336–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura I., Fernandez-Barrena M. G., Ortiz-Ruiz M. C., Almada L. L., Hu C., Elsawa S. F., Mills L. D., Romecin P. A., Gulaid K. H., Moser C. D., Han J. J., Vrabel A., Hanse E. A., Akogyeram N. A., Albrecht J. H., Monga S. P., Sanderson S. O., Prieto J., Roberts L. R., Fernandez-Zapico M. E. (2013) Activation of the transcription factor GLI1 by WNT signaling underlies the role of SULFATASE 2 as a regulator of tissue regeneration. J. Biol. Chem. 288, 21389–21398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katoh Y., Katoh M. (2009) Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 9, 873–886 [DOI] [PubMed] [Google Scholar]
- 41. Stecca B., Ruiz I Altaba A. (2010) Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell. Biol. 2, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heldin C. H., Moustakas A. (2012) Role of Smads in TGFβ signaling. Cell Tissue Res. 347, 21–36 [DOI] [PubMed] [Google Scholar]
- 43. Xu P., Liu J., Derynck R. (2012) Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 586, 1871–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joost S., Almada L. L., Rohnalter V., Holz P. S., Vrabel A. M., Fernandez-Barrena M. G., McWilliams R. R., Krause M., Fernandez-Zapico M. E., Lauth M. (2012) GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 72, 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosenfeld M. G., Lunyak V. V., Glass C. K. (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428 [DOI] [PubMed] [Google Scholar]
- 46. Love I. M., Sekaric P., Shi D., Grossman S. R., Androphy E. J. (2012) The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 11, 2458–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel J. H., Du Y., Ard P. G., Phillips C., Carella B., Chen C. J., Rakowski C., Chatterjee C., Lieberman P. M., Lane W. S., Blobel G. A., McMahon S. B. (2004) The c-Myc oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24, 10826–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xenaki G., Ontikatze T., Rajendran R., Stratford I. J., Dive C., Krstic-Demonacos M., Demonacos C. (2008) PCAF is an HIF-1α cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 27, 5785–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim J. H., West K. L., Rubinstein Y., Bergel M., Postnikov Y. V., Bustin M. (2005) Chromosomal protein HMGN1 enhances the acetylation of lysine 14 in histone H3. EMBO J. 24, 3038–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poux A. N., Marmorstein R. (2003) Molecular basis for Gcn5/PCAF histone acetyltransferase selectivity for histone and nonhistone substrates. Biochemistry 42, 14366–14374 [DOI] [PubMed] [Google Scholar]
- 51. Malatesta M., Steinhauer C., Mohammad F., Pandey D. P., Squatrito M., Helin K. (2013) Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 73, 6323–6333 [DOI] [PubMed] [Google Scholar]
- 52. Mazzà D., Infante P., Colicchia V., Greco A., Alfonsi R., Siler M., Antonucci L., Po A., De Smaele E., Ferretti E., Capalbo C., Bellavia D., Canettieri G., Giannini G., Screpanti I., Gulino A., Di Marcotullio L. (2013) PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 20, 1688–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]