

Background: Activation of SplB protease requires precise N-terminal processing, but the molecular mechanism remains unknown.
Results: The new N-terminal Glu-1 forms a distinctive H-bond network essential for full catalytic activity.
Conclusion: Changes in protein dynamics rather than a direct effect on the active site are of crucial importance in SplB activation.
Significance: A novel serine protease activation mechanism was uncovered.
Keywords: Crystal Structure, Enzyme Inactivation, Protease, Serine Protease, Staphylococcus aureus, Signal Peptide, Zymogen Activation
Abstract
Staphylococcal SplB protease belongs to the chymotrypsin family. Chymotrypsin zymogen is activated by proteolytic processing at the N terminus, resulting in significant structural rearrangement at the active site. Here, we demonstrate that the molecular mechanism of SplB protease activation differs significantly and we characterize the novel mechanism in detail. Using peptide and protein substrates we show that the native signal peptide, or any N-terminal extension, has an inhibitory effect on SplB. Only precise N-terminal processing releases the full proteolytic activity of the wild type analogously to chymotrypsin. However, comparison of the crystal structures of mature SplB and a zymogen mimic show no rearrangement at the active site whatsoever. Instead, only the formation of a unique hydrogen bond network, distant form the active site, by the new N-terminal glutamic acid of mature SplB is observed. The importance of this network and influence of particular hydrogen bond interactions at the N terminus on the catalytic process is demonstrated by evaluating the kinetics of a series of mutants. The results allow us to propose a consistent model where changes in the overall protein dynamics rather than structural rearrangement of the active site are involved in the activation process.
Introduction
Proteolytic enzymes fulfill a multitude of crucial physiological functions ranging from unspecific catabolism to precisely targeted signaling events. At the same time, the production of proteases by living cells is associated with a serious risk of unwanted degradation of cellular components. Therefore, the expression of proteases is meticulously controlled at multiple levels. The majority of proteases are synthesized as inactive precursors (zymogens) activated only at the desired site of action. Tightly controlled compartmentalization targets the proteases at desired substrates. Ubiquitous proteinaceous protease inhibitors further modulate proteolysis. Moreover, protease specific regulatory mechanisms have been described including glycosylation, cofactor binding, formation of disulphides or pH-dependent activation among others (1, 2).
Protease zymogens are often composed of a profragment, the mature protease, and frequently contain additional targeting signals. The profragment precludes proteolytic activity by distorting the critical elements of catalytic machinery, physically blocking substrate access, or both. In addition to the primary inhibitory role, the profragments are often involved in folding (3, 4) or stability (5). In most cases, the profragment is located at the N-terminal part of the protein, less frequently is an insertion within the protease domain (6), but no examples of C-terminal profragments are available to our knowledge. This is most likely associated with the direction of protein synthesis; it is crucial that the inhibitory part is formed before the protease domain to preclude unwanted proteolysis. Profragments differ in size ranging from several amino acids (e.g. chymotrypsinogen) to independently folding domains, sometimes larger than the protease domain itself (e.g. prothrombin). Correspondingly, the mechanistic details of inhibition differ significantly between the examples. Despite those differences, the conversion of inactive zymogen into the active enzyme is most often associated with proteolytic removal of the profragment (7).
SplB belongs to the chymotrypsin family of serine proteases (family S1) (8). The activation of the chymotrypsin protease zymogen (chymotrypsinogen) has been described in great detail. Although chymotrypsinogen differs from α-chymotrypsin (a final product of trypsin catalyzed chymotrypsinogen activation) by as little as four amino acids, it is almost devoid of proteolytic activity. Analysis of crystal structures of the precursor and the mature form demonstrate that the substrate binding cavity is not fully formed, and the oxyanion hole is in non-productive conformation in chymotrypsinogen compared with chymotrypsin (9, 10). Proteolytic processing of the zymogen releases a new N terminus, which turns inward and forms a buried salt bridge with Asp-194. This results in a structural rearrangement at the active site and formation of a functional S1 pocket and an oxyanion hole, releasing proteolytic activity.
Within the S1 family of serine proteases, SplB belongs to a small subfamily S1B (encompassing staphylococcal V8 protease, epidermolytic toxins and Spl proteases, and several additional enzymes from other species). The activating role of N terminus processing and the activation mechanism differ significantly between subfamily members and from that described for chymotrypsin (S1A subfamily). V8 protease, the type protease of subfamily S1B, is synthesized as a zymogen and activated by proteolytic processing; however, the molecular interactions at the liberated N terminus differ from those observed for chymotrypsin. Epidermolytic toxins are synthesized with an N-terminal extension as compared with mature chymotrypsin; however, they are active as such, without proteolytic processing. Similar to the above-mentioned S1B subfamily representatives, Spl proteases are synthesized at the ribosome containing an N-terminal secretion signal. The secretion signal is removed by a signal peptidase, but in the case of all Spl proteases, a mature enzyme is liberated rather than a zymogen of any kind. In this study, we argue that the signal peptide of SplB fulfills a dual role: (i) targeting the protease into extracellular compartment and (ii) inhibiting protease activity in the case the enzyme is misdirected into the intracellular compartment. To determine the inhibitory role of SplB protease signal peptide and to explain the activation process mechanistically, we use a combination of enzymology, crystallography, and targeted mutagenesis.
EXPERIMENTAL PROCEDURES
Construction of Expression Vectors
A gene encoding signal peptide containing SplB (sp-SplB) and its fragment encoding mature SplB protease were PCR-amplified from Staphylococcus aureus 8325-4 genomic DNA and cloned into pGEX-5T (11) using XhoI and BamHI restriction sites, resulting in pSplB(T) and p(sp-SplB(T)) partially as described previously (12). The thrombin site was exchanged in both plasmids into that recognized by factor Xa by site-directed mutagenesis, resulting in pSplB(X) and p(sp-SplB(X)). Catalytic triad serine mutants (S157A) were obtained by site-directed mutagenesis of pSplB(X), pSplB(T), and p(sp-SplB(X)). Expression constructs encoding other mutants utilized in this study (Table 1) were obtained by site-directed mutagenesis of pSplB(X).
TABLE 1.
Proteolytic activity of SplB protease and its variants against synthetic peptide and protein substrates
Amino acids are abbreviated in a single letter code. Ac, acetyl group; AMC, 7-amino-4-methylcoumarin; ACC, 7-amino-4-carbamoylmethylcoumarin.
For construction of expression vector encoding fluorescence quenched protein substrate of SplB, the genes encoding enhanced cyan and enhanced yellow fluorescent proteins (CFP3 and YFP, respectively) were amplified from pECFP-N1 and pEYFP-N1 (Clontech) and cloned into pET28a (Novagen) using NcoI and BamHI sites resulting in pET28a_cfp and pET28a_yfp, respectively. The reverse primer used for cloning cfp contained additional sequence coding for a polypeptide linker (GSWELQGS; consensus SplB cleavage site underlined). yfp was PCR-amplified from pET28a_yfp and cloned at XhoI and BamHI restriction sites into pET28a_cfp, resulting in pET28a_cfp_yfp encoding CFP-GSWELQGS-YFP substrate. All plasmids used in this study were sequenced to confirm the presence of desired inserts and mutations and to ensure that no random nucleotide changes were introduced during manipulations.
Protein Expression and Purification
SplB and its variants were produced in Escherichia coli BL21(DE3) as GST fusion proteins. Bacteria carrying appropriate plasmid were cultured at 37 °C until A600 reached 0.8 when the expression of recombinant protein was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and temperature was decreased to 22 °C. Cells were collected 3 h after induction and lysed by sonication in PBS. Recombinant GST fusion protein was recovered on glutathione-Sepharose (GE Healthcare). After elution, GST tag was separated either with thrombin (Sigma-Aldrich) or factor Xa (Merck) as appropriate for particular constructs. GST was separated from the protease of interest on SourceS (GE Healthcare) in 50 mm sodium acetate, pH 5.0. Gel filtration on Superdex s75 (GE Healthcare) in 50 mm Tris-HCl, pH 8.0, was used as a final polishing step. The quality of obtained preparations was verified using SDS-PAGE. Only preparations of >95% purity were used for further experiments.
Fluorescence-quenched protein substrate was produced as His6-tagged protein in E. coli BL21(DE3). The bacteria were cultured at 37 °C until A600 reached 0.6 when the expression of recombinant protein was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and temperature was decreased to 22 °C. The culture was continued overnight. Cells were harvested and lysed by sonication in 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 10 mm imidazole. Recombinant protein was recovered on chelating Sepharose (GE Healthcare). The final preparations were prepared in 50 mm Tris-HCl, pH 8.0, by overnight dialysis. On average, 20 mg of purified protein was obtained from 1 liter of starting culture.
GS-SplB and L-SplB Processing by Aminopeptidase
GS-SplB and L-SplB were incubated with aminopeptidase from Aeromonas proteolytica (Sigma; 1 unit per 5 μg of GS-SplB and 1 unit per 200 μg of L-SplB) at 37 °C in 50 mm Tris-HCl, pH 8.0. Samples were removed from the reaction mixture at various time points, and the reaction was terminated with bestatin (Sigma). The activity against Ac-WELQ-ACC was assessed. Protein integrity was evaluated by SDS-PAGE. Aminopeptidase stability during the reaction was evaluated using Leu-pNa (Sigma). Selected samples were analyzed by N-terminal Edman degradation sequencing (BioCentrum).
Activity Assays and Determination of Enzyme Kinetics
Commercially available Ac-VEID-AMC (Merck) was initially used to assess the proteolytic activity of SplB. More efficient substrates (Ac-WELD-ACC and Ac-WELQ-ACC) were later synthesized based on the consensus sequence recognized and cleaved by SplB as determined previously (13). Moreover, FRET protein substrates based on the SplB consensus sequence were used for assessing the kinetics of mutants. All reactions were performed at 37 °C in 50 mm Tris-HCl, pH 8.0. Enzymatic activity was monitored as an increase in fluorescence at λex = 350 nm and λem = 460 nm for ACC and AMC substrates and λex = 440 nm, λem = 485 nm, and 528 nm for FRET protein substrates. For synthetic substrates, the final concentration of dimethyl sulfoxide in the reaction mixture did not exceed 5%. All measurements were performed at least in triplicate.
Km and kcat values were determined only for Ac-VEID-AMC, due to a limited amount of available substrates (Ac-WELD-ACC, Ac-WELQ-ACC) or problems in solubility (FRET protein substrate, Km exceeding 100 μm). For the determination of Km and kcat values, Ac-VEID-AMC (concentration range of 45 to 800 μm) was incubated with 3 μm enzyme, and fluorescence was monitored. Quantitative kinetic data were derived by hyperbolic regression analysis using program Hyper. For other substrates, the kcat/Km values were determined under pseudo-first-order conditions in which the substrate concentration is far below the estimated Km (14). Fluorescent protein substrates (1 μm) or synthetic substrates (25 μm) were incubated in the presence of increasing enzyme concentration (25, 50, 100, 200, and 500 nm) and changes in fluorescence intensity (Int) were recorded for 60 min. Using OriginPro software, progress curves were fitted to the equation Int = I0 + Imax(1 − exp(−[E]kcat/Kmt)), where I0 is the initial fluorescence of the substrate (uncleaved substrate), Imax is the maximum fluorescence intensity (fully hydrolyzed substrate), and [E] is the total enzyme concentration.
Protein Crystallization and X-ray Diffraction Data Collection
Purified GS-SplB was concentrated to ∼35 mg/ml in the crystallization buffer (5 mm Tris-HCl, pH 8.0, containing 50 mm NaCl) and used for screening at room temperature. Crystals appeared after a week in Index solution 45 (Hampton Research; 0.1 m Tris-HCl, pH 8.5, 25% (w/v) PEG 3350) and Crystal Screen II solution 27 (Hampton Research; 0.1 m MES-NaOH, pH 6.5, 0.01 m zinc sulfate, 25% (v/v) PEG monomethyl ether 550). Single monocrystals were used for measurements without further optimization. Crystals were cryo-cooled in liquid nitrogen. The diffraction data were collected at 100 K on MPG/GBF beamline BW6 at DESY (Hamburg, Germany).
Structure Determination
Data were indexed and integrated with Mosflm (15, 16). Crystals obtained in Index solution 45 belonged to the P41212 space group, whereas those obtained in Crystal Screen II solution 27 belonged to the C2 space group. The following steps were performed using programs contained in the CCP4 software package. Data were scaled with Scala (17, 18). Molecular replacement was performed using Phaser (19) and an alanine search model based on the structure of SplA (PDB code 2W7S). The structures were refined in multiple rounds of manual model building, and restrained refinement was performed, respectively, with COOT (20) and Refmac (version 5.0) (21). Throughout the refinement, 5% of the reflections were used for cross-validation analysis (22), and the behavior of Rfree was employed to monitor the refinement strategy. At the last steps of refinement, water molecules were added using Arp/Warp (23) and manually inspected. The final models were deposited in the PDB, codes 4K1S and 4K1T. Data collection and refinement statistics are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics
Statistics for the highest shell are listed in parentheses.
| Data collection | ||
| PDB code | 4K1T1 | 4K1S |
| Space group | C2 | P41212 |
| Cell constants | a = 134.74, b = 77.76, and c = 95.79 Å; α = γ = 90.00°, β = 131.81° | a = 117.90, b = 117.90, and c = 73.33 Å; α = β = γ = 90.00° |
| Wavelength (Å) | 1.050 | 1.050 |
| B factor (Wilson) (Å2) | 13.80 | 19.8 |
| Resolution range (Å) | 18.91–1.60 | 22.97–1.96 |
| Completeness (%) | 83.6 (90.9) | 86.1 (90.3) |
| Rmerge (%) | 0.060 (0.128) | 0.049 (0.217) |
| Rmeas (%) | 0.075 (0.165) | 0.051 (0.229) |
| Observed reflections | 192,823 (28,372) | 391,791 (37,487) |
| Unique reflections | 81,007 (12,854) | 32,322 (3969) |
| I/σ(I) | 9.0 (3.9) | 32.1 (9.6) |
| Average multiplicity | 2.4 (2.2) | 12.1 (9.4) |
| Refinement | ||
| Resolution (Å) | 18.91–1.60 | 22.97–1.96 |
| No. of reflections used | 76,698 | 30,556 |
| R factor (%) | 18.4 | 19.2 |
| Rfree (%) | 22.4 | 24.7 |
| Average B (Å2) | ||
| Protein | 11.62 | 21.6 |
| SO42− | 23.38 | |
| Zn2+ | 9.49 | |
| Cl− | 31.93 | |
| r.m.s.d. from ideal values | ||
| Bond length (Å) | 0.023 | 0.020 |
| Bond angles | 2.312° | 2.042° |
| Ramachandran statistics (%) | ||
| Most favored regions | 96.9 | 96.0 |
| Additionally allowed regions | 3.1 | 3.5 |
| Generously allowed regions | 0.0 | 0.5 |
| Content of asymmetric unit | ||
| No. of protein molecules/residues/atoms | 3/607/4669 | 2/395/3021 |
| No. of SO42− molecules/atoms | 8/40 | |
| No. of Zn2+ atoms | 10 | |
| No. of Cl− atoms | 3 | |
| No. of solvent molecules | 837 | 390 |
RESULTS
Precise Processing at the N terminus Regulates the Activity of SplB Protease
Popowicz and co-workers (12) have previously reported that precise N-terminal trimming is fundamental for enzymatic activity of SplB protease. Of the recombinant protease variants tested, only those with the N terminus corresponding to the natural protein but not those containing amino acids artificially added in cloning, demonstrated activity in a zymography assay (12). Using a more sensitive and quantitative assay, we show here that the activity of a full-length, 36-amino acid signal peptide containing SplB protease variant (sp-SplB) is not fully abolished, but decreased by more than an order of magnitude compared with that of the mature protease (SplB). Moreover, the inhibitory effect of the signal peptide is closely mimicked by a short N-terminal extension of two amino acids (GS-SplB). For both modifications of the N terminus, the rate of substrate hydrolysis (kcat) decreases by an order of magnitude. At the same time, an increase in Km is observed (Table 1).
The catalytic triad serine mutant (S157A) of sp-SplB protease was inactive, demonstrating that the observed residual activity of sp-SplB was specific to this variant of the protease and not a result of possible sample contamination with other proteases. Similar results were obtained for a GS-SplB S157A mutant. As expected, SplB S157A mutant was also inactive.
Removal of the N-terminal Extension Activates GS-SplB and L-SplB Protease Variants
To exclude the possibility that the N-terminal extension affects the overall protein folding rather than the protease activity itself, we analyzed the influence of in situ removal of Gly-(−2)-Ser-(−1) residues from the GS-SplB protein on proteolytic activity. To this end, GS-SplB was incubated with a roughly equimolar amount of aminopeptidase from Aeromonas proteolytica (AAP) (24), and the proteolytic activity characteristic of SplB (but not AAP) was monitored. AAP was selected for this experiment because it was reported to remove most amino-terminal residues except glutamic acid, the exact residue found at the N terminus of native, mature SplB. The proteolytic activity characteristic for SplB gradually increased during the experiment (Fig. 1A). This correlated with gradual conversion of GS-SplB through S-SplB into SplB as determined by Edman degradation sequencing of the reaction products. Importantly, the fraction of recovered activity roughly corresponded to the extent of conversion of GS-SplB into S-SplB and SplB. Although these results strongly support the notion that removal of N-terminal extension releases proteolytic activity of SplB protease, their quantitative interpretation is complicated by incomplete conversion and already increased activity of S-SplB (Table 1). To circumvent limited activity of AAP toward substrates containing glycine and serine (24), we constructed SplB variant having additional leucine at the N terminus (L-SplB), a residue most preferred by AAP. L-SplB exhibited proteolytic activity comparable with sp-SplB and GS-SplB (Table 1). Incubation with catalytic amount of AAP resulted in efficient recovery of proteolytic activity to the level of equal amount of mature SplB. The observed increase in activity was associated with removal of the N-terminal leucine as evidenced by Edman degradation sequencing (Fig. 1B). These results demonstrate that decreased activity of GS-SplB and L-SplB are directly associated with N-terminal extension rather than improper folding of extended variants and further suggest that exact shaping of the N terminus is necessary for full proteolytic activity.
FIGURE 1.

Removal of artificial N-terminal extension releases the proteolytic activity of GS-SplB and l-SplB. A, GS-SplB was incubated with APP, and the proteolytic activity against Ac-WELQ-ACC (specific for SplB) was monitored. The activity is represented as a percentage of the maximum activity observed for the same concentration of SplB protease as the initial concentration of GS-SplB. Bestatin inhibits AAP activity. Samples were collected at indicated time points and subjected to N-terminal Edman degradation sequencing (results shown at the bottom of the panel). Activity of AAP remained unchanged during the experiment as monitored using Leu-pNa (not shown). B, same as in A but for L-SplB.
Activity of SplB Protease Variants toward Consensus Sequence Substrates
The results described above were obtained using a peptide substrate (Ac-WELQ-ACC) based on the consensus sequence recognized and cleaved by SplB protease (WELQ↓; ↓ indicates the cleavage site) as established previously (13). To determine whether the differences in the activity of SplB protease variants constitute a general property of the enzyme, rather than being substrate-specific, we tested further two fluorescent peptide substrates and developed a protein-based FRET substrate. We determined the kinetic parameters of their hydrolysis by different variants of SplB protease. Similarly to Ac-WELQ-ACC, both in the case of Ac-WELD-ACC and Ac-VEID-AMC and in the case of CFP-GSWELQGS-YFP, the substrate hydrolysis rate by N-terminally extended variants of SplB was lower than that observed for the mature form of SplB. For all tested substrates, the kcat/Km values for sp-SplB, GS-SplB, and L-SplB were comparable for a particular substrate, whereas the ratio of kcat/Km for SplB and N-terminally extended variants were ∼20, whereas for Ac-WELQ-ACC, the ratio was ∼50. These results confirm that N-terminal extensions decrease the proteolytic activity of SplB protease regardless the sequence of the substrate or its chemical identity (peptide versus protein).
Crystal Structure Provides Insights into the Molecular Mechanism of Protease Activation
To reveal the molecular basis of the activating effect of N-terminal processing in GS-SplB, we determined the x-ray structure of the protein and compared it with a previously determined structure of SplB (PDB code 2VID) (13). Crystals of GS-SplB were obtained from two different conditions and belonged to different space groups. One of the conditions and the arrangement of molecules in the crystal are identical to those at which SplB was previously crystallized (P41212 space group), and this structure is discussed further below. The second structure (C2 space group) is discussed only as a reference because, in the region of interest, it exhibits features that are induced by crystal specific interactions and unlikely to be preserved in solution (see below). The data collection and refinement statistic are summarized in Table 2. The P41212 structure of GS-SplB comprises of two molecules in the asymmetric unit, which are essentially identical to each other (r.m.s.d. of 0.53 Å). GS-SplB adopts a characteristic chymotrypsin-like fold. The catalytic triad residues His-39, Asp-77, and Ser-157 adopt a canonical conformation and overlay with corresponding residues of SplB with r.m.s.d. of 0.06 Å. Identical to what was previously observed in the structure of SplB, the essential component of the catalytic machinery, the oxyanion hole, is found in a non-productive, closed conformation. The Ser-154–Gly-155 peptide bond is flipped ∼180° compared with the productive conformation. As a result, the carbonyl oxygen of Ser-154 occupies the position normally reserved for the oxyanion. This conformation is stabilized via hydrogen bonds between carbonyl oxygen of Ser-154 and both amide and side chain hydroxyl of Ser-157, exactly as that found in SplB protease. Overall, the structures of GS-SplB and SplB are almost identical with r.m.s.d. of 0.3 Å for all atoms. The canonical disposition of catalytic triad residues and the closed conformation of the oxyanion hole are repeated exactly in all three molecules contained in the asymmetric unit of the C2 structure.
The only notable structural differences between the P41212 structures of GS-SplB and SplB are found in the direct vicinity of the N terminus (Fig. 2). In the structure of SplB, the N-terminal glutamic acid is firmly positioned on the surface of the molecule through a network of hydrogen bonds connecting the side chain of Glu-1 with the side chain of Arg-115 and the N-terminal amine group of Glu-1 and the side chain hydroxyl and backbone carbonyl of Ser-149. Moreover, it is possible that the N-terminal amine interacts transiently with loop 2, but the loop is poorly defined by electron density, which makes this conclusion ambiguous. Furthermore, the carbonyl oxygen of Glu-1 is hydrogen bonded with the backbone amide of His-151 and the side chain of Asn-3 forms hydrogen bonds with Thr-135. Adjacent water molecules further contribute to the hydrogen bond network at the N terminus. The interactions described for Glu-1 and Asn-3 define the disposition of Asn-2, which, by itself, is not involved in hydrogen bond contacts. In the GS-SplB P41212 structure, residues from Gly-(−2) to Asn-2 are not defined by electron density suggesting flexibility of this region. This implies that the artificially elongated N terminus does not allow for anchoring of Glu1 in a manner similar to that found in SplB. Asn-3 is still poorly defined, but in one of the two molecules in the asymmetric unit, it already adopts a conformation found in SplB. In the C2 structure of GS-SplB, the N-terminal Gly-(−2) coordinates a zinc ion together with residues belonging to adjacent molecules. This stabilizes the disposition of the entire N terminus, which is thus well defined by electron density. Although fixed, the disposition of Glu-1 and Asn-2 are different than that found in the structure of SplB. We believe that the disposition of the N terminus in C2 structure is induced by crystal packing (zinc ion coordination) and is not retained in solution where the Gly-(−2)–Asn-2 fragment most probably remains flexible as found in the P41212 structure. Apart from the above-described differences, starting from Val-4, the P41212 and C2 GS-SplB structures are virtually identical with each other and with the structure of SplB. Thus, the significant structural rearrangement at the active site observed upon activation of chymotrypsinogen to chymotrypsin is not repeated during activation of SplB protease precursor (Fig. 3). In the latter case, the structural differences between the precursor and the fully active protease are limited only to the direct vicinity of the N terminus and do not reach anywhere near the active site of the protease. The only structural difference located close to the active site observed between the structure of mature protease and the precursor mimic is the interaction of the N-terminal amine with loop 2 (Gly-170–Tyr-189). In the structure of SplB, the interaction of Glu-1 with Ser-149 positions the latter residue such that it forms hydrogen bonds with loop 2 (backbone oxygen of Arg-183), whereas such an interaction is missing in GS-SplB. Moreover, the N-terminal amine of Glu-1 seems to transiently interact directly with loop 2 in SplB but not GS-SplB. Because loop 2 forms a part of the SplB protease active site being involved in substrate recognition (13), one may expect some influence on the process. However, because a large part of loop 2 is poorly defined by electron density and the entire region is rather flexible, this conclusion is highly ambiguous.
FIGURE 2.
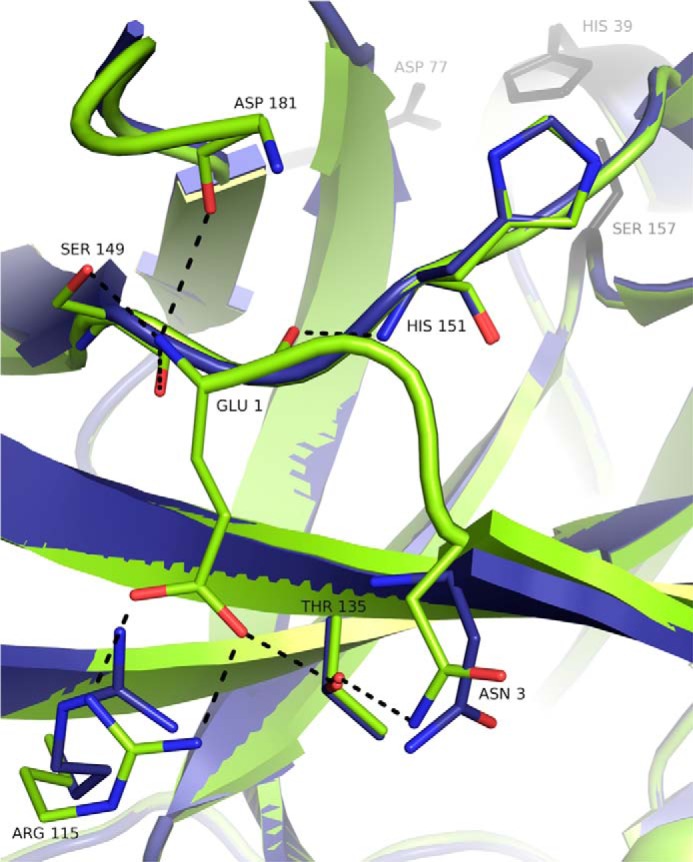
Hydrogen bond network involving N-terminal residues of mature SplB protease is absent in GS-SplB precursor mimic. Activation of SplB protease precursor mimic (blue, GS-SplB; PDB code 4K1S) by proteolytic processing is associated with formation of an extended hydrogen bond network at the newly formed N terminus of mature SplB protease (green; PDB code 2VID). Residues Gly-(−2) through Asn-2 of the precursor mimic are not defined by electron density and therefore not present in the model. Hydrogen bonds in SplB are indicated as black dotted lines. Water molecules are not shown for clarity.
FIGURE 3.

Activation of SplB protease takes place without broad structural rearrangement in the vicinity of the active site contrary to the activation of chymotrypsinogen. Overlays of active and zymogen forms of serine proteases are shown. Regions of pronounced structural rearrangements are highlighted in color. Catalytic triad residues are highlighted gray. A, overlay of GS-SplB (PDB code 4K1S) and SplB (PDB code 2VID) crystal structures. N-terminal residues are depicted in red and yellow, respectively. Gly-(−1)–Asn-2 are undefined by electron density in the structure of GS-SplB. B, overlay of chymotrypsinogen (PDB code 2CGA) and chymotrypsin (PDB code 1YPH) crystal structures. Regions removed during proteolytic activation are highlighted black. N-terminal peptide of the precursor (red) is processed by trypsin to release a new N terminus (green). Regions of the “activation domain” rearranged upon precursor activation are highlighted yellow and blue.
Mutational Analysis of N-terminal Hydrogen Bond Network
Comparison of the crystal structures of GS-SplB and SplB in the light of their proteolytic activity highlighted the importance of a hydrogen bond network involving the N-terminal glutamic acid residue in the expression of full proteolytic activity. However, this observation did not allow an unambiguous mechanistic explanation because the observed differences are distant from the active site. To determine the influence of particular hydrogen bonds formed by Glu-1 on the activity of SplB protease, a series of mutants was generated, and their proteolytic activity was determined against a panel of substrates. The mutants are divided into three groups: (i) those designed to evaluate the interaction of the side chain carboxyl of Glu-1 with the side chain guanidinium of Arg-115 (E1A; E1D; E1Q; R115A; E1A, R115A), (ii) those designed to evaluate the interaction of N-terminal amine with the side chain of Ser-149 (S149A), and (iii) those designed to evaluate the combined contributions of above interactions (E1A, S149A; R115A, S149A; E1A, R115A, S149A). All mutations negatively affected the catalytic efficiency (kcat/Km) of SplB protease, although each affected it to a different extent. The E1A mutant, which presumably retains interactions of the N-terminal amine group with Ser-149 but is unable to form a salt bridge with Arg-115, exhibits roughly 2-fold decrease in activity (kcat). The effect of the E1D mutation is comparable with that of E1A presumably because the side chain of aspartic acid is too short to interact with Arg-115 in a manner similar to Glu-1, but the N-terminal amine group interactions are likely unaffected. The activities of the R115A mutant and E1A,R115A double mutant are comparable with both E1A and E1D. All of the discussed mutations have no significant influence on Km, and thus, we predict that substrate recognition remains intact. In contrast, the S149A mutation, which disrupts the hydrogen bond network of the N-terminal amine, decreases the affinity toward the substrate (increases Km) to a level observed for sp-SplB. In parallel substitution of Ser-149 with alanine results in an almost 3-fold decrease in kcat compared with the wild type. The E1Q mutation affects the activity (kcat) stronger than either of the mutations only influencing the interaction with Arg-115 (E1A, E1D, or R115A). This is most probably due to repealing interactions with the side chain of Arg-115, which presumably result in partial distortion of the positioning of the N-terminal amine as suggested by parallel influence on Km. Evaluated double mutants (E1A,S149A and R115A,S149A) demonstrate kinetics comparable to S149A contrary to the expected additive effect; however, the activity of a triple mutant (E1A,R115A,S149A) is significantly lower than that of either of single mutants as expected. Yet, it is still higher than that of N-terminally extended variants (sp-SplB, GS-SplB, and L-SplB). We believe that the lack of expected additive effect in double mutants and the fact that the triple mutant does not fully recapitulate the activity of GS-SplB is likely associated with unspecific effects of mutations on the overall dynamics of the molecule (see “Discussion”).
In the light of the above results, we further tested the hypothesis concerning the importance of hydrogen bond network at the N terminus in regulating SplB protease activity by verifying the activity of SplB (ΔE1) variant where the central residue of the H-bond network (Glu-1) was removed. Expectedly, this variant demonstrated the kinetics identical to that of N-terminally extended variants.
DISCUSSION
Protease Zymogens Assure Protection against Unwanted Proteolysis
The activity of proteolytic enzymes is tightly controlled to avoid unwanted degradation of cellular components. Cells direct proteases to appropriate compartments. However, the targeting systems are leaky to a certain extent. Therefore, proteases are usually synthesized as inactive zymogens, the proteolytic activity of which is only liberated at the desired site of action. To further control proteolysis, protease inhibitors are often present at non-target compartments (25). For example chymotrypsinogen, a precursor of a digestive enzyme and a type protease of the S1 family is stored within the cell in membrane-bound zymogen granules that contain pancreatic protease inhibitors, primarily targeting any prematurely activated trypsin, but also capable of inhibiting chymotrypsin to a certain extent (26). The hazard of misdirecting a secretory protease into the cytoplasm of bacteria is clearly exemplified by staphylococcal cysteine proteases (staphopains). Staphopains are secreted to the extracellular compartment and contain a propeptide, but their safe production still requires dedicated cytoplasmic inhibitors (27, 28).
Contrary to the above examples, SplB protease is devoid of a propeptide, and no inhibitors of serine proteases have been identified in the staphylococcal genome. One possible explanation is that efficient targeting to the extracellular compartment or restricted substrate specificity of SplB or both are enough to ensure acceptable level of toxicity or no toxicity. However, in this study, we argue for another explanation: a novel mechanism where the secretion signal has a parallel function in restricting protease activity in a manner similar to that observed for S1 family protease propeptides. We demonstrate that the activity of a signal peptide-containing form of SplB protease (sp-SplB) is more than an order of magnitude lower compared with mature SplB, both against synthetic peptides and protein substrates. This phenomenon is not attributed to the specific properties of the signal peptide, but rather, modification of the native N terminus of SplB results in comparable decrease in proteolytic activity. Removal of the N-terminal extension from GS-SplB and L-SplB by aminopeptidase restores proteolytic activity. We believe that activation of GS-SplB and L-SplB by aminopeptidase closely mimics the activation of sp-SplB by signal peptidase. Therefore, any SplB accidentally misdirected into the cytoplasm would contain the signal peptide, possess low activity and thus low toxicity, especially that the protease exhibits high substrate specificity. Only upon secretion, the signal peptidase would remove the signal peptide releasing full proteolytic activity of SplB. In fact, SplB purified from culture supernatants of S. aureus does not contain the signal peptide (12).
The Mechanistic Details of SplB Protease Maturation
The mechanistic details of protease zymogen activation by proteolytic processing of a propeptide have been described in detail in the chymotrypsin family. Crystallographic analysis of chymotrypsinogen revealed distorted conformation of the S1 subsite and oxyanion hole, crucial elements of the catalytic machinery of serine proteases (9). The removal of a short zymogen profragment allows interaction of the newly liberated N terminus with a buried side chain of Asp-194. This induces a structural rearrangement at the active site (9), which results in formation of functional S1 pocket and oxyanion hole, thus liberating the proteolytic activity. A comparable activation mechanism is repeated in multiple mammalian proteases of family S1.
Although V8 serine protease from Staphylococcus aureus is closely related in structure to chymotrypsin, its activation mechanism differs significantly. The inhibitory profragment of V8 protease is 39 residues long and is removed in a multistep process (29, 30). The newly formed N terminus becomes directly a part of the S1 specificity pocket contrary to the N terminus of chymotrypsin, which only influences the folding of the polypeptide chain constituting the S1 pocket. The description of further details of structural rearrangements upon V8 protease zymogen activation awaits determination of a crystal structure of the zymogen.
SplB protease is closely related to V8 protease and adopts a similar overall structure as chymotrypsin. However, analysis of structural rearrangements upon activation of SplB protease precursor demonstrates that the molecular mechanism of activation differs from both that of chymotrypsin and V8 protease. Contrary to chymotrypsinogen, the disposition of catalytic machinery is identical in the structures of GS-SplB and SplB. The only significant difference between the structure of precursor mimic (GS-SplB) and mature SplB is found in the direct vicinity of the N terminus. The N-terminal extension Gly-(−2)-Ser-(−1) as well as the two further amino acids Glu-1–Asn-2 are flexible and not defined by electron density in the structure of precursor mimic. Only the removal of the N-terminal extension allows the newly released N terminus to form characteristic hydrogen bond network (interaction of side chains of Glu-1 and Arg-115 and of N-terminal amine group with the side chain of Ser-149). Glu-1 is thus firmly positioned at the surface of mature SplB, which is reflected in clearly defined electron density of this residue. However, Glu-1 in SplB is relatively distant from the catalytic machinery compared with the N termini of either chymotrypsin or V8 protease, and most importantly, no significant rearrangement of the catalytic machinery or in its direct vicinity is associated with the conversion of GS-SplB into fully active SplB protease.
Another unique feature of SplB protease is that the oxyanion hole adopts a closed conformation both in GS-SplB as well as in the mature protease (SplB). In chymotrypsin, the activation process is associated with rearrangement of the closed conformation of the oxyanion hole observed in the zymogen into an open, canonical conformation found in the active enzyme. According to the standard mechanism of catalysis of serine proteases the closed conformation of the oxyanion hole in mature SplB should preclude enzymatic activity, but such activity was demonstrated experimentally. A similar phenomenon was described previously in the case of epidermolytic toxins and factor VIIa, which are active serine proteases despite closed conformation of the oxyanion hole observed in x-ray structures. In all of the cases, it was suggested that the rearrangement within the oxyanion hole of the mature enzyme is induced upon consensus substrate binding, thus increasing the substrate specificity of the enzyme (31–33), but no direct experimental evidence has been provided as of yet.
The comparison of crystallographic structures of mature SplB protease and a mimic of its zymogen (GS-SplB) does not mechanistically explain the observed differences in proteolytic activity. The crucial elements of catalytic machinery are already present in the structure of SplB zymogen, and no significant rearrangements are observed in this region upon zymogen processing. The hydrogen bond network at the N terminus in the mature protease is relatively distant from the active site contrary to what is observed in chymotrypsin and V8 protease. Even though both the structures of SplB and GS-SplB were determined at relatively high resolution, not even subtle differences were noted in the vicinity of the active site. We believe that the interactions at the N terminus affect the dynamics of the whole molecule influencing substrate recognition and hydrolysis, but such subtle dynamic differences are hard to explain comprehensively based on static crystallographic structures. To gain additional information, we have attempted to separate the influence of hydrogen bonds of the amino group of Glu-1 and those of the side chain carboxyl of that residue. It turns out that the interaction of the side chain of Glu-1 with that of Arg-115 influences the rate of hydrolysis (kcat), whereas the interactions of the N-terminal amine group influence both the rate of hydrolysis and substrate recognition (Km). Interaction with Arg-115 is responsible for about half of the difference observed in kcat between SplB and GS-SplB, whereas the other half is contributed by the interactions of the N-terminal amine. The observed difference in substrate affinity (Km) is contributed solely by the interactions of the N-terminal amine. Such simple analysis is however only partially relevant because it does not take into account the influence of introduced mutations on the dynamics of nearby residues, tightly coordinated water molecules, and especially the structure of water envelope surrounding the N terminus. This is especially evident when comparing the activity of L-SplB and S-SplB. Hydrophobic residue, likely to strongly influence the water structure in its vicinity, abolishes the protease activity to the level of sp-SplB, whereas a smaller polar residue results only in partial decrease in activity. We speculate that similar effects are involved when tested double mutants are concerned. Such complex interactions are however beyond analysis by simple mutagenesis. Nonetheless, low activity of SplB (ΔE1), corresponding to that of sp-SplB clearly demonstrates the overall importance of the hydrogen bond network at the N terminus.
The above observations, however, still do not explain how do the interactions distant from the catalytic machinery affect the catalytic activity and we only hypothesize that changes in the overall tumbling dynamics of the entire molecule are involved. Such effects are however extremely hard to study with currently available scientific methodology.
This work was supported by the Foundation for Polish Science-PARENT-BRIDGE Programme (to K. P.) and in part by Grants N N301 032834 (to G. D.) from the Polish Ministry of Science and Higher Education, UMO-2011/01/D/NZ1/01169 (to G. D.) and UMO-2011/01/N/NZ1/00208 (to M. Z.) from the National Science Center. The research was carried out with the equipment purchased from support of the European Union structural funds (Grants POIG.02.01.00-12-064/08 and POIG.02.01.00-12-167/08).
The atomic coordinates and structure factors (codes 4K1S and 4K1T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- PDB
- Protein Data Bank
- AAP
- aminopeptidase from Aeromonas proteolytica
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Władyka B., Pustelny K. (2008) Regulation of bacterial protease activity. Cell. Mol. Biol. Lett. 13, 212–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. López-Otín C., Bond J. S. (2008) Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strausberg S., Alexander P., Wang L., Schwarz F., Bryan P. (1993) Catalysis of a protein folding reaction: thermodynamic and kinetic analysis of subtilisin BPN′ interactions with its propeptide fragment. Biochemistry 32, 8112–8119 [DOI] [PubMed] [Google Scholar]
- 4. Baker D., Shiau A. K., Agard D. A. (1993) The role of pro regions in protein folding. Curr. Op. Cell Biol. 5, 966–970 [DOI] [PubMed] [Google Scholar]
- 5. Winther J. R., Sørensen P. (1991) Propeptide of carboxypeptidase Y provides a chaperon-like functions as well as inhibition of enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 88, 9330–9334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rudenko G., Bonten E., d'Azzo A., Hol W. G. (1995) 3-dimensional structure of the human protective protein: structure of the precursor form suggests a complex activation mechanism. Structure 3, 1249–1259 [DOI] [PubMed] [Google Scholar]
- 7. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rawlings N. D., Barrett A. J., Bateman A. (2010) MEROPS: the peptidase database. Nucleic Acids Res. 38, D227–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Freer S. T., Kraut J., Robertus J. D., Wright H. T., Xuong N. H. (1970) Chymotrypsinogen: 2.5A crystal structure, comparison with α-chymotrypsin and implications for zymogen activation. Biochemistry 9, 1997–2009 [DOI] [PubMed] [Google Scholar]
- 10. Wang D., Bode W., Huber R. (1985) Bovine chymotrypsinogen a X-ray crystal structure analysis and refinement of a new crystal form at 1.8 A resolution. J. Mol. Biol. 185, 595–624 [DOI] [PubMed] [Google Scholar]
- 11. Berthold H., Frorath B., Scanarini M., Abney C. C., Ernst B., Northemann W. (1992) Plasmid pGEX-5T - and alternative system for expression and purification of recombinant proteins. Biotechnol. Lett. 14, 245–250 [Google Scholar]
- 12. Popowicz G. M., Dubin G., Stec-Niemczyk J., Czarny A., Dubin A., Potempa J., Holak T. A. (2006) Functional and structural characterization of Spl proteases from Staphylococcus aureus. J. Mol. Biol. 358, 270–279 [DOI] [PubMed] [Google Scholar]
- 13. Dubin G., Stec-Niemczyk J., Kisielewska M., Pustelny K., Popowicz G. M., Bista M., Kantyka T., Boulware K. T., Stennicke H. R., Czarna A., Phopaisarn M., Daugherty P. S., Thøgersen I. B., Enghild J. J., Thornberry N., Dubin A., Potempa J. (2008) Enzymatic activity of the Staphylococcus aureus SpIB serine protease is induced by substrates containing the sequence Trp-Glu-Leu-Gin. J. Mol. Biol. 379, 343–356 [DOI] [PubMed] [Google Scholar]
- 14. Fersht A. (1985) Enzyme Structure and Mechanism, 2nd Ed., Freeman, New York [Google Scholar]
- 15. Leslie A. G. (1992) Recent changes to the MOSFLM package for processing film and image plate data, Join CCP4 and ESF-EAMCB Newsletter on Protein Crystallography 26 [Google Scholar]
- 16. Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 17. Evans P. (1997) Scala. Join CCP4 and ESF-EAMCB Newsletter 33, 22–24 [Google Scholar]
- 18. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D. Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 19. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 21. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D. Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 22. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures, Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 23. Lamzin V. S., Wilson K. S. (1993) Automated refinement of protein models. Acta Crystallogr. D. Biol. Crystallogr. 49, 129–147 [DOI] [PubMed] [Google Scholar]
- 24. Wilkes S. H., Bayliss M. E., Prescott J. M. (1973) Specificity of aeromonas aminopeptidase toward oligopeptides and polypeptides. Eur. J. Biochem. 34, 459–466 [DOI] [PubMed] [Google Scholar]
- 25. Neurath H., Walsh K. A. (1976) Role of proteolytic-enzymes in biological regulation. Proc. Natl. Acad. Sci. U.S.A. 73, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steer M. L., Meldolesi J. (1987) The cell biology of experimental pancreatitis. N. Engl. J. Med. 316, 144–150 [DOI] [PubMed] [Google Scholar]
- 27. Filipek R., Szczepanowski R., Sabat A., Potempa J., Bochtler M. (2004) Prostaphopain B structure: A comparison of proregion-mediated and staphostatin-mediated protease inhibition. Biochemistry 43, 14306–14315 [DOI] [PubMed] [Google Scholar]
- 28. Rzychon M., Sabat A., Kosowska K., Potempa J., Dubin A. (2003) Staphostatins: an expanding new group of proteinase inhibitors with a unique specificity for the regulation of staphopains, Staphylococcus spp. cysteine proteinases. Mol. Microbiol. 49, 1051–1066 [DOI] [PubMed] [Google Scholar]
- 29. Nickerson N. N., Prasad L., Jacob L., Delbaere L. T., McGavin M. J. (2007) Activation of the SspA serine protease zymogen of Staphylococcus aureus proceeds through unique variations of a trypsinogen-like mechanism and is dependent on both autocatalytic and metalloprotease-specific processing. J. Biol. Chem. 282, 34129–34138 [DOI] [PubMed] [Google Scholar]
- 30. Drapeau G. R. (1978) Role of a metalloprotease in activation of precursor of staphylococcal protease. J. Bacteriol. 136, 607–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vath G. M., Earhart C. A., Rago J. V., Kim M. H., Bohach G. A., Schlievert P. M., Ohlendorf D. H. (1997) The structure of the superantigen exfoliative toxin A suggests a novel regulation as a serine protease. Biochemistry 36, 1559–1566 [DOI] [PubMed] [Google Scholar]
- 32. Papageorgiou A. C., Plano L. R., Collins C. M., Acharya K. R. (2000) Structural similarities and differences in Staphylococcus aureus exfoliative toxins A and B as revealed by their crystal structures. Protein Sci. 9, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bajaj S. P., Schmidt A. E., Agah S., Bajaj M. S., Padmanabhan K. (2006) High resolution structures of p-aminobenzamidine- and benzamidine-VIIa/soluble tissue factor: unpredicted conformation of the 192–193 peptide bond and mapping of Ca2+, Mg2+, Na+, and Zn2+ sites in factor VIIa. J. Biol. Chem. 281, 24873–24888 [DOI] [PubMed] [Google Scholar]