

Background: The mechanism for rapid biosynthesis of platelet-activating factor (PAF), a potent proinflammatory lipid mediator, is unclear.
Results: Phosphorylation of lysophosphatidylcholine acyltransferase 2 (LPCAT2) at Ser-34 via PKCα enhanced rapid PAF biosynthesis following PAF or ATP stimulation.
Conclusion: The molecular basis for rapid PAF biosynthesis in response to inflammatory stimuli is elucidated.
Significance: These data suggest a better understanding of PAF production in early-phase inflammation.
Keywords: Enzyme Catalysis, Inflammation, Macrophages, Membrane Lipids, Phosphorylation, Protein Kinase C (PKC), Platelet-activating Factor, Lysophospholipid Acyltransferase
Abstract
Platelet-activating factor (PAF), a potent proinflammatory lipid mediator, is synthesized rapidly in response to extracellular stimuli by the activation of acetyl-CoA:lyso-PAF acetyltransferase (lyso-PAFAT). We have reported previously that lyso-PAFAT activity is enhanced in three distinct ways in mouse macrophages: rapid activation (30 s) after PAF stimulation and minutes to hours after LPS stimulation. Lysophosphatidylcholine acyltransferase 2 (LPCAT2) was later identified as a Ca2+-dependent lyso-PAFAT. However, the mechanism of rapid lyso-PAFAT activation within 30 s has not been elucidated. Here we show a new signaling pathway for rapid biosynthesis of PAF that is mediated by phosphorylation of LPCAT2 at Ser-34. Stimulation by either PAF or ATP resulted in PKCα-mediated phosphorylation of LPCAT2 to enhance lyso-PAFAT activity and rapid PAF production. Biochemical analyses showed that the phosphorylation of Ser-34 resulted in augmentation of Vmax with minimal Km change. Our results offer an answer for the previously unknown mechanism of rapid PAF production.
Introduction
Platelet-activating factor (PAF,3 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a potent proinflammatory lipid mediator that triggers various cellular functions through binding to its G protein-coupled receptor, PAF receptor (PAFR), resulting in pleiotropic biological effects such as platelet activation, airway constriction, hypotension, and systemic anaphylaxis (1–5). Production of lipid mediators is an important step for regulating inflammation and immune responses. The most important feature of lipid mediators is that they are synthesized rapidly from precursor biomembrane phospholipids, adapting to the environment around cells. Specific enzymes synthesize most lipid mediators on demand in a Ca2+-dependent manner. In response to extracellular stimuli, PAF is synthesized rapidly in the remodeling pathway from its precursor membrane phospholipid, 1-O-alkyl-2-acyl-sn-glycero-3-phosphocholine (1-alkyl-PC). Under inflammatory conditions, 1-alkyl-PC is hydrolyzed in a Ca2+-dependent manner by the action of cytosolic phospholipase A2 to produce polyunsaturated fatty acid and lyso-PAF, a lysophosphatidylcholine (LPC) derivative with an alkyl ether bond at the sn-1 position. Biosynthetic enzymes of various lipid mediators subsequently utilize polyunsaturated fatty acid and lyso-PAF as substrates. Our previous studies showed that macrophages and neutrophils from cytosolic phospholipase A2α-deficient mice are unable to produce PAF in response to extracellular stimuli (6, 7). PAF is synthesized from lyso-PAF by acetyl-CoA:lyso-PAF acetyltransferase (lyso-PAFAT (EC: 2.3.1.67)) (2). Endogenous lyso-PAFAT activity has been reported initially in 1980 and partially characterized (8). Our laboratory identified a molecular entity of lyso-PAFAT, lysophosphatidylcholine acyltransferase 2 (LPCAT2) (9). This enzyme is a 60-kDa protein with three putative transmembrane domains and several EF-hand motifs and is localized in endoplasmic reticulum (1). Although the biosynthesis of PAF is understood poorly, its degradation has been well characterized. Deacetylation of PAF is catalyzed by PAF acetyl hydrolase and leads to the loss of its activity (10–12).
LPCAT2 also possesses lysophosphatidylcholine acyltransferase (LPCAT (EC: 2.3.1.23)) activity that catalyzes the incorporation of arachidonoyl-CoA into membrane phosphatidylcholine (PC). In fact, 1-alkyl-PC represents approximately half of the phospholipid content in the biomembranes of hematopoietic cells (13). Thus, LPCAT2 functions in the biosynthetic pathway of phospholipids known as the Lands cycle or remodeling pathway, which is responsible for generating the diversity of phospholipid species in biomembranes (14). LPCAT2 also influences the fatty acid content of cellular membranes and the availability of substrates for cellular lipid mediator production.
There are a number of reports showing that lyso-PAFAT activity is enhanced by several distinct mechanisms. We reported previously that, in mouse macrophages, lyso-PAFAT activity was increased within 30 s after PAFR stimulation (15) and within 30 min or 16 h after LPS stimulation (9, 16). LPS stimulation for 16 h increased mRNA of LPCAT2 (9). LPS stimulation for 30 min activated LPCAT2 by Ser-34 phosphorylation via the p38 MAPK/MAPK-activated protein kinase 2 (MK2)-dependent pathway (16). However, the mechanism of lyso-PAFAT activation by PAF stimulation on the order of seconds was unclear. Several studies have reported that activation of endogenous lyso-PAFAT activity is dependent on protein kinase A, PKC, or p38 MAPK pathways (17–19), but the regulatory mechanisms and the gene responsible for the rapid lyso-PAFAT activity in response to PAF have not been clarified.
In this study, we identified phosphorylation at Ser-34 of LPCAT2 via PKCα signaling as the underlying mechanism that led to rapid enhancement of lyso-PAFAT activity following PAF- or ATP-stimulation. Enzymological analyses showed that phosphorylation increased the Vmax value of LPCAT2, whereas the Km value was almost unchanged. Our report identifies the enzyme and signaling pathway that mediate rapid activation of PAF production, which is critical in the early stages of inflammatory responses.
EXPERIMENTAL PROCEDURES
Materials
PC from frozen egg yolk, LPS from Salmonella minnesota, ATP, anti-FLAG M2 antibody, and anti-β-actin antibody were from Sigma. Deuterium-labeled d4-16:0 lyso-PAF, d4-16:0 PAF, 16:0 lyso-PAF, and 16:0 methylcarbamyl PAF (mcPAF, a non-hydrolyzed analog of PAF (20)) were from Cayman Chemical Co. (Ann Arbor, MI). Arachidonoyl-CoA was from Avanti Polar Lipids (Alabaster, AL). [3H]Acetyl-CoA (129.5 GBq/mmol) and horseradish peroxidase-linked anti-rabbit or anti-mouse IgGs were from GE Healthcare (Buckinghamshire, UK). [1-14C]Arachidonoyl-CoA (2.22 GBq/mmol) was from Moravec Biochemicals (Brea, CA). TLC silica gel plates (type 5721) were from Merck (Darmstadt, Germany). Acetyl-CoA, dimethyl sulfoxide, acrylamide, and Phos tag acrylamide were from Wako (Osaka, Japan). U73122 (PLCβ inhibitor), U73343 (an inactive analog of U73122), bisindolylmaleimide I (BIM-I, a PKC inhibitor), Ro-31-8220, Ro-31-8425, bisindolylmaleimide V (BIM-V, an inactive analog of BIM-I), and 12-O-tetradecanoylphorbol-13-acetate (TPA) were from Calbiochem. The siRNAs (ON-TARGETplus Non-targeting Pool D-001810-10−20, ON-TARGETplus SMARTpool L-040348-00-0005 (PKCα), and L-052470-01-0005 (LPCAT2)) were from Thermo Scientific (Waltham, MA). The cell line Nucleofector kit V was from Lonza (Basel, Switzerland). Anti-MK2, anti-phospho-Thr-334-MK2, and anti-PKCα antibodies were from Cell Signaling Technology (Beverly, MA). The protease inhibitor mixture, EDTA-free Complete, was from Roche. The anti-LPCAT2 and anti-phospho-LPCAT2 antibodies and FLAG-LPCAT1, FLAG-LPCAT2, S34A, and S34D plasmids have been described previously (16).
Mice
C57BL/6J mice were obtained from Clea Japan, Inc. (Tokyo, Japan). Mice were maintained in a light-dark cycle with lights on from 08:00–20:00 h at 22 °C. Mice were fed with a standard laboratory diet and water ad libitum. All animal studies were conducted in accordance with the guidelines for animal research at The University of Tokyo and were approved by The University of Tokyo Ethics Committee for Animal Experiments.
Isolation of Mouse Peritoneal Macrophages
Thioglycolate-induced mouse peritoneal macrophages were isolated as described previously (15). Cells were cultured for 16 h before inhibitor treatment or mcPAF stimulation.
Preparation of Cell Lysates
Cells were pretreated with the following reagents for the indicated times: 10 μm BAPTA/AM, 10 μm U73122, or 10 μm U73343 for 10 min; 1–20 μm of BIM-I, 1–10 μm of Ro-31-8220, 1–10 μm of Ro-31-8425, and 10 μm of BIM-V for 30–60 min; and 100 nm TPA for 24 h. Cells were then stimulated with 200 nm mcPAF or 250 μm ATP for 30 s. After stimulation, cells (peritoneal macrophages, CHO-S cells stably expressing PAFR (21), non-transfected RAW264.7 cells, and RAW264.7 cells stably expressing the vector or FLAG-LPCAT2 (16)) were washed with ice-cold buffer containing 20 mm Tris-HCl (pH 7.4), 0.3 m sucrose, and 1 mm sodium orthovanadate and collected in buffer containing 20 mm Tris-HCl (pH 7.4), 1 mm sodium orthovanadate, 5 mm 2-mercaptoethanol, and 1× EDTA-free complete protease inhibitor mixture. Subsequently, cells were sonicated twice on ice for 30 s each time and centrifuged at 9000 × g for 10 min at 4 °C to remove cellular debris, intact cells, and mitochondria.
For peritoneal macrophages, the resultant supernatant from the centrifugation at 9000 × g was separated at 100,000 × g for 1 h at 4 °C. The resultant pellet was resuspended with ice-cold buffer containing 20 mm Tris-HCl (pH 7.4), 1 mm sodium orthovanadate, 5 mm 2-mercaptoethanol, and 1× EDTA-free Complete protease inhibitor mixture. The concentration of each protein was measured by the Bradford method (22) using protein assay solution (Bio-Rad). Bovine serum albumin (fraction V, fatty acid-free, Sigma) served as a standard.
Transfection of CHO-S Cells Stably Expressing PAFR
CHO-S cells stably expressing PAFR (CHO-S-PAFR cells) (21) were seeded in 10-cm collagen-coated dishes 24 h before transfection. Empty vector, FLAG-LPCAT1, FLAG-LPCAT2, S34A, and S34D plasmids (15 μg of each) were transfected into CHO-S-PAFR cells using 30 μl of Lipofectamine 2000 (catalog no. LF2000, Invitrogen). Twenty-four hours after transfection, cells from each 10-cm dish were divided into three 6-cm dishes and incubated at 37 °C in 5% CO2 for 2 h. The medium was then changed to DMEM containing 0.5% BSA. Cells were cultured without serum for 24 h and then stimulated with 200 nm mcPAF for 30 s.
For the knockdown of PKCα, CHO-S-PAFR cells were seeded in 6-cm collagen-coated dishes 24 h before transfection of 180 pmol siRNA using 17 μl of RNAi MAX (Invitrogen). The cells were incubated for 24 h and subsequently cotransfected with 4 μg of FLAG-LPCAT2. After another 24 h of incubation, cells from each 6-cm dish were divided into two 6-cm dishes and incubated for 2 h. The medium was changed to DMEM containing 0.5% BSA. The cells were cultured without serum for 24 h and then stimulated with 200 nm mcPAF for 30 s.
Transfection of RAW264.7 Cells Stably Expressing FLAG-LPCAT2
RAW264.7 cells stably expressing FLAG-LPCAT2 were suspended in 100 μl of Nucleofector kit V and mixed with 30 pmol PKCα siRNA or negative control siRNA. The mixture in the cuvette was set onto the Amaxa Nucleofector and electroporated with program D-032. Then, cells were seeded onto 6-cm dishes. Forty-eight hours after transfection, cells were stimulated with 250 μm ATP for 30 s.
Western Blot Analysis
Western blot analyses were performed as described previously (23). To detect band shifts that represent phosphorylated proteins, SDS-PAGE gels containing 50 μm Phos tag acrylamide and 100 μm manganese ions (Mn2+) were used. The Phos tag forms a complex with two Mn2+ ions and serves as a phosphate-binding molecule (24). The complex is used for phosphate affinity SDS-PAGE, which results in the upward mobility shift of phosphorylated proteins. Reacted proteins were detected using the LAS4000 or LAS500 imaging systems (GE Healthcare).
Assay of Lyso-PAFAT and LPCAT
The lyso-PAFAT and LPCAT assays were performed using radioisotopes, as described previously (9, 25), or LC-MS/MS. The Km and Vmax of LPCAT2 and phospho-LPCAT2 activities were measured under the following conditions: 5 μg/ml microsomal proteins (pellet after centrifugation at 100,000 × g for 1 h) were incubated in buffer containing 100 mm Tris-HCl (pH 7.4), 0.015% Tween 20, 1 μm CaCl2, deuterium-labeled lyso-alkyl-PC (d4-16:0 lyso-PAF), and acetyl-CoA or arachidonoyl-CoA at 37 °C for 5 min. The concentrations of each substrate are described in Fig. 3, D–G. Other assays were performed under the following conditions: 10 μg/ml proteins (supernatant after centrifugation at 9000 × g for 10 min) were incubated in buffer containing 100 mm Tris-HCl (pH 7.4), 0.015% Tween 20, 1 μm CaCl2, 5 μm d4-16:0 lyso-PAF and 1 mm acetyl-CoA at 37 °C for 5 min. Lipids were extracted with methanol containing dimyristoyl phosphatidylcholine (14:0/14:0 PC) or 17:0 LPC as an internal standard and subsequently analyzed by LC-MS/MS.
FIGURE 3.

Site-directed mutagenesis of LPCAT2 and effect of LPCAT2 phosphorylation on enzymatic activities. CHO-S-PAFR cells transfected with vector, WT, or mutant (S34A and S34D) LPCAT2 were stimulated with 200 nm mcPAF for 30 s. A, phosphorylation was examined by Phos tag Western blot analysis. An upward-shifted band was detected in WT but not S34A or S34D mutants following mcPAF stimulation. Lyso-PAFAT (B) and LPCAT (C) activities were analyzed by measuring the incorporation of radiolabeled acyl-CoA ([3H]acetyl-CoA or [14C]arachidonoyl-CoA) into PAF or PC, respectively. WT LPCAT2 showed enhanced lyso-PAFAT and LPCAT activities following mcPAF stimulation. The S34A mutant did not show enhanced activities following stimulation, whereas the S34D showed enhanced basal activity compared with the WT but was not increased following mcPAF stimulation. Results are expressed as the mean + S.D. of an experiment performed in triplicate. Four independent experiments were performed with similar results. **, p < 0.01; ***, p < 0.001; analysis of variance and Tukey's multiple comparison test. Lyso-PAF acetyltransferase assays were performed using 5 μm deuterium-labeled lyso-PAF and the indicated concentrations of acetyl-CoA (D) or 1 mm acetyl-CoA and increasing concentrations of lyso-PAF (E). LPCAT assays were conducted with 5 μm deuterium-labeled lyso-PAF and the indicated concentrations of arachidonoyl-CoA (F) or 2.5 μm arachidonoyl-CoA and increasing concentrations of deuterium-labeled lyso-PAF (G). Phosphorylation of LPCAT2 affected the Vmax value of its catalytic activities. The data represent the mean ± S.D. of the difference between LPCAT2- and vector-transfected samples in triplicate measurements. The units of Vmax and Km are nanomoles/minute/milligram and micromolar, respectively. The results are representative of two independent experiments with similar results.
Extraction and Purification of Cellular PAF Product
For endogenous PAF analysis, lipids from cells grown on 35-mm dishes were extracted with 500 μl of methanol containing d4–16:0 PAF as an internal standard and purified with Oasis HLB cartridges (Waters, Milford, MA) as described previously (26, 27).
Analysis of Enzyme Assay Products and Endogenous PAF Production by LC-MS/MS
Chromatography was performed on an AcquityTM UPLC BEH C8 column (1.7 μm, 2.1 × 30 mm, Waters) using an AcquityTM ultra-performance LC system (Waters) as described previously (28). Lipids were measured by a TSQ Vantage Triple Stage Quadrupole mass spectrometer (Thermo Scientific) equipped with a HESI-II electrospray ionization source, as described previously (29). Endogenous PAF from peritoneal macrophages and RAW264.7 cells was measured using LC-MS/MS. Chromatography was carried out on a Kinetex C8 column (2.1 × 150 mm, Phenomenex, Torrance, CA). Nexera UHPLC (Shimadzu, Kyoto, Japan) was connected to an LCMS-8040 (Shimadzu) mass spectrometer (25). Deuterium-labeled d4–16:0 PAF and non-labeled PAF were monitored by selected reaction monitoring transition (m/z 528.4–184.1 and 524.4–184.1, respectively) in positive ion scan mode. Deuterium-labeled 1-alkyl-PC was monitored by selected reaction monitoring transition (m/z 832.6–303.2) in negative ion scan mode. Signal intensities were determined relative to the internal standard or the percentage of the positive control. Mass spectra were processed using Xcalibur 2.0 software (Thermo Scientific) or LabSolutions 5.53 software (Shimadzu).
Quantitative Real-time PCR
Total RNAs were prepared using the RNeasy mini kit (Qiagen), and first strand cDNA was subsequently synthesized using Superscript III (Invitrogen). The PCRs were performed using Fast SYBR Green Master Mix and the StepOnePlusTM real-time PCR system (Applied Biosystems). The following primers were used: Lpcat2, a 64-bp fragment, CCCTGCCAATACAGAAGAGATCA (forward) and GCCGTCCTCATCAACATCAA (reverse); Prkca, a 172-bp fragment, GTGTCCTACCCCAAATCCTTGTC (forward) and GTTGGATCTCCCTGTTCTCCAGT (reverse); and Rplp0, a 136-bp fragment, CTGAGATTCGGGATATGCTGTTG (forward) and AAAGCCTGGAAGAAGGAGGTCTT (reverse).
Software
All statistical calculations were performed using Prism 5 (GraphPad Software, La Jolla, CA).
RESULTS
Activation and Phosphorylation of LPCAT2 Following mcPAF Stimulation
There are two known enzymes that possess lyso-PAFAT activity in vitro: LPCAT1 and LPCAT2 (9, 25). In inflammatory cells, LPCAT2 is the predominant lyso-PAFAT to produce PAF in response to several stimuli (9). To examine the different characteristics of the two lyso-PAFATs, FLAG-tagged LPCAT1 and LPCAT2 were transiently transfected into CHO-S cells stably expressing PAFR (CHO-S-PAFR cells). CHO-S-PAFR cells can respond to PAF by several signaling events, including Ca2+ response (21). Cells were stimulated with mcPAF for 30 s, and lyso-PAFAT activities in the 9000 × g centrifugation supernatants were examined by incorporation of [3H]-labeled acetyl-CoA into PAF. Although the activity of LPCAT1 was unchanged after mcPAF stimulation, that of LPCAT2 was enhanced (Fig. 1A). Lyso-PAFAT activity was not detected in vector-transfected samples.
FIGURE 1.
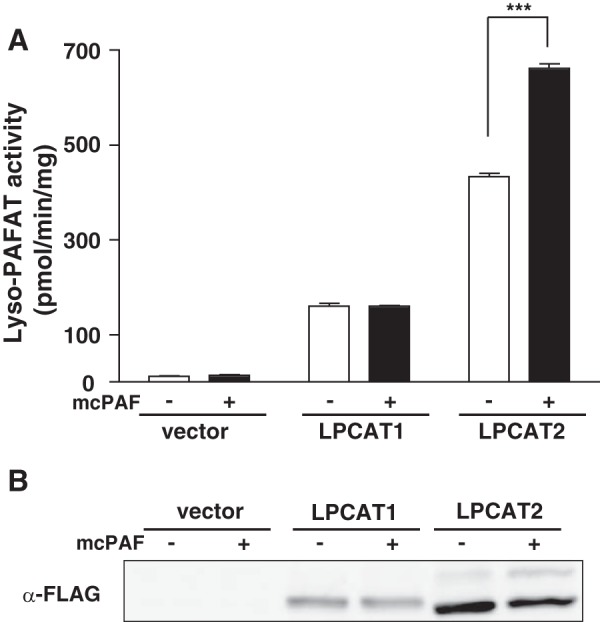
LPCAT2 activation and phosphorylation. CHO-S-PAFR cells transfected with FLAG-LPCAT1, FLAG-LPCAT2, or empty vector were stimulated with 200 nm mcPAF for 30 s. A, mcPAF enhanced the activity of LPCAT2 but not LPCAT1. Results are expressed as the mean + S.D. of an experiment performed in triplicate. Three independent experiments showed similar results. B, the phosphorylation of LPCAT1 and LPCAT2 was examined by Phos tag acrylamide gel electrophoresis and Western blot analysis. LPCAT2 showed an upward shifted band indicative of phosphorylation, which was increased after mcPAF stimulation. ***, p < 0.001; analysis of variance and Tukey's multiple comparison test.
Previously, we observed that phosphorylation of LPCAT2 at a single residue, Ser-34, occurs 30 min after LPS stimulation and leads to an increase of lyso-PAFAT activity (16). To investigate the mechanism of rapid LPCAT2 activation, we examined the phosphorylation status of LPCAT2 after 30-s exposure to mcPAF by Phos tag acrylamide gel electrophoresis and Western blot analysis. An upward shifted band indicative of phosphorylation was detected for FLAG-LPCAT2 but not FLAG-LPCAT1, indicating that LPCAT2 is rapidly phosphorylated after mcPAF stimulation to enhance its enzymatic activity (Fig. 1B).
Different Pathways Mediate LPCAT2 Phosphorylation Following mcPAF and LPS Stimulation
To determine the phosphorylation site of LPCAT2 following mcPAF stimulation, peritoneal macrophages were stimulated with 100 ng/ml LPS for 30 min or 200 nm mcPAF for 30 s, and the microsomal proteins (100,000 × g pellet) were analyzed by Western blot analysis using antibodies to detect total or phospho-Ser-34 LPCAT2. Phosphorylated LPCAT2 was barely detectable in untreated cells but detected readily following either 30 min of LPS or 30 s of mcPAF stimulation. To examine whether phosphorylation of LPCAT2 following mcPAF stimulation is also MK2-dependent, the samples were probed with antibodies that recognize total or activated MK2. Phosphorylated MK2 was observed following LPS but not mcPAF stimulation (Fig. 2A), indicating that the rapid phosphorylation of LPCAT2 in response to mcPAF is independent of MK2 signaling and occurs via a separate signaling pathway.
FIGURE 2.
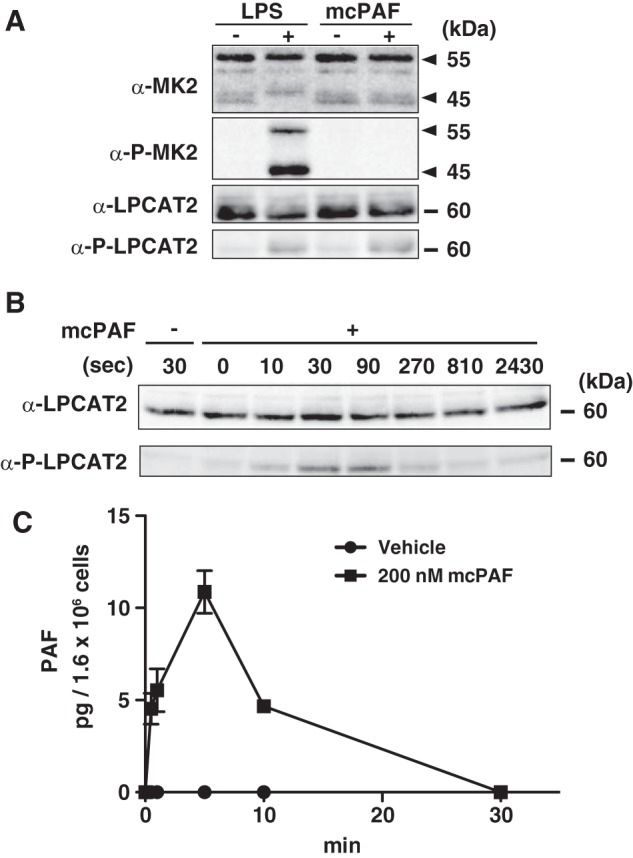
Signaling pathway for LPCAT2 phosphorylation. Peritoneal macrophages were stimulated with 100 ng/ml LPS for 30 min or 200 nm mcPAF for 30 s. A, phosphorylation of MK2 and LPCAT2 was detected by Western blot analysis. Phosphorylation of LPCAT2 at Ser-34 occurred after stimulation with either compound. Phospho-MK2 reactive bands were detected following LPS but not mcPAF stimulation. Arrowheads indicate MK2 splice variants. B, the time course of LPCAT2 phosphorylation following mcPAF stimulation was determined by Western blot analysis. Phosphorylation of LPCAT2 at Ser-34 reached a maximum within 30 s. Three independent experiments were performed with similar results (A and B). C, PAF production from peritoneal macrophages was measured by LC-MS/MS. Lipids were extracted with methanol containing deuterium-labeled PAF as an internal standard. Results are expressed as the mean + S.E. of three experiments.
Time Course of LPCAT2 Phosphorylation and PAF Production Following mcPAF Stimulation
To investigate the rapid time course of LPCAT2 phosphorylation, peritoneal macrophages were stimulated with 200 nm mcPAF for varying times (0–2430 s), and microsomal proteins were analyzed by Western blot analysis using antibodies to detect total or phospho-Ser-34 LPCAT2. The amount of total LPCAT2 was similar in all samples. Phosphorylated LPCAT2 was not detected in vehicle-treated cells, but stimulation with mcPAF rapidly induced phosphorylation of LPCAT2 in as little as 10 s, reached a maximum by 30–90 s, and was decreased by 270 s (Fig. 2B), consistent with our previous study (15). Next, we examined whether endogenous PAF production correlated with the phosphorylation of LPCAT2 following mcPAF stimulation. A rapid and transient increase in PAF occurred within 30 s and was at a maximum at 5 min but undetectable by 30 min, consistent with its rapid synthesis and degradation (Fig. 2C).
Ser-34 Phosphorylation Is Required for mcPAF-induced LPCAT2 Activation
To further examine the role of Ser-34 phosphorylation following mcPAF-stimulation, two FLAG-tagged mutant constructs (S34A and S34D) of LPCAT2 were transiently expressed in CHO-S-PAFR cells. The cells were stimulated with mcPAF for 30 s, and the cellular proteins were analyzed by Phos tag acrylamide gel electrophoresis and Western blot analysis. An upward mobility shift in LPCAT2 indicative of phosphorylation was detected in WT LPCAT2 following mcPAF-stimulation but not in the S34A or S34D mutant proteins (Fig. 3A). Next, we examined the effect of phosphorylation on the dual activities of LPCAT2 (lyso-PAFAT and LPCAT) by measuring the incorporation of radiolabeled acyl-CoA ([3H]acetyl-CoA or [14C]arachidonoyl-CoA) into PAF or PC, respectively. Both the lyso-PAFAT and LPCAT activities of WT LPCAT2 were enhanced by mcPAF stimulation (Fig. 3, B and C). Basal S34A activities were similar to WT but not increased by mcPAF stimulation. Conversely, basal activities of LPCAT2 containing the S34D mutation, which functions as a phosphomimetic, were higher than in the WT, but enhancement by mcPAF stimulation was not observed (Fig. 3B). Because the expression level of each construct was similar (Fig. 3A), our results indicate that both the lyso-PAFAT and LPCAT activities of LPCAT2 are enhanced by Ser-34 phosphorylation in response to mcPAF stimulation.
Effect of Ser-34 Phosphorylation on the Enzymatic Activities of LPCAT2
Next, we investigated how Ser-34 phosphorylation affects enzymatic activities of LPCAT2 using microsomal proteins from CHO-S-PAFR cells overexpressing LPCAT2. Proteins from mcPAF-treated cells exhibited higher Vmax values than control proteins in both lyso-PAFAT and LPCAT activities in vitro (Fig. 3, D–G), potentially because of a conformational change in LPCAT2, which is phosphorylated at Ser-34. Changes in the Km values were minimal following mcPAF stimulation.
Signaling Pathways That Mediate LPCAT2 Phosphorylation following mcPAF Stimulation
Although the phosphorylation of LPCAT2 by 30 min of LPS stimulation was dependent on MK2, mcPAF stimulation for 30 s utilized a different pathway (Fig. 2A). Therefore, we examined signaling pathway activation after mcPAF stimulation.
It has been shown that, following PAFR stimulation, intracellular Ca2+ concentration is increased rapidly via the Gq/11-PLCβ-inositol 1,4,5-trisphosphate pathway (30–32). We also detected an increased intracellular Ca2+ concentration in peritoneal macrophages and CHO-S-PAFR cells within 30 s of mcPAF stimulation (data not shown). Therefore, we examined the effect of pretreatment with a cytosolic Ca2+ chelator (BAPTA/AM) (33, 34) as well as a PLCβ inhibitor (U73122) or an inactive analog (U73343) (35) on LPCAT2 phosphorylation. Peritoneal macrophages were pretreated with each compound for 10 min and then stimulated with mcPAF for 30 s. The phosphorylation status of LPCAT2 was analyzed by Western blot analysis. Both inhibitors abolished mcPAF-induced LPCAT2 phosphorylation (Fig. 4A), indicating that this phosphorylation requires PLCβ activation and intracellular Ca2+ elevation.
FIGURE 4.

Conventional PKC-dependent LPCAT2 phosphorylation. Peritoneal macrophages were preincubated with or without inhibitors and subsequently stimulated with 200 nm mcPAF for 30 s. Microsomal proteins were subjected to Western blot analysis. A, pretreatment with 10 μm BAPTA/AM or 10 μm U73122, but not an inactive analog, U73343, abolished LPCAT2 phosphorylation. DMSO, dimethyl sulfoxide. B, pretreatment with the PKC inhibitor BIM-I inhibited mcPAF-induced LPCAT2 phosphorylation in a dose-dependent manner. 5 μm BIM-I was sufficient to achieve maximum inhibition. C, CHO-S-PAFR cells transfected with FLAG-LPCAT2 were preincubated with or without PKC inhibitors for 1 h and stimulated with 200 nm mcPAF for 30 s. Western blot analysis showed that each of the three different PKC inhibitors abolished the phosphorylation in a dose-dependent manner. Pretreatment with BIM-V, an inactive analog of BIM-I, had no effect. D, CHO-S-PAFR cells transfected with FLAG-LPCAT2 were pretreated with 100 nm TPA for 24 h and stimulated with mcPAF. TPA decreased PKCα protein and phosphorylation of LPCAT2 by mcPAF. E, pretreatment with TPA also resulted in reduced lyso-PAFAT activation following mcPAF stimulation, as measured by incorporation of acetyl-CoA into deuterium-labeled PAF. F and G, PKCα siRNA and FLAG-LPCAT2 were cotransfected into CHO-S-PAFR cells. Reduction of PKCα protein expression by its siRNA transfection was associated with reduced mcPAF-induced phosphorylation of LPCAT2 and lyso-PAFAT activation following mcPAF stimulation. Results are expressed as the mean + S.D. of an experiment performed in triplicate. NC, negative control. ***, p < 0.001; analysis of variance and Tukey's multiple comparison test. Three (A–C) or four (D--G) independent experiments were performed with similar results.
Next, we investigated kinase activation following PAFR signaling. Because previous reports showed that PKC is activated rapidly after PAFR stimulation (36, 37), the involvement of PKC in LPCAT2 phosphorylation was examined using the PKC inhibitor BIM-I (38). Peritoneal macrophages were pretreated with various concentrations of BIM-I (1–20 μm) for 30 min and then stimulated with mcPAF for 30 s. Microsomal proteins were analyzed by Western blot analysis using antibodies against total or phospho-LPCAT2. mcPAF-induced LPCAT2 phosphorylation was diminished by BIM-I pretreatment in a dose-dependent manner. Pretreatment with 5 μm BIM-I was sufficient to obtain this effect (Fig. 4B).
Other PKC-specific inhibitors (Ro-31-8220 and Ro-31-8425) also showed dose-dependent inhibition of LPCAT2 phosphorylation in CHO-S-PAFR cells transiently expressing FLAG-LPCAT2, whereas pretreatment with BIM-V, an inactive analog of BIM-I, had no effect (Fig. 4C). These compounds selectivity inhibit conventional PKC (cPKC) isozymes (39) over the Ca2+-independent PKC isozymes (40). We conclude that Ca2+ cPKC signaling regulates the phosphorylation of LPCAT2 following mcPAF stimulation.
Next, CHO-S-PAFR cells transiently expressing FLAG-LPCAT2 were pretreated with 100 nm TPA for 24 h. Consistent with previous reports (41, 42), prolonged exposure to TPA caused down-regulation of PKCα protein, whereas expression of LPCAT2 was not affected (Fig. 4D). The phosphorylation of LPCAT2 by mcPAF-stimulation was reduced by TPA treatment, accompanied by a decrease in lyso-PAFAT activity in vitro (Fig. 4E).
To further confirm the involvement of PKCα in phosphorylation of LPCAT2 at Ser-34, PKCα siRNA and FLAG-LPCAT2 were cotransfected into CHO-S-PAFR cells. PKCα protein was reduced by the knockdown, and mcPAF-induced phosphorylation of LPCAT2 was also decreased (Fig. 4F). We then examined the effect of PKCα siRNA on lyso-PAFAT activity by measuring the incorporation of acetyl-CoA into deuterium-labeled PAF. Knockdown of PKCα resulted in a significant decrease of lyso-PAFAT activation induced by mcPAF (Fig. 4G). From these data, we concluded that PKCα mediated the Ser-34 phosphorylation of LPCAT2 following mcPAFstimulation.
Activation and Phosphorylation of LPCAT2 by ATP Stimulation in RAW264.7 Cells Stably Expressing LPCAT2
In addition to mcPAF, we investigated the ability for other G protein-coupled receptor ligands to mediate phosphorylation of LPCAT2 in RAW264.7 cells. Treatment with ATP led to phosphorylation of endogenous LPCAT2 within 30 s (Fig. 5). To confirm that LPCAT2 mediates PAF production in these cells, LPCAT2 was knocked down in RAW264.7 cells stably expressing either vector (RAW-VEC) or LPCAT2 (RAW-LPCAT2) (16). LPCAT2 mRNA and protein were decreased 48 h after transfection of LPCAT2 siRNA (Fig. 5, A and B). Endogenous expression of LPCAT2 in RAW264.7 cells was much lower than FLAG-LPCAT2. Phosphorylation of LPCAT2 following ATP stimulation was diminished correspondingly in cells transfected with LPCAT2 siRNA (Fig. 5A). Lyso-PAFAT activity in cellular lysates was measured by incorporation of acetyl-CoA into deuterium-labeled PAF. RAW-LPCAT2 cells had higher basal lyso-PAFAT activity than RAW-VEC cells, and ATP-stimulation increased the activity (Fig. 5C). When LPCAT2 was knocked down, both RAW-VEC and RAW-LPCAT2 showed lower lyso-PAFAT activity (Fig. 5C).
FIGURE 5.

LPCAT2 is the responsible enzyme for rapid PAF production. RAW264.7 cells stably expressing vector (RAW-VEC) and FLAG-LPCAT2 (RAW-LPCAT2) were transfected with LPCAT2 siRNA by electroporation and incubated for 48 h. The cells were subsequently stimulated with 250 μm ATP for 30 s (B and C) or 1 min (D). A, the amount of mRNA of LPCAT2 was decreased by siRNA. NC, negative control. B, Western blot analysis showed decreased expression of both endogenous and FLAG-LPCAT2 by LPCAT2 siRNA. Signals from phospho-LPCAT2 were also reduced. C, lyso-PAFAT activity was measured by incorporation of acetyl-CoA into deuterium-labeled PAF. LPCAT2 siRNA reduced lyso-PAFAT activity in both RAW-VEC and RAW-LPCAT2. D, endogenous PAF production from each cellular extract was quantified using LC-MS/MS. Reduction of LPCAT2 protein expression by the siRNA was associated with decreased PAF production following ATP stimulation. Results are expressed as the mean + S.E. of three experiments (A and D) or mean + S.D. of an experiment performed in triplicate (C). *, p < 0.05; **, p < 0.01; ***, p < 0.001; analysis of variance and Tukey's multiple comparison test. The results (C) are representative of three independent experiments.
PAF production from RAW-VEC and RAW-LPCAT2 after 1 min of ATP stimulation was quantified by LC-MS/MS. The amount of PAF was elevated in RAW-LPCAT2 and increased upon ATP stimulation for 1 min. Knockdown of LPCAT2 by siRNA led to decreased LPCAT2 phosphorylation and PAF production (Fig. 5, B and D), compatible with lyso-PAFAT activity in vitro (Fig. 5C). From these data, we conclude that LPCAT2 is the enzyme that catalyzes the rapid PAF production following ATP stimulation.
PKCα Regulated the Phosphorylation of LPCAT2 following ATP Stimulation in RAW 264.7 Cells
To examine whether the PKC pathway also mediates ATP-induced LPCAT2 phosphorylation, non-transfected RAW264.7 cells were pretreated with PKC inhibitors and subsequently stimulated with 250 μm ATP for 30 s. The proteins were analyzed by Western blot analysis (Fig. 6A). Similar to the experiments using CHO-S-PAFR cells (Fig. 4), the inhibitors diminished phosphorylation of LPCAT2, which indicated that mcPAF and ATP utilize the same signaling pathway.
FIGURE 6.

PKCα mediates the phosphorylation of LPCAT2 following ATP stimulation. A, non-transfected RAW264.7 cells were pretreated with 10 μm BIM-I, Ro-31-8220, Ro-31-8425, or BIM-V, an inactive analog of BIM-I, and stimulated with 250 μm ATP for 30 s. The cellular proteins were analyzed by Western blot analysis. Pretreatment with BIM-I, Ro-31-8220, or Ro-31-8425, but not BIM-V, abolished LPCAT2 phosphorylation. DMSO, dimethyl sulfoxide. B, RAW-LPCAT2 cells were treated with 100 nm TPA for 24 h and subsequently stimulated with ATP. C, lyso-PAFAT activity was measured by incorporation of acetyl-CoA into deuterium-labeled PAF. Pretreatment with TPA decreased the activation of lyso-PAFAT activity. D, the cellular proteins were analyzed by Western blot analysis. siRNA-mediated knockdown of PKCα protein was associated with decreased phosphorylation of LPCAT2 following ATP-stimulation. NC, negative control. E, decreased PKCα expression resulted in reduced lyso-PAFAT activation following ATP stimulation. Results are expressed as the mean + S.D. of an experiment performed in triplicate. ***, p < 0.001; analysis of variance and Tukey's multiple comparison test. The results are representative of three independent experiments with similar results.
To down-regulate cPKC, RAW-LPCAT2 cells were treated with 100 nm TPA for 24 h and subsequently stimulated with 250 μm ATP for 30 s. Prolonged exposure to TPA treatment decreased the amount of PKCα protein (Fig. 6B). The phosphorylation of LPCAT2 by ATP stimulation was also reduced, accompanied by decrease in lyso-PAFAT activity in vitro (Fig. 6C). These two pharmacological experiments showed that PKCα mediated Ser-34 phosphorylation of LPCAT2.
We targeted PKCα by siRNA to examine the involvement of this isoform. Forty-eight hours after transfection with PKCα siRNA, the cells were treated with ATP for 30 s. PKCα protein was decreased by transfection of PKCα siRNA (Fig. 6D). Knockdown of PKCα led to reduced phosphorylation of LPCAT2 after ATP stimulation. However, some residual phosphorylation was still detected, possibly because of residual PKCα protein still present in the cell because of incomplete knockdown (Fig. 6D).
We then examined the lyso-PAFAT activity of LPCAT2 by measuring incorporation of acetyl-CoA into deuterium-labeled PAF. Knockdown of PKCα resulted in significant decrease of lyso-PAFAT activation after ATP stimulation (Fig. 6E). These data demonstrate that PKCα mediates the phosphorylation of LPCAT2 following ATP stimulation. However, a partial redundancy of other kinases cannot be fully excluded.
DISCUSSION
This study establishes that rapid PAF biosynthesis in response to mcPAF or ATP stimulation requires PKCα-dependent Ser-34 phosphorylation of LPCAT2 (Figs. 4 and 6), whereas phosphorylation of the same residue following 30 min of LPS stimulation is mediated by MK2, as reported previously (16).
Our results from stable expression and knockdown of LPCAT2 confirmed that this enzyme is responsible for rapid PAF production (Fig. 5). Our experiments using PKC inhibitors and TPA showed that activation of cPKC is essential for the Ser-34 phosphorylation of LPCAT2 by mcPAF or ATP (Figs. 4 and 6). Among the three PKC subfamilies (39), only cPKC requires Ca2+ for its activation, whereas neither novel PKC nor atypical PKC exhibit Ca2+ dependence. Our knockdown experiments established the positive involvement of PKCα on Ser-34 phosphorylation. However, a partial redundancy of other isoforms or kinases cannot be fully excluded in this study (Figs. 4 and 6).
Previously, we demonstrated that PAF production by LPCAT2 is up-regulated following LPS stimulation by two modes: phosphorylation of LPCAT2 at Ser-34 by MK2 occurs within 30 min (16), and LPCAT2 mRNA is increased by 16 h (9). LPS is a major component of the outer membrane of Gram-negative bacteria, and PAF production in response to bacterial infection likely functions in the proinflammatory reaction of host defense. Here, we established a new third pathway and mechanism to enhance PAF biosynthesis following stimulation by endogenous G protein-coupled receptor ligands within a shorter time period (30 s). It is possible that G protein-coupled receptor signaling works as the direct trigger for PAF production, whereas LPS-stimulation serves as preconditioning for the subsequent inflammatory state. Besides its many intracellular and intercellular functions, ATP functions as a damage-associated molecular pattern molecule and modulates immune responses following release from the cell during cellular stress or tissue injury (43). The novel pathway described here suggests roles of PAF during intrinsic events such as necrosis, apoptosis, and tissue damage by ischemia-reperfusion (44). Because PAF works as a chemoattractant (30), we hypothesize that rapid production of PAF by phosphorylated LPCAT2 in tissue macrophages recruits more immune and stromal cells to the lesion to repair damaged tissue (20). Because LPCAT2 was activated by its own product, PAF, a positive feedback mechanism for PAF production that may function at the onset of inflammation is indicated.
The regulation of LPCAT2 in macrophages implies stage-specific roles of PAF in a wide variety of proinflammatory responses from the innate to the adaptive immune systems. Rapid production of PAF by phosphorylated LPCAT2 may trigger inflammatory reactions such as increased blood vessel permeability or chemical mediator release at the onset of infections or type I allergy (30). PAF-stimulated LPCAT2 phosphorylation and endogenous PAF production decrease rapidly when activated in vitro (Fig. 2, B and C), suggesting that this role of PAF is required for a very short period, consistent with the rapid degradation of PAF by PAF acetyl hydrolase in the cytosol and plasma (10–12). Production and degradation of PAF seem strictly controlled within a limited area and time span, indicating that PAF may function as a trigger for subsequent events during pathophysiological circumstances.
On the other hand, the LPCAT activity of LPCAT2 was also increased during the same rapid time course (Fig. 3C). LPCAT2 selectively catalyzes the incorporation of polyunsaturated fatty acids, such as arachiconic acid, into 1-alkyl-PC at the sn-2 position. 1-alkyl-PC containing arachidonic acid is a major membrane phospholipid of inflammatory cells (14). The physiological roles of 1-alkyl-PC production by LPCAT2 remain to be elucidated, but it possibly regulates the properties of membrane phospholipids in response to the inflammatory stimuli. It is unclear whether incorporation of arachidonic acid into the cellular membranes either increases or decreases its bioavailability for subsequent eicosanoid production.
The data obtained in this study reveal a novel mechanism for PAF production that may function at the onset or early stages of inflammation. Because LPCAT2 may be a key enzyme balancing pro- and anti-inflammatory lipid metabolism, additional studies are necessary to understand its functions at various stages of inflammation, which may elucidate potential targets for medical intervention. For example, selective inhibition of the lyso-PAFAT activity of phosphorylated LPCAT2 might ameliorate excessive inflammation in response to exogenous or endogenous triggers. Thus, further in vivo studies are required to elucidate the roles of phosphorylated LPCAT2 under inflammatory conditions.
Acknowledgments
We thank Drs. K. Waku (Teikyo University); Drs. T. Hashidate, Y. Hishikawa, D. Hishikawa, T. Harayama, K. Yanagida, and W. Valentine (National Center for Global Health and Medicine); and M. Yamada (The University of Tokyo) for suggestions. We also thank Y. Takahashi (National Center for Global Health and Medicine) for technical support, Dr. W. Valentine for critical reading of the manuscript, and Dr. J. Miyazaki (Osaka University) for pCXN2.
This work was supported by a grant-in-aid for Scientific Research (S) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; by National Center for Global Health and Medicine grant 24-001, 25-201; and by the Takeda Science Foundation (to T. S.).
- PAF
- platelet-activating factor
- PAFR
- platelet-activating factor receptor
- 1-alkyl-PC
- 1-O-alkyl-2-acyl-sn-glycero-3-phosphocholine
- lyso-PAF
- lyso-PAF, 1-O-alkyl-sn-glycero-3-phosphocholine
- LPC
- lysophosphatidylcholine
- lyso-PAFAT
- acetyl-CoA:1-O-alkyl-sn-glycero-3-phosphocholine acetyltransferase
- LPCAT
- lysophosphatidylcholine acyltransferase
- PC
- phosphatidylcholine
- mcPAF
- methylcarbamyl platelet-activating factor
- BIM
- bisindolylmaleimide
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- cPKC
- conventional PKC
- MS/MS
- tandem mass spectrometry.
REFERENCES
- 1. Shimizu T. (2009) Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 49, 123–150 [DOI] [PubMed] [Google Scholar]
- 2. Prescott S. M., Zimmerman G. A., McIntyre T. M. (1990) Platelet-activating factor. J. Biol. Chem. 265, 17381–17384 [PubMed] [Google Scholar]
- 3. Tsujimura Y., Obata K., Mukai K., Shindou H., Yoshida M., Nishikado H., Kawano Y., Minegishi Y., Shimizu T., Karasuyama H. (2008) Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28, 581–589 [DOI] [PubMed] [Google Scholar]
- 4. Jönsson F., Mancardi D. A., Kita Y., Karasuyama H., Iannascoli B., Van Rooijen N., Shimizu T., Daëron M., Bruhns P. (2011) Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 121, 1484–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jönsson F., Mancardi D. A., Zhao W., Kita Y., Iannascoli B., Khun H., van Rooijen N., Shimizu T., Schwartz L. B., Daëron M., Bruhns P. (2012) Human FcγRIIA induces anaphylactic and allergic reactions. Blood 119, 2533–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uozumi N., Kume K., Nagase T., Nakatani N., Ishii S., Tashiro F., Komagata Y., Maki K., Ikuta K., Ouchi Y., Miyazaki J., Shimizu T. (1997) Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390, 618–622 [DOI] [PubMed] [Google Scholar]
- 7. Shindou H., Ishii S., Uozumi N., Shimizu T. (2000) Roles of cytosolic phospholipase A(2) and platelet-activating factor receptor in the Ca-induced biosynthesis of PAF. Biochem. Biophys. Res. Commun. 271, 812–817 [DOI] [PubMed] [Google Scholar]
- 8. Wykle R. L., Malone B., Snyder F. (1980) Enzymatic synthesis of 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine, a hypotensive and platelet-aggregating lipid. J. Biol. Chem. 255, 10256–10260 [PubMed] [Google Scholar]
- 9. Shindou H., Hishikawa D., Nakanishi H., Harayama T., Ishii S., Taguchi R., Shimizu T. (2007) A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J. Biol. Chem. 282, 6532–6539 [DOI] [PubMed] [Google Scholar]
- 10. Karasawa K., Kuge O., Kawasaki K., Nishijima M., Nakano Y., Tomita M., Yokoyama K., Setaka M., Nojima S. (1996) Cloning, expression and characterization of plasma platelet-activating factor-acetylhydrolase from guinea pig. J. Biochem. 120, 838–844 [DOI] [PubMed] [Google Scholar]
- 11. Hattori M., Adachi H., Aoki J., Tsujimoto M., Arai H., Inoue K. (1995) Cloning and expression of a cDNA encoding the β-subunit (30-kDa subunit) of bovine brain platelet-activating factor acetylhydrolase. J. Biol. Chem. 270, 31345–31352 [DOI] [PubMed] [Google Scholar]
- 12. Tjoelker L. W., Eberhardt C., Unger J., Trong H. L., Zimmerman G. A., McIntyre T. M., Stafforini D. M., Prescott S. M., Gray P. W. (1995) Plasma platelet-activating factor acetylhydrolase is a secreted phospholipase A2 with a catalytic triad. J. Biol. Chem. 270, 25481–25487 [DOI] [PubMed] [Google Scholar]
- 13. Yamashita A., Sugiura T., Waku K. (1997) Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J. Biochem. 122, 1–16 [DOI] [PubMed] [Google Scholar]
- 14. Shindou H., Shimizu T. (2009) Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 284, 1–5 [DOI] [PubMed] [Google Scholar]
- 15. Shindou H., Ishii S., Yamamoto M., Takeda K., Akira S., Shimizu T. (2005) Priming effect of lipopolysaccharide on acetyl-coenzyme A:lyso-platelet-activating factor acetyltransferase is MyD88 and TRIF independent. J. Immunol. 175, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 16. Morimoto R., Shindou H., Oda Y., Shimizu T. (2010) Phosphorylation of lysophosphatidylcholine acyltransferase 2 at Ser34 enhances platelet-activating factor production in endotoxin-stimulated macrophages. J. Biol. Chem. 285, 29857–29862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nieto M. L., Velasco S., Sanchez Crespo M. (1988) Biosynthesis of platelet-activating factor in human polymorphonuclear leukocytes. Involvement of the cholinephosphotransferase pathway in response to the phorbol esters. J. Biol. Chem. 263, 2217–2222 [PubMed] [Google Scholar]
- 18. Nieto M. L., Velasco S., Sanchez Crespo M. (1988) Modulation of acetyl-CoA:1-alkyl-2-lyso-sn-glycero-3-phosphocholine (lyso-PAF) acetyltransferase in human polymorphonuclears. The role of cyclic AMP-dependent and phospholipid sensitive, calcium-dependent protein kinases. J. Biol. Chem. 263, 4607–4611 [PubMed] [Google Scholar]
- 19. Nixon A. B., O'Flaherty J. T., Salyer J. K., Wykle R. L. (1999) Acetyl-CoA:1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase is directly activated by p38 kinase. J. Biol. Chem. 274, 5469–5473 [DOI] [PubMed] [Google Scholar]
- 20. Kume K., Shimizu T. (1997) Platelet-activating factor (PAF) induces growth stimulation, inhibition, and suppression of oncogenic transformation in NRK cells overexpressing the PAF receptor. J. Biol. Chem. 272, 22898–22904 [DOI] [PubMed] [Google Scholar]
- 21. Ishii S., Kihara Y., Shimizu T. (2005) Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J. Biol. Chem. 280, 9083–9087 [DOI] [PubMed] [Google Scholar]
- 22. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 23. Hishikawa D., Shindou H., Kobayashi S., Nakanishi H., Taguchi R., Shimizu T. (2008) Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. U.S.A. 105, 2830–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 25. Harayama T., Shindou H., Ogasawara R., Suwabe A., Shimizu T. (2008) Identification of a novel noninflammatory biosynthetic pathway of platelet-activating factor. J. Biol. Chem. 283, 11097–11106 [DOI] [PubMed] [Google Scholar]
- 26. Kita Y., Takahashi T., Uozumi N., Shimizu T. (2005) A multiplex quantitation method for eicosanoids and platelet-activating factor using column-switching reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Biochem. 342, 134–143 [DOI] [PubMed] [Google Scholar]
- 27. Tokuoka S. M., Kita Y., Shindou H., Shimizu T. (2013) Alkylglycerol monooxygenase as a potential modulator for PAF synthesis in macrophages. Biochem. Biophys. Res. Commun. 436, 306–312 [DOI] [PubMed] [Google Scholar]
- 28. Koeberle A., Shindou H., Harayama T., Shimizu T. (2010) Role of lysophosphatidic acid acyltransferase 3 for the supply of highly polyunsaturated fatty acids in TM4 Sertoli cells. FASEB J. 24, 4929–4938 [DOI] [PubMed] [Google Scholar]
- 29. Koeberle A., Shindou H., Harayama T., Yuki K., Shimizu T. (2012) Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J. 26, 169–180 [DOI] [PubMed] [Google Scholar]
- 30. Ishii S., Shimizu T. (2000) Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog. Lipid. Res. 39, 41–82 [DOI] [PubMed] [Google Scholar]
- 31. Rhee S. G. (2001) Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70, 281–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singer W. D., Brown H. A., Sternweis P. C. (1997) Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 66, 475–509 [DOI] [PubMed] [Google Scholar]
- 33. Tsien R. Y. (1980) New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry 19, 2396–2404 [DOI] [PubMed] [Google Scholar]
- 34. Tsien R. Y. (1981) A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290, 527–528 [DOI] [PubMed] [Google Scholar]
- 35. Thompson A. K., Mostafapour S. P., Denlinger L. C., Bleasdale J. E., Fisher S. K. (1991) The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation. J. Biol. Chem. 266, 23856–23862 [PubMed] [Google Scholar]
- 36. Bussolino F., Silvagno F., Garbarino G., Costamagna C., Sanavio F., Arese M., Soldi R., Aglietta M., Pescarmona G., Camussi G. (1994) Human endothelial cells are targets for platelet-activating factor (PAF). Activation of α and β protein kinase C isozymes in endothelial cells stimulated by PAF. J. Biol. Chem. 269, 2877–2886 [PubMed] [Google Scholar]
- 37. James-Kracke M. R., Sexe R. B., Shukla S. D. (1994) Picomolar platelet-activating factor mobilizes Ca to change platelet shape without activating phospholipase C or protein kinase C: simultaneous fluorometric measurement of intracellular free Ca concentration and aggregation. J. Pharmacol. Exp. Ther. 271, 824–831 [PubMed] [Google Scholar]
- 38. Martiny-Baron G., Kazanietz M. G., Mischak H., Blumberg P. M., Kochs G., Hug H., Marmé D., Schächtele C. (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J. Biol. Chem. 268, 9194–9197 [PubMed] [Google Scholar]
- 39. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilkinson S. E., Parker P. J., Nixon J. S. (1993) Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J. 294, 335–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ballester R., Rosen O. M. (1985) Fate of immunoprecipitable protein kinase C in GH3 cells treated with phorbol 12-myristate 13-acetate. J. Biol. Chem. 260, 15194–15199 [PubMed] [Google Scholar]
- 42. Ohno S., Konno Y., Akita Y., Yano A., Suzuki K. (1990) A point mutation at the putative ATP-binding site of protein kinase C α abolishes the kinase activity and renders it down-regulation-insensitive. A molecular link between autophosphorylation and down-regulation. J. Biol. Chem. 265, 6296–6300 [PubMed] [Google Scholar]
- 43. Rock K. L., Kono H. (2008) The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bitencourt C. S., Bessi V. L., Huynh D. N., Ménard L., Lefebvre J. S., Lévesque T., Hamdan L., Sohouhenou F., Faccioli L. H., Borgeat P., Marleau S. (2013) Cooperative role of endogenous leucotrienes and platelet-activating factor in ischaemia-reperfusion-mediated tissue injury. J. Cell Mol. Med. 17, 1554–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]