

Background: Actively respiring brain mitochondria can consume H2O2 through thioredoxin/peroxiredoxin (Trx/Prx).
Results: Inhibition of nicotinamide nucleotide transhydrogenase (Nnt) decreases NADPH levels, decreases Trx and Trx reductase activity, and increases toxicity to oxidative stress.
Conclusion: Nnt links mitochondrial respiration and antioxidant activity in brain mitochondria.
Significance: Nnt may be a therapeutic target to increase the antioxidant activity in cells.
Keywords: Mitochondria, Oxidative Stress, Parkinson Disease, Reactive Oxygen Species (ROS), Thioredoxin Reductase, Nicotinamide Nucleotide Transhydrogenase
Abstract
Mitochondrial reactive oxygen species are implicated in the etiology of multiple neurodegenerative diseases, including Parkinson disease. Mitochondria are known to be net producers of ROS, but recently we have shown that brain mitochondria can consume mitochondrial hydrogen peroxide (H2O2) in a respiration-dependent manner predominantly by the thioredoxin/peroxiredoxin system. Here, we sought to determine the mechanism linking mitochondrial respiration with H2O2 catabolism in brain mitochondria and dopaminergic cells. We hypothesized that nicotinamide nucleotide transhydrogenase (Nnt), which utilizes the proton gradient to generate NADPH from NADH and NADP+, provides the link between mitochondrial respiration and H2O2 detoxification through the thioredoxin/peroxiredoxin system. Pharmacological inhibition of Nnt in isolated brain mitochondria significantly decreased their ability to consume H2O2 in the presence, but not absence, of respiration substrates. Nnt inhibition in liver mitochondria, which do not require substrates to detoxify H2O2, had no effect. Pharmacological inhibition or lentiviral knockdown of Nnt in N27 dopaminergic cells (a) decreased H2O2 catabolism, (b) decreased NADPH and increased NADP+ levels, and (c) decreased basal, spare, and maximal mitochondrial oxygen consumption rates. Nnt-deficient cells possessed higher levels of oxidized mitochondrial Prx, which rendered them more susceptible to steady-state increases in H2O2 and cell death following exposure to subtoxic levels of paraquat. These data implicate Nnt as the critical link between the metabolic and H2O2 antioxidant function in brain mitochondria and suggests Nnt as a potential therapeutic target to improve the redox balance in conditions of oxidative stress associated with neurodegenerative diseases.
Introduction
Mitochondrial reactive oxygen species (ROS)2 play important roles in physiological cell signaling as well as pathological degeneration associated with neurodegenerative diseases such as Parkinson disease (PD) (1–3). Brain mitochondria have multiple antioxidant pathways that help maintain the balance between ROS production (for signaling pathways) and consumption/catabolism (to prevent oxidative damage). Mitochondrial superoxide (O2⨪) radicals are efficiently converted to hydrogen peroxide (H2O2) by spontaneous dismutation and/or enzymatic reaction by manganese superoxide dismutase (1, 4, 5). Additionally, mitochondrial H2O2 can be produced by citric acid cycle enzymes such as aconitase and α-ketoglutrate dehydrogenase (6–8). H2O2 is detoxified to water by the thioredoxin/peroxiredoxin (Trx/Prx) pathway or by glutathione peroxidase, which is part of the glutathione (GSH) pathway. Although mitochondria are well known to produce oxygen radicals (2, 9, 10) and nitric oxide (11, 12), studies in the literature, including our laboratory, have demonstrated that isolated brain mitochondria are capable of consuming exogenously added H2O2 in a respiration-dependent manner (13, 14). We demonstrated that brain mitochondria remove H2O2 when the respiration substrates malate and glutamate (which feed NADH into complex I of the electron transport chain) and to a lesser extent succinate (feeds FADH2 to complex II) were present (14). In contrast, isolated liver mitochondria do not require respiration substrates for H2O2 removal (14). We demonstrated for the first time that ∼80% of the mitochondrial H2O2 catabolism occurred via the Trx/Prx pathway and only ∼25% by the GSH pathway, which was previously implicated as the main detoxification pathway in this process (13, 14). It was also a curious finding that H2O2 catabolism by brain, but not liver mitochondria, required respiration substrates.
The specific goal of this study was to determine the mechanistic link that couples respiration/substrate utilization with mitochondrial H2O2 antioxidant activity via Trx/Prx. A prime candidate for such a link would preferably generate NADPH because H2O2 detoxification via either GSH or Trx/Prx utilizes NADPH to maintain the pathways in a reduced state but catalase (present in liver, but not brain mitochondria) and manganese superoxide dismutase do not (4, 15). Approximately 45% of NADPH in the mitochondria is produced by nicotinamide nucleotide transhydrogenase (Nnt) and the remainder comes from NADP+-isocitrate dehydrogenase 2 or mitochondrial NAD(P)-malic enzyme (16, 17). What separates Nnt from the other mechanisms of NADPH production is it utilizes the proton gradient generated through the citric acid cycle and the electron transport chain to convert NADP+ into NADPH (18, 19). The role of Nnt in mitochondrial redox regulation has been demonstrated previously in studies, which showed that Nnt inhibition decreased GSH redox status and increased sensitivity to oxidative stress; however, its role in modulating the Trx/Prx system in brain mitochondria remains unknown (16, 20–22).
We hypothesized that Nnt activity links the ability of the mitochondria to remove H2O2 in a substrate- and respiration-dependent manner to the Trx/Prx antioxidant system. This hypothesis predicts that the presence of Nnt is essential for mitochondrial defense against normal and low levels of oxidative stress as supported by previous research, demonstrating that defects in the Nnt gene could be a potential risk for increased mitochondrial vulnerability (19–21). Here, we show that inhibition of Nnt results in decreased 1) mitochondrial H2O2 removal rates, 2) cellular NADPH levels, 3) TrxR and Trx activity in isolated mitochondria, and increased 4) mitochondrial Prx oxidation and 5) sensitivity to ROS production and oxidative cell death. These data implicate Nnt as the link between mitochondrial respiration and H2O2 catabolism via the Trx/Prx antioxidant pathway.
EXPERIMENTAL PROCEDURES
Chemical Reagents
All chemicals and reagents were obtained from Sigma-Aldrich unless otherwise noted.
Isolation of Pure Rat Liver and Brain Mitochondria
All animal experiments were performed with approval from the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus. Mitochondria were isolated from adult male Sprague-Dawley rats as described previously through a Percoll density gradient centrifugation (14, 23, 24). For liver mitochondria, a crude isolation was conducted first as described previously by Salvi et al. (25) and then subjected to the Percoll gradient similar to brain mitochondria. Purity of mitochondrial and cytosolic fractions was confirmed by Western blotting for actin, lamin B, and complex IV (see Fig. 1A). Protein levels were determined by the Bradford assay.
FIGURE 1.

Pharmacological inhibition of Nnt results in substrate-dependent decreases in TrxR and Trx activity in isolated brain mitochondria. A, a representative Western blot to indicate the purity of isolated mitochondria conducted in these studies. Trx (B) or TrxR (C) activity assay was conducted as described under “Experimental Procedures.” There was a significant decrease in TrxR and Trx activity when respiration substrates malate and glutamate (M&G) were present, and Nnt was pharmacologically inhibited with palmitoyl-CoA (Palm). Data are represented as mean ± S.E. (n = 4–11). *, p < 0.05; **, p < 0.005; ***, p < 0.001 as determined by one-way ANOVA.
Isolation of Crude Rat Brain Mitochondria
Adult male Sprague-Dawley rat brains were homogenized in isolation buffer, and the homogenate was centrifuged at 3,000 rpm at 4 °C for 10 min. Supernatant was removed and centrifuged at 13,000 rpm at 4 °C for 10 min. Supernatant was removed and saved as the cytosolic fraction, and mitochondrial pellet was resuspended in desired assay buffer, and protein levels were determined by Bradford protein assay.
Cell Culture
N27 immortalized rat dopaminergic cells were a kind gift from Drs. Curt Freed and Kedar Prasad at the University of Colorado, Anschutz Medical Campus (26). Cell culture reagents were purchased from Invitrogen. N27 cells were grown and plated as reported previously (26, 27). All experiments were conducted in cell passages 3–10.
Nnt Knockdown
Nnt levels were inhibited in N27 cells using SMART vector 2.0 lentiviral shRNA particles according to the manufacturer's instructions (Thermo Scientific Dharmacon, Lafayette, CO) and as outlined previously in Lopert et al. (28). Dharmacon provided three predesigned gene-specific shRNA lentiviral particles, and all three were screened for transfection efficiency. The best lentiviral particle was used for the remainder of the experiments (GGAGTATCCACATTTCGCA).
Real-time PCR
According to the manufacturer's instructions, RNA from transfected N27 cells was isolated using the RNeasy kit® (Qiagen, Valencia, CA). RNA was quantified through 260/280 wavelength measurement by a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Pure RNA was reverse transcribed using the high capacity cDNA reverse transcription kit according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). Real-time PCR was performed on an Applied Biosystems 7500 Fast Real-time PCR system. Primers and probes for rat 18 S, Nnt, TrxR2, and TrxR1 were purchased from Applied Biosystems.
Isocitrate Dehydrogenase Activity Assay
Isocitrate dehydrogenase 2 activity was determined as described previously (29, 30). Cells were collected and lysed in 0.01% (v/v) Triton X-100, and protein levels were determined by Bradford protein assay. 500 μg of cell solution was used to initiate the reaction, and the reduction of NADP+ to NADPH was measured in a 1-ml cuvette spectophotometrically at 25 °C for 3 min.
TrxR and Trx Activity Assay
TrxR and Trx activity was measured in isolated pure rat brain mitochondria using an insulin reduction assay in the presence of Escherichia coli Trx or rat TrxR as described previously by Arnér et al. (31) with slight modification. 25 μg of isolated mitochondria were plated and exposed to respiration substrates, palmitoyl-CoA or water for 15 min. After incubation, mitochondria were lysed with 0.01% (v/v) Triton X-100 added to the assay buffer without exogenously added NADPH. After a 1-h incubation, the number of reduced thiols was determined on a Versamax microplate reader (Molecular Devices, Sunnyvale, CA).
Polarographic Measurement of Exogenous H2O2 Removal
H2O2 removal rates were measured in 1 × 106 cells per sample or 100 μg of isolated pure rat brain or liver mitochondria (as determined by Bradford assay) using a 100 μm Clark-type electrode with an Apollo 4000 Free Radical Analyzer (World Precision Instruments, Inc., Sarasota, FL). Measurements were conducted as described previously by Drechsel et al. (14). Briefly, respiration substrates and/or pharmacological inhibitors were added, and a stable H2O2 removal rate was measured. Next, mitochondria or cells were added to the chamber, and H2O2 removal rates were calculated based on the linear signal decay after the addition of mitochondria or cells compared with rates with respiration substrates or pharmacological inhibitors.
Nnt Activity Assay
Nnt activity was measured on Shimadzu UV-2401PC UV-visible recording spectrophotometer (Kyoto, Japan) as described previously by Rydström and Shimomura et al. (32, 33). Briefly isolated mitochondria or N27 cells were suspended in an activity buffer, and 300 μm APAD and NADPH plus pharmacological inhibitors were added. Nnt activity was measured over a 3-min linear decay period with a dual wavelength of 375 nm and 400 nm (reference) in a 1-ml cuvette at 37 °C.
HPLC to Measure NADH, NAD+, NADPH, and NADP+
The concentration of NADP+, NAD+, NADH, and NADPH in cells was measured using a HPLC system with spectrophotometer detection (Elite LaChrom System; Hitachi) following the method described previously (34) with a slight modification. The reduced forms were extracted by 0.25 m KOH (basic condition) and then passed through a Millipore ultrafree Eppendorf filtration system (Millipore, Bedford, MA) to eliminate proteins then 1 m KH2PO4 (one-fourth volume) was added to neutralize pH. The oxidized forms were extracted by 0.1 n perchloric acid (acidic condition) and centrifuged at 16,000 × g for 10 min at 4 °C to precipitate protein. The pyridine dinucleotides were then separated with a YMC-Pack ODS-A column (4.6 × 250 mm, 5 μm, Waters, Milford, MA), and the mobile phase was 100 mm potassium phosphate (pH 6.0) and 5% (v/v) methanol for the reduced forms and 100 mm potassium phosphate (pH 5.0) and 5% (v/v) methanol for the oxidized forms. NADPH and NADH were measured by the absorbance at 340 nm and NADP+ and NAD+ at 254 nm.
HPLC to Determine GSH and Glutathione Disulfide (GSSG) Levels
GSH assay was performed with an ESA 5600 CoulArray HPLC equipped with eight electrochemical cells as described previously by Liang and Patel (35). The potentials of the electrochemical cells were set, and analyte separation was conducted on a TOSOHAAS (Montgomeryville, PA) reverse-phase ODS 80-TM C-18 analytical column (4.6 mm × 250 cm; 5-μm particle size). A two-component gradient elution system was used with component A of the mobile phase composed of 50 mm NaH2PO4, pH 3.2, and component B composed of 50 mm NaH2PO4 and 40% methanol, pH 3.2. An aliquot (40 μl) of the supernatant was injected into the HPLC.
Western Blot
Total protein levels were measured using standard SDS-PAGE Western blots with 15 μg of mitochondria, cytosol, or cell extract. Actin was purchased from Abcam (Cambridge, MA) and used at 1:10,000. Lamin B was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and complex IV was purchased from Mitosciences (Eugene, OR) and diluted to 1:1,000. Goat anti-rabbit secondary was purchased from Abcam and used at 1:10,000. Antibodies were detected using enhanced chemiluminescence (Thermo Fisher Scientific), and bands were imaged on a Storm Optical Scanner (Molecular Dynamics, Inc., Sunnyvale, CA).
Peroxiredoxin 3 (Prx3) Redox Blot
The redox state of Prx3 was conducted as described previously by Cox et al. (36) and optimized in rat N27 cells. Prx3 primary antibody was purchased from Abcam, and secondary anti-mouse antibody was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA) and used at 1:2,500 and 1:10,000, respectively. Bands were quantified using densitometry with NIH ImageJ software (Bethesda, MD).
Measurement of Oxygen Consumption Rate (OCR)
Oxygen consumption rates were determined using a Seahorse XF24 analyzer (Seahorse Biosciences, North Billerica, MA) and conducted as outlined previously in Cantu et al. (27) and Lopert et al. (28).
Mitochondrial Membrane Potential Measurement
Tetramethylrhodamine ethyl ester (Invitrogen) was conducted as described in the manufacturer's instructions (Abcam) with a slight modification. 2.0 × 104 cells/well were seeded and allowed to grow overnight. Medium was removed and replaced with RPMI along with 20 μm carbonyl cyanide-p-trifluoromethoxyphenylhydrazone as a positive control. After 10 min of incubation, 1 μm tetramethylrhodamine ethyl ester was added and incubated for 20 min. After incubation cells were washed three times with warmed PBS supplemented with 0.2% (w/v) BSA and immediately read on SynergyTM multi-mode microplate reader. All values were normalized to protein as determined by a Bradford assay.
Detection of H2O2
H2O2 levels were measured using the Amplex Red fluorescence assay (Invitrogen) (27, 28) using a SynergyTM multi-mode microplate reader (Biotek).
Cell Death Assessment
Cells were plated in 96-well plate at 2.0 × 104 cells/well. Medium and cell lysis (Tris/NaCl + 0.1% (v/v) Triton X-100) samples were collected, and lactate dehydrogenase levels was measured by measuring the release of lactate dehydrogenase enzyme activity as described by Bergmeyer et al. (37)and published previously (28).
Statistical Methods
Data were analyzed in GraphPad Prism software (version 5; San Diego, CA). One-way ANOVA was used to test differences between multiple groups with a Bonferroni post test. To determine statistical significance between two groups a two-tailed Student's t test was utilized.
RESULTS
H2O2 Removal Is Linked to Nnt Activity in Isolated Brain Mitochondria
To implicate Nnt in mitochondrial H2O2 catabolism, we asked whether pharmacological inhibition of Nnt directly affected the rates of H2O2 removal in isolated mitochondria and whether this occurred in a respiration-dependent manner. A polarographic method using a Clark-type electrode was used to measure real-time H2O2 levels in isolated brain mitochondria. Isolated pure brain mitochondria (Fig. 1A) had Nnt inhibited with the pharmacological inhibitor palmitoyl-CoA (38) and exposed to exogenous H2O2 with and without respiration substrates present. As outlined in Table 1, isolated brain mitochondria with no addition of respiration substrates (state 1) had very little H2O2 removal. However, stimulation with the respiration substrates malate and glutamate (state 2) resulted in a significant increase in the ability of mitochondria to remove exogenous H2O2. When 100 μm palmitoyl-CoA was added to actively respiring mitochondria, there was a significant decrease in the rate of H2O2 removal back to the rates of non-stimulated mitochondria. Addition of palmitoyl-CoA had no additional effect on respiration when mitochondria were in state 1. These data demonstrate that Nnt activity controls respiration-dependent H2O2 catabolism by isolated brain mitochondria.
TABLE 1.
Decreased H2O2 removal rates in isolated brain mitochondria but not liver mitochondria with Nnt inhibited pharmacologically
H2O2 removal/catabolism rates were determined with an H2O2-sensitive Clark-type electrode. Isolated brain mitochondria required respiration substrates to consume H2O2, whereas liver mitochondria did not. Addition of palmitoyl-CoA affected brain but did not affect liver mitochondrial H2O2 removal. Data are represented as mean ± S.E. (n = 3–11).
| Substrate/Condition | Rate (nmol/min) |
|---|---|
| Brain mitochondria | |
| No substrates | 3.6 ± 0.6 |
| Malate/glutamate | 19.8 ± 0.9a |
| 100 μm palmitoyl CoA | 2.3 ± 0.7 |
| Malate/glutamate + 100 μm Palmitoyl CoA | 2.6 ± 1.0b |
| Liver mitochondria | |
| No substrates | 128 ± 4 |
| Malate/glutamate | 121 ± 4 |
| 100 μm palmitoyl CoA | 124 ± 4 |
| Malate/glutamate + 100 μm palmitoyl CoA | 119 ± 4 |
a p < 0.0001 versus no substrates.
b p < 0.0001 versus malate + glutamate only as determined by one-way ANOVA.
Nnt Activity Is Not Required for H2O2 Removal in Liver Mitochondria
We have shown previously that in contrast to brain mitochondria, isolated liver mitochondria remove H2O2 in a non-respiration-dependent manner (14). To further establish Nnt activity as a mechanistic link between respiration-dependent H2O2 removal in brain mitochondria, we sought to determine the consequence of Nnt inhibition on H2O2 removal by liver mitochondria. We hypothesized that because liver mitochondria remove H2O2 in a respiration- or substrate-independent manner, Nnt inhibition would not alter the rate of removal. As outlined in Table 1, isolated liver mitochondria alone consumed H2O2 at the same rate as when malate and glutamate were present. Additionally, addition of palmitoyl-CoA did not decrease H2O2 removal. As a control aminotriazole, an inhibitor of catalase (that is not NADPH-dependent), was added, and there was a significant decrease (∼45%) in activity (data not shown). The inability of Nnt inhibition to alter H2O2 removal rates in liver mitochondria confirms the substrate independence of this process and suggests that Nnt activity selectively plays a critical role in actively respiring brain ability of mitochondria to remove exogenous H2O2.
Pharmacological Inhibition of Nnt Results in Respiration-dependent Decreases in Trx and TrxR Activity
As indicated previously, isolated pure brain mitochondria primarily consume H2O2 by the Trx/Prx system (14). To determine whether the decrease in H2O2 removal in isolated mitochondria following pharmacological inhibition of Nnt was due to a decrease in Trx/TrxR activity, Trx and TrxR activity was measured in isolated brain mitochondria incubated with malate, glutamate, and palmitoyl-CoA. As indicated in Fig. 1, addition of malate and glutamate caused a significant increase in TrxR activity (∼300%). When 100 μm palmitoyl-CoA was added, there was no change in Trx or TrxR activity when mitochondria were in state 1. However, when malate and glutamate were present and Nnt was inhibited, there was a significant decrease in Trx and TrxR activity (∼83 and ∼95%, respectively), indicating Trx and TrxR activity is linked to Nnt activity in actively respiring mitochondria. Additionally, the purity of mitochondria was confirmed by Western blot for complex IV (mitochondrial), lamin B (nuclear), and actin (cytosol) as depicted as a representative blot in Fig. 1A.
Pharmacological Inhibition of Nnt in N27 Cells Results in Decreased H2O2 Removal Rates
Based on the results obtained in isolated mitochondria, the next step was to determine the effects of pharmacological inhibition of Nnt in intact cells. N27 dopaminergic cells exposed to 100 μm palmitoyl-CoA had a ∼48% decrease in Nnt activity (Fig. 2A). The decrease in Nnt activity resulted in a significant decrease in the ability of the cell to remove exogenous H2O2 by 22 ± 2% (Fig. 2B). This indicates that pharmacological inhibition of Nnt in a dopaminergic cellular model also results in a decrease in H2O2 catabolism.
FIGURE 2.

N27 cells with Nnt pharmacologically inhibited results in a decrease in H2O2 catabolism. N27 cells were exposed to 100 μm palmitoyl-CoA, and H2O2 removal rate was measured. A, there was a significant decrease in Nnt activity (n = 7–9). B, N27 cells exposed to 100 μm palmitoyl-CoA had a significant decrease in the ability to consume 3 μm exogenous H2O2 (n = 5–6). Data are represented as mean ± S.E. *, p < 0.05; ***, p < 0.001 as determined by Student's t test.
Constitutive Knockdown of Nnt in N27 Cells
To confirm the role of Nnt without the use of pharmacological agents with potential off-target effects in a cell model, N27 cells were transfected with a shRNA construct knocking down Nnt expression (Nnt-deficient) or a non-targeting control (mock). As indicated in Fig. 3, transfection with a shRNA targeting vector for Nnt resulted in a 94.1 ± 0.4% decrease in Nnt mRNA levels (Fig. 3A) corresponding to a 74.4 ± 5.7% decrease in Nnt activity (Fig. 3B).
FIGURE 3.

Generation of Nnt-deficient cell line. N27 cells were transfected with Nnt shRNA (Nnt-deficient) and compared with mock-transfected cells (mock). A, Nnt mRNA expression in Nnt-deficient cells had an ∼95% decrease in Nnt mRNA compared with mock-transfected cells (n = 3). B, Nnt activity was measured in mock control and Nnt-deficient cells, and there was an ∼75% decrease in activity (n = 9). All bars represent mean ± S.E.; ***, p < 0.0001 (Student's t test).
Nnt-deficient Cells Have Decreased NADPH and Increased NADP+ Levels Resulting in Decreased H2O2 Removal Rates and GSH Levels
To determine whether decreased Nnt activity resulted in a decrease in NADPH levels (the reducing equivalent used by TrxR to help detoxify H2O2 and generated by Nnt) NADPH and NADP+ levels were measured in cells via a HPLC method. Fig. 4 shows that Nnt-deficient cells had a significant decrease in NADPH levels (∼25%), which correlated with a significant increase in NADP+ levels (∼30%) compared with mock control cells. The increase in NADP+ levels paired with the decrease in NADPH levels resulted in a significant increase in NADP+/NADPH ratios by ∼75%, indicating the Nnt-deficient cells have a more oxidized NADP+/NADPH intracellular environment (Fig. 4C). To determine whether the effects observed with pharmacological inhibition in N27 cells was specific to Nnt inhibition and that the decrease in NADPH levels in Nnt-deficient cells resulted in changes in H2O2 catabolism, Nnt-deficient cells were tested for exogenous H2O2 removal rates compared with mock controls. As depicted in Fig. 4D, Nnt-deficient cells showed significant decreases (∼40%) in their ability to remove H2O2 compared with the mock controls. The magnitude of the decrease was similar to that observed in isolated mitochondria and N27 cells treated with palmitoyl-CoA. Additionally, GSH and GSSG levels were measured by an HPLC method to determine whether the NADPH deficiency in Nnt KO cells resulted in changes in the GSH system (another NADPH-utilizing pathway). Indeed, Nnt-deficient cells had significantly higher basal GSSG and concomitantly decreased GSH levels compared with mock controls (Fig. 4, E and F), consistent with the decrease in NADPH levels.
FIGURE 4.

Nnt-deficient N27 cells have increased NADP+ and decreased NADPH levels, H2O2 removal rates, and GSH levels. NADP+ levels (A) and NADPH levels (B) were determined in Nnt-deficient cells and compared with mock control via HPLC. There was a significant increase in NADP+ levels coupled with a decrease in NADPH levels. Each individual NADP+ value was divided by the average NADPH value to determine the ratio (C), which was significantly oxidized in the Nnt-deficient cells (n = 4–9). D, Nnt-deficient cells showed an ∼40% decrease in the ability to remove exogenous H2O2 (n = 10). Additionally, Nnt-deficient cells showed a decrease in GSH levels (E) and an increase in GSSG level (F; n = 3). G, there was no change in isocitrate dehydrogenase 2 (IDH) activity (n = 5–8). *, p < 0.05; **, p < 0.005; ***, p < 0.0005 (Student's t test). Data are represented as mean ± S.E.
To determine whether Nnt knockdown and resultant decrease of NADPH levels caused alterations in enzymatic activities of other NADPH-producing enzymes, we measured isocitrate dehydrogenase 2 activity. No changes in isocitrate dehydrogenase 2 activity was observed in Nnt knockdown compared with mock controls (Fig. 4G).
Nnt-deficient Cells Have More Oxidized Mitochondrial Prx3 Than Mock Controls
To determine whether the decrease in NADPH levels in Nnt-deficient cells resulted in a more oxidized Trx/Prx system, a redox blot for Prx3 was conducted in mock and Nnt-deficient cells. As shown in Fig. 5, Nnt-deficient cells had significantly more (∼40%) oxidation of Prx3 compared with mock controls. This indicates that the decrease in TrxR activity in the Nnt-deficient cells results in more oxidized Prx3. Total Prx3 levels was measured in Nnt deficient and mock cells to determine whether the increased oxidation was due to a decrease in total protein levels. As shown in Fig. 5D, total Prx3 levels did not differ significantly between control and Nnt-deficient cells.
FIGURE 5.
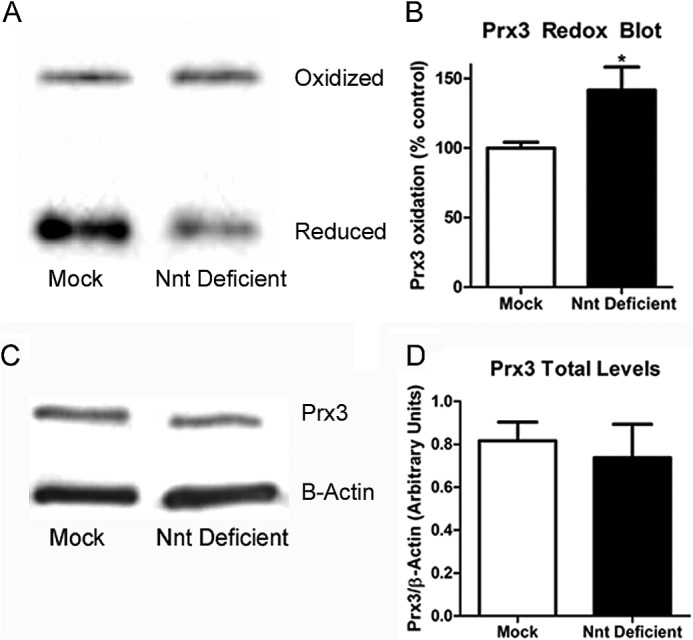
Nnt-deficient N27 cells had a significant increase in oxidation of Prx3 compared with mock controls. A, a representative oxidized versus reduced Western blot. B, quantification of the redox blot for mitochondrial Prx3 from five separate samples. C, a representative SDS-PAGE blot for total Prx3 levels in mock and Nnt-deficient cells. D, quantification from four to five separate samples normalized to β-actin levels for total levels of Prx3. *, p < 0.05 as determined by Student's t test. Bars represent mean ± S.E.
Decreased Mitochondrial OCR Occurs in Nnt-deficient Cells
To examine whether Nnt inhibition, which results in decreased NADPH levels and more oxidized Prx3, leads to mitochondrial dysfunction, we measured mitochondrial OCR as well as other bioenergetic parameters in Nnt-deficient cells and mock controls using an XF24 analyzer (Seahorse Biosciences). As outlined in the summary trace in Fig. 6 and detailed in Table 2, Nnt-deficient cells showed significant decreases in baseline respiration, ATP turnover, respiratory capacity, reserve respiratory capacity, and glycolysis. These data suggest that Nnt knockdown per se results in mitochondrial dysfunction based on decreased baseline OCR as well as maximal and reserve respiratory capacity.
FIGURE 6.

Summarized trace of decreased OCR in Nnt deficient cells compared with mock cells. Using a Seahorse 24XF analyzer, mitochondrial OCR was measured in Nnt-deficient and mock control N27 cells under different respiratory parameters. Data are represented as mean ± S.E. (n = 18–19). Oligo, oligonucleotide; FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; Anti-A, antimyicin A.
TABLE 2.
Summarized table of OCR parameters in Nnt-deficient cells and mock controls
Nnt-deficient cells had significant decreases in baseline OCR rates along with ATP turnover, reserve respiratory capacity, and maximal respiratory capacity compared with mock controls (n = 18–19). *, p < 0.05; ***, p < 0.0001 as determined by Student's t test. Data are represented as mean ± S.E.
| Parameter OCR (pmol/min)/(mg/ml) | Mock | Nnt-deficient |
|---|---|---|
| Baseline | 399 ± 22 | 228 ± 22*** |
| ATP turnover | 345 ± 17 | 154 ± 17*** |
| Proton leak | 52 ± 9 | 75 ± 11 |
| Reserve capacity | 443 ± 20 | 343 ± 37* |
| Respiratory capacity | 841 ± 36 | 558 ± 56*** |
Nnt-deficient Cells Are More Susceptible to Oxidative Stress
To determine whether the changes in mitochondrial antioxidant activity and resulting mitochondrial function leads to increased susceptibility to oxidative stress, the mitochondrial membrane potential was measured. Additionally, to test the role of Nnt in an in vitro model of PD, cells were exposed to various levels of the known PD toxicant paraquat. Nnt-deficient cells had a significantly altered mitochondrial membrane potential (Fig. 7A), which indicates that these cells have increased mitochondrial dysfunction. Indeed, as indicated in Fig. 7B, baseline H2O2 production in Nnt-deficient cells was increased compared with control (∼35% more). When incubated with 100, 600, and 1000 μm paraquat for 24 h, Nnt-deficient cells produced significantly more H2O2 compared with mock controls (∼40, 35, and 40% increase, respectively). After 48 h of paraquat incubation (Fig. 7C), Nnt-deficient cells had significantly increased lactate dehydrogenase release at 300, 600, and 1,000 μm compared with controls (∼12, 16, and 18% increased release, respectively). Cell death was not observed following 24 h of paraquat exposure, indicating the increase in H2O2 production at this time point occurred prior to overt increases in cell death (data not shown).
FIGURE 7.

Decreased mitochondrial membrane potential leading to increased susceptibility to paraquat toxicity in Nnt-deficient compared with mock control N27 cells. A, mitochondrial membrane potential was measured via tetramethylrhodamine ethyl ester assay, and there was a significant decrease in the membrane potential in the Nnt-deficient cells compared with mock control N27 cells (n = 35–36). B, Nnt-deficient and mock control cells were exposed to various concentrations of paraquat for 24 h, and the amount of H2O2 produced was measured via Amplex Red assay. Nnt cells produced significantly more H2O2 alone and when exposed to various concentrations of paraquat (n = 10). As indicated in C, there was a significant shift in lactate dehydrogenase (LDH) released in Nnt-deficient cells exposed to various concentrations of paraquat (PQ) compared with mock after 48 h of exposure (n = 5–10). *, p < 0.05; **, p < 0.01; ***, p < 0.001 as determined by two-way ANOVA. Data are represented as mean ± S.E.
DISCUSSION
Here, we identify Nnt activity as a critical link between respiration- and substrate-dependent H2O2 catabolism by the Trx/Prx antioxidant system in isolated brain mitochondria and N27 dopaminergic cells. Three principal observations support the role of Nnt as the link between respiration and H2O2 detoxification by the Trx/Prx antioxidant pathway. First, pharmacological inhibition of Nnt in actively respiring brain mitochondria significantly decreased H2O2 removal rates and Trx/TrxR activity. Furthermore, Nnt inhibition failed to alter H2O2 removal in liver mitochondria consistent with the ability of brain but not liver mitochondria to utilize respiration substrates for H2O2 catabolism. Second, deficiency of Nnt either pharmacologically or by shRNA in N27 dopaminergic cells, resulted in a significant decrease in NADPH levels along with a decrease in H2O2 removal rates and an increase in oxidized Prx3, with no change in Prx3 protein expression. Finally, Nnt-deficient cells showed decreased mitochondrial reserve and maximal respiratory capacity, increased steady-state H2O2 levels, and increased cell death when exposed to low levels of the redox cycling agent paraquat. Together, these results highlight the importance of Nnt as a link between mitochondrial respiration and H2O2 catabolism in isolated brain mitochondria and dopaminergic cells (Fig. 8).
FIGURE 8.

A proposed model of Nnt activity in linking respiration-dependent H2O2 removal by the Trx/Prx antioxidant system in isolated brain mitochondria. When respiration substrates are present the citric acid cycle (TCA) and electron transport chain (ETC) can actively generate NADH and maintain the proton (H+) gradient. Nnt will then utilize the H+ gradient and NADH to create NADPH, which will then be utilized by TrxR to keep the Trx/Prx antioxidant system in a reduced state to detoxify H2O2. Thus, Nnt links the energy side of the schematic to the redox side.
Nnt activity provides a novel mechanistic explanation to our previous observation that isolated brain mitochondria catabolize H2O2 by the Trx/Prx system when actively respiring (14). Interestingly, liver mitochondria do not need to be actively respiring to catabolize H2O2 as they primarily use the antioxidant catalase that is not present in isolated brain mitochondria (5, 13, 14, 39, 40). Catalase does not require the reducing equivalent NADPH to detoxify H2O2 and can work independently of the activity of the electron transport chain (1). Although liver mitochondria have Nnt as well as other sources of NADPH (isocitrate dehydrogenase 2, malic enzyme, etc.), H2O2 catabolism may be favored by mitochondrial catalase rather than dependence on respiration substrates.
The main antioxidant systems in the brain mitochondria (Trx/Prx and GSH) require the presence of NADPH as the reducing equivalents (41). The requirement of mitochondrial respiration for antioxidant activity in the brain is likely due to the unique function of neurons to generate neuronal action potentials. Neurons have activity-dependent ion fluxes that require a significant demand for ATP (generated by the mitochondria) to maintain the membrane potential through ATP-dependent sodium/potassium pumps, whereas liver mitochondria are not subjected to such dramatic ion fluxes. Indeed, Nnt inhibition in actively respiring brain mitochondria resulted in a significant decrease in TrxR, Trx activity, and H2O2 removal. However, there was no effect of substrates or Nnt inhibition in isolated liver mitochondria, indicating that Nnt activity is critical in coupling substrate-dependent respiration and H2O2 removal in isolated brain but not liver mitochondria by the Trx/Prx pathway.
Of the different types of neuronal cells, dopaminergic cells have an increased risk for ROS production presumably due to increased iron levels, monoamine oxidase activity, dopaminergic-forming quinones and semi-quinones, a high metabolic activity, and the pace-making ability of dopaminergic neurons utilizing Ca2+ channels (42–44). Trx/Prx activity is critically important in mitochondria to maintain the delicate balance of ROS production for signaling activities versus catabolism to prevent detrimental oxidative stress. The Trx/Prx and GSH systems require NADPH, which can be generated through multiple pathways in neuronal cells (45–47), and therefore, it was important to determine the link between Nnt activity to mitochondrial function and antioxidant activity in intact ROS-sensitive dopaminergic cells. Previous work in cell and animal models suggests that Nnt inhibition by siRNA transfection results in a more oxidized NADP+/NADPH ratio accompanied by a more oxidized GSH/GSSG ratio (20, 22, 48). Indeed, Nnt inhibition by shRNA transfection resulted in a more oxidized NADP+/NADPH ratio and an increase in GSSG and decrease in GSH levels (Fig. 4). Additionally, the decrease in NADPH production in Nnt-deficient cells was not compensated by altering isocitrate dehydrogenase 2 activity. Thus, Nnt deficiency appears to decrease the activities of NADPH-utilizing antioxidant enzymatic pathways rather than NADPH-producing enzymes such as NADP+-dependent isocitrate dehydrogenase 2. However, our data do not rule out the role of additional enzymes that may compensate to replenish mitochondrial NADPH in Nnt-deficient cells.
Notably, the role of Nnt activity and the Trx/Prx system in dopaminergic cells has not been studied in depth. Results from N27 dopaminergic cells in this study confirmed that Nnt inhibition via shRNA transfection resulted in significant decreases in NADPH levels and consequently a decrease in the H2O2 removal rates and more oxidized Prx3 (Figs. 4 and 5). This suggests a critical role of Nnt in NADPH production in brain mitochondria and further validates the link of Nnt between substrate-driven H2O2 catabolism and NADPH-dependent Trx/Prx activity in N27 dopaminergic cells as depicted in Fig. 8. Given the critical role of oxidative stress in neurodegenerative diseases such as PD, these results also suggest that alterations in Nnt activity may be an important checkpoint or control mechanism for changes in steady-state H2O2 levels during disease pathogenesis. As stated previously, dopaminergic cells have an increased risk for detrimental ROS production and postmortem studies have shown increased oxidative stress in the substantia nigra pars compacta in patients with PD (49, 50). Additionally pesticide exposure, decreased complex I activity and aging have been implicated in the pathogenesis of sporadic PD (44, 51–53). The finding that Nnt inhibition resulted in decreased OCR and mitochondrial membrane potential resulting in increased ROS production and cell death validates the idea that Nnt activity in dopaminergic cells plays a critical role in the cells ability to detoxify ROS through the Trx/Prx system when cells are exposed to subtoxic levels of oxidative stress. This is consistent with our previous work showing increased vulnerability of N27 cells to inhibition of TrxR activity in the presence of sub-toxic levels paraquat or 6-hydroxydopamine (28).
The role of Nnt activity as a risk factor for rendering dopaminergic neurons vulnerable to age-related neurodegeneration is novel and has not been noted in the literature to date. However, the precedence for the ability of Nnt to control disease pathogenesis exists. Nnt mutations have been linked to multiple disease states, including diabetes, obesity, failing myocardium, and most recently, homozygous Nnt knock-out in a family showed a link between Nnt knock-out and glucocorticoid deficiency (16, 33, 54–57). Additionally, the Nnt gene has been identified as a modifier gene in manganese superoxide dismutase-deficient mice that show increased mitochondrial oxidative stress (58). Specifically, it was demonstrated that manganese superoxide dismutase-deficient mice without Nnt had a shorter lifespan than manganese superoxide dismutase-deficient mice with the Nnt gene present (58).
Improving mitochondrial antioxidant function has been an area of interest for the treatment of PD; however, to date, no pharmacological drugs targeting this area have been FDA-approved (59, 60). Thus, increasing Nnt activity or levels may improve the redox status of a critical antioxidant system such as Trx/Prx and potentially lead to new therapies in PD.
Acknowledgments
We thank Li-Ping Liang for help with the HPLC methodology to measure NADP+/NADPH levels and GSH and GSSG levels. We also thank James Roede for critical help with reagents and equipment in regards to the Prx3 redox blot.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1NS04748 (to M. P.).
- ROS
- reactive oxygen species
- Nnt
- nicotinamide nucleotide transhydrogenase
- Trx
- thioredoxin
- Prx
- peroxiredoxin
- TrxR
- thioredoxin reductase
- PD
- Parkinson disease
- GSH
- Glutathione
- Prx
- Peroxiredoxin
- GSSG
- glutathione disulfide
- OCR
- oxygen consumption rates
- ANOVA
- analysis of variance.
REFERENCES
- 1. Halliwell B., Gutteridge J. (2007) Free Radicals in Biology and Medicine, 4th Ed., pp. 25–110, Oxford University Press, New York [Google Scholar]
- 2. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tait S. W., Green D. R. (2012) Mitochondria and cell signalling. J. Cell Sci. 125, 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fukai T., Ushio-Fukai M. (2011) Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox. Signal 15, 1583–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mavelli I., Rigo A., Federico R., Ciriolo M. R., Rotilio G. (1982) Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem. J. 204, 535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gardner P. R., Fridovich I. (1991) Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 266, 19328–19333 [PubMed] [Google Scholar]
- 7. Tretter L., Adam-Vizi V. (2000) Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [α]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 20, 8972–8979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Starkov A. A., Fiskum G., Chinopoulos C., Lorenzo B. J., Browne S. E., Patel M. S., Beal M. F. (2004) Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boveris A., Chance B. (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nohl H., Hegner D. (1978) Do mitochondria produce oxygen radicals in vivo? Eur. J. Biochem. 82, 563–567 [DOI] [PubMed] [Google Scholar]
- 11. Giulivi C., Poderoso J. J., Boveris A. (1998) Production of nitric oxide by mitochondria. J. Biol. Chem. 273, 11038–11043 [DOI] [PubMed] [Google Scholar]
- 12. Ghafourifar P., Cadenas E. (2005) Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 26, 190–195 [DOI] [PubMed] [Google Scholar]
- 13. Zoccarato F., Cavallini L., Alexandre A. (2004) Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+. J. Biol. Chem. 279, 4166–4174 [DOI] [PubMed] [Google Scholar]
- 14. Drechsel D. A., Patel M. (2010) Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 285, 27850–27858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mishra S., Imlay J. (2012) Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch. Biochem. Biophys. 525, 145–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheeran F. L., Rydström J., Shakhparonov M. I., Pestov N. B., Pepe S. (2010) Diminished NADPH transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. Biochim. Biophys. Acta 1797, 1138–1148 [DOI] [PubMed] [Google Scholar]
- 17. Rydström J. (2006) Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726 [DOI] [PubMed] [Google Scholar]
- 18. Bizouarn T., Fjellström O., Meuller J., Axelsson M., Bergkvist A., Johansson C., Göran Karlsson B., Rydström J. (2000) Proton translocating nicotinamide nucleotide transhydrogenase from E. coli. Mechanism of action deduced from its structural and catalytic properties. Biochim. Biophys. Acta 1457, 211–228 [DOI] [PubMed] [Google Scholar]
- 19. Yap L. P., Garcia J. V., Han D., Cadenas E. (2009) The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 61, 1283–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arkblad E. L., Tuck S., Pestov N. B., Dmitriev R. I., Kostina M. B., Stenvall J., Tranberg M., Rydström J. (2005) A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic. Biol. Med. 38, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 21. Huang T. T., Naeemuddin M., Elchuri S., Yamaguchi M., Kozy H. M., Carlson E. J., Epstein C. J. (2006) Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum. Mol. Genet. 15, 1187–1194 [DOI] [PubMed] [Google Scholar]
- 22. Yin F., Sancheti H., Cadenas E. (2012) Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 1817, 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sims N. R., Anderson M. F. (2008) Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 3, 1228–1239 [DOI] [PubMed] [Google Scholar]
- 24. Castello P. R., Drechsel D. A., Patel M. (2007) Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 282, 14186–14193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salvi M., Battaglia V., Brunati A. M., La Rocca N., Tibaldi E., Pietrangeli P., Marcocci L., Mondovì B., Rossi C. A., Toninello A. (2007) Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 282, 24407–24415 [DOI] [PubMed] [Google Scholar]
- 26. Prasad K. N., Carvalho E., Kentroti S., Edwards-Prasad J., La Rosa F. G., Kumar S., Freed C. R., Vernadakis A. (1994) Production of terminally differentiated neuroblastoma cells in culture. Restor. Neurol. Neurosci. 7, 13–19 [DOI] [PubMed] [Google Scholar]
- 27. Cantu D., Fulton R. E., Drechsel D. A., Patel M. (2011) Mitochondrial aconitase knockdown attenuates paraquat-induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2. J. Neurochem. 118, 79–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lopert P., Day B. J., Patel M. (2012) Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS One 7, e50683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ronchi J. A., Figueira T. R., Ravagnani F. G., Oliveira H. C., Vercesi A. E., Castilho R. F. (2013) A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 63, 446–456 [DOI] [PubMed] [Google Scholar]
- 30. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., Velculescu V. E., Vogelstein B., Bigner D. D. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arner E. S., Holmgren A. (2001) Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. Chapter 7, Unit 7 4 [DOI] [PubMed] [Google Scholar]
- 32. Rydström J. (1979) Assay of nicotinamide nucleotide transhydrogenases in mammalian, bacterial, and reconstituted systems. Methods Enzymol. 55, 261–275 [DOI] [PubMed] [Google Scholar]
- 33. Shimomura K., Galvanovskis J., Goldsworthy M., Hugill A., Kaizak S., Lee A., Meadows N., Quwailid M. M., Rydström J., Teboul L., Ashcroft F., Cox R. D. (2009) Insulin secretion from beta-cells is affected by deletion of nicotinamide nucleotide transhydrogenase. Methods Enzymol. 457, 451–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalhorn T. F., Thummel K. E., Nelson S. D., Slattery J. T. (1985) Analysis of oxidized and reduced pyridine dinucleotides in rat liver by high-performance liquid chromatography. Anal. Biochem. 151, 343–347 [DOI] [PubMed] [Google Scholar]
- 35. Liang L. P., Patel M. (2006) Seizure-induced changes in mitochondrial redox status. Free Radic. Biol. Med. 40, 316–322 [DOI] [PubMed] [Google Scholar]
- 36. Cox A. G., Winterbourn C. C., Hampton M. B. Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol. 474, 51–66 [DOI] [PubMed] [Google Scholar]
- 37. Bergmeyer H. U., Bernt E. (1983) Lactate Dehydrogenase. UV Assay with Pyruvate and NADH in Methods of Enzymatic Analysis, 3rd Ed., pp. 118–125, Wiley, Hoboken, NJ [Google Scholar]
- 38. Rydstrom J. (1972) Site-specific inhibitors of mitochondrial nicotinamide-nucleotide transhydrogenase. Eur. J. Biochem. 31, 496–504 [DOI] [PubMed] [Google Scholar]
- 39. Radi R., Turrens J. F., Chang L. Y., Bush K. M., Crapo J. D., Freeman B. A. (1991) Detection of catalase in rat heart mitochondria. J. Biol. Chem. 266, 22028–22034 [PubMed] [Google Scholar]
- 40. Sinet P. M., Heikkila R. E., Cohen G. (1980) Hydrogen peroxide production by rat brain in vivo. J. Neurochem. 34, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 41. Arnér E. S., Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 42. Surmeier D. J., Schumacker P. T. (2013) Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J. Biol. Chem. 288, 10736–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guzman J. N., Sánchez-Padilla J., Chan C. S., Surmeier D. J. (2009) Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jenner P. (2003) Oxidative stress in Parkinson's disease. Ann. Neurol. 53, S26–36; discussion S36–38 [DOI] [PubMed] [Google Scholar]
- 45. Ying W. (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox. Signal 10, 179–206 [DOI] [PubMed] [Google Scholar]
- 46. Yankner B. A., Lu T., Loerch P. (2008) The aging brain. Annu. Rev. Pathol. 3, 41–66 [DOI] [PubMed] [Google Scholar]
- 47. Lam P. Y., Yin F., Hamilton R. T., Boveris A., Cadenas E. (2009) Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radic. Res. 43, 431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogel R., Wiesinger H., Hamprecht B., Dringen R. (1999) The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci. Lett. 275, 97–100 [DOI] [PubMed] [Google Scholar]
- 49. Jenner P., Schapira A. H., Marsden C. D. (1992) New insights into the cause of Parkinson's disease. Neurology 42, 2241–2250 [DOI] [PubMed] [Google Scholar]
- 50. Jenner P., Olanow C. W. (1996) Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 47, S161–170 [DOI] [PubMed] [Google Scholar]
- 51. Dinis-Oliveira R. J., Remião F., Carmo H., Duarte J. A., Navarro A. S., Bastos M. L., Carvalho F. (2006) Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology 27, 1110–1122 [DOI] [PubMed] [Google Scholar]
- 52. Jenner P., Olanow C. W. (1998) Understanding cell death in Parkinson's disease. Ann. Neurol. 44, S72–84 [DOI] [PubMed] [Google Scholar]
- 53. Zhang Y., Dawson V. L., Dawson T. M. (2000) Oxidative stress and genetics in the pathogenesis of Parkinson's disease. Neurobiol. Dis. 7, 240–250 [DOI] [PubMed] [Google Scholar]
- 54. Heiker J. T., Kern M., Kosacka J., Flehmig G., Stumvoll M., Shang E., Lohmann T., Dreßler M., Kovacs P., Blüher M., Klöting N. (2013) Nicotinamide nucleotide transhydrogenase mRNA expression is related to human obesity. Obesity 21, 529–534 [DOI] [PubMed] [Google Scholar]
- 55. Wong N., Blair A. R., Morahan G., Andrikopoulos S. (2010) The deletion variant of nicotinamide nucleotide transhydrogenase (Nnt) does not affect insulin secretion or glucose tolerance. Endocrinology 151, 96–102 [DOI] [PubMed] [Google Scholar]
- 56. Meimaridou E., Kowalczyk J., Guasti L., Hughes C. R., Wagner F., Frommolt P., Nürnberg P., Mann N. P., Banerjee R., Saka H. N., Chapple J. P., King P. J., Clark A. J., Metherell L. A. (2012) Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 44, 740–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamaguchi R., Kato F., Hasegawa T., Katsumata N., Fukami M., Matsui T., Nagasaki K., Ogata T. (2013) A novel homozygous mutation of the nicotinamide nucleotide transhydrogenase gene in a Japanese patient with familial glucocorticoid deficiency. Endocr. J. 60, 855–859 [DOI] [PubMed] [Google Scholar]
- 58. Kim A., Chen C. H., Ursell P., Huang T. T. (2010) Genetic modifier of mitochondrial superoxide dismutase-deficient mice delays heart failure and prolongs survival. Mamm. Genome 21, 534–542 [DOI] [PubMed] [Google Scholar]
- 59. Smith R. A., Murphy M. P. (2010) Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N.Y. Acad. Sci. 1201, 96–103 [DOI] [PubMed] [Google Scholar]
- 60. Gilgun-Sherki Y., Melamed E., Offen D. (2001) Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 40, 959–975 [DOI] [PubMed] [Google Scholar]