

Background: Scml2 is a subunit of the Polycomb repressive complex 1 (PRC1) responsible for epigenetic regulation of gene expression.
Results: A conserved DNA-binding domain within Scml2 has been identified, and its structure determined by NMR revealed a novel, previously uncharacterized fold.
Conclusion: A new Scml2 domain may prove important for PRC1 recognition of target genes.
Significance: Characterization of Scml2 is crucial for understanding the mechanism of PRC1-dependent gene silencing.
Keywords: DNA-binding Protein, Epigenetics, NMR, Polycomb, Protein Structure
Abstract
Scml2 is a member of the Polycomb group of proteins involved in epigenetic gene silencing. Human Scml2 is a part of a multisubunit protein complex, PRC1 (Polycomb repressive complex 1), which is responsible for maintenance of gene repression, prevention of chromatin remodeling, preservation of the “stemness” of the cell, and cell differentiation. Although the majority of PRC1 subunits have been recently characterized, the structure of Scml2 and its role in PRC1-mediated gene silencing remain unknown. In this work a conserved protein domain within human Scml2 has been identified, and its structure was determined by solution NMR spectroscopy. This module was named Scm-like embedded domain, or SLED. Evolutionarily, the SLED domain emerges in the first multicellular organisms, consistent with the role of Scml2 in cell differentiation. Furthermore, it is exclusively found within the Scm-like family of proteins, often accompanied by malignant brain tumor domain (MBT) and sterile α motif (SAM) domains. The domain adopts a novel α/β fold with no structural analogues found in the Protein Data Bank (PDB). The ability of the SLED to bind double-stranded DNA was also examined, and the isolated domain was shown to interact with DNA in a sequence-specific manner. Because PRC1 complexes localize to the promoters of a specific subset of developmental genes in vivo, the SLED domain of Scml2 may provide an important link connecting the PRC1 complexes to their target genes.
Introduction
The Polycomb group (PcG)2 is a group of conserved proteins that function as chromatin regulators in multicellular organisms (1). They are crucial for epigenetic gene silencing, maintenance of the pluripotent state of stem cells, and cell differentiation. PcG proteins form a few distinct multisubunit nuclear complexes, among which Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) are the best studied (2). Both PRC1 and PRC2 covalently modify histones and work together to accomplish their goal: silencing of a group of developmental genes (3). The PRC2 complex acts as a lysine methyltransferase that methylates histone 3 at Lys-27 (H3K27). The PRC1 complex contains an E3 ubiquitin ligase that recognizes methylated H3K27 and ubiquitinates Lys-119 of histone H2A (H2AK119). The resulting H3K27 and H2AK119 histone modifications are the hallmarks of chromatin compaction and gene silencing. However, the molecular mechanisms of PRC1 targeting to the subset of developmental genes and the mechanism of PRC1-mediated gene silencing are poorly understood (3, 4).
PRC1 is a multisubunit protein complex that consists of three homologous RING domain-containing subunits: Ring1B, Ring1A, and PCGF, as well as the proteins Cbx and PHC (2). The Ring1B/PCGF heterodimer endows the PRC1 complex with E3 ubiquitin ligase activity, whereas the Cbx subunit is responsible for H3K27 recognition. The roles of the Ring1A and the PHC subunits are unclear. Unlike PRC1 in Drosophila, human PRC1 has an additional level of complexity because every PRC1 subunit has numerous homologues that may constitute distinct PRC1-like complexes (2).
Scml2 (sex comb on midleg-like 2) is an additional subunit of the PRC1 complex. It is a transcriptional repressor and a member of the PcG group of proteins (2, 4–6). Previous biochemical and functional studies have shown that Scml2 co-localizes with both PRC1 and PRC2 in vivo to the Polycomb-response elements of PcG target genes (4). Scml2 is found associated with the PRC1 complex in nonstoichiometric amounts during co-immunoprecipitation (7) and can directly associate with the PHC1 subunit of PRC1 (8, 9). Despite the fact that Scml2 itself is not part of the PRC1 core, it is just as crucial for Hox gene silencing in Drosophila as any other subunit of PRC1 and PRC2. For example, mutations in the Scm gene of Drosophila result in severe developmental malformation and are lethal at embryonic stage (5). The human SCML2 gene, located in the short arm of the X chromosome, was identified in an initial effort to create a transcription map of the Xp22 region associated with multiple human genetic diseases (6). Although Scml2 clearly plays an important role in PcG-mediated gene silencing, its function remains largely unknown.
Structural characterization of Scml2 may provide vital clues to its function. Human Scml2 is a 700-amino acid protein that consists of two N-terminal malignant brain tumor domains (MBTs) and a C-terminal sterile α motif (SAM) domain. Additionally, an unidentified domain is located between the second MBT and the SAM domain as described below (see Fig. 1). The MBT domains are ∼100-amino acid modules that are often found in multiple copies arranged in MBT repeats. They are known to bind methylated lysine residues (10), which suggests that Scm-like proteins may be able to recognize methylated histones. Indeed, a number of structures of MBT domains were solved in complexes with peptides containing methylated lysine residues (11–18), including structures of two MBT domains from Scml2. The structures of MBT domains and biochemical analyses of their binding specificities revealed that they weakly interact with mono- and dimethylated lysine residues through a conserved site with affinities ranging from 30 μm to 1 mm (18). Most MBT domains are promiscuous binders that associate with mono- and dimethylated lysines regardless of their sequence context, whereas some have a preference for a specific methylated substrate (18). However, in almost all cases, MBT repeats recognize only lower methylation states of the histone lysines. Thus, the MBT domains in Scml2 likely serve as “sensors” of the degree of histone methylation. The C terminus of Scml2 contains a SAM domain, which belongs to a group of small helical domains that can form head-to-tail homo-and hetero-dimers leading to SAM polymerization. Notably, the PHC1 subunit of PRC1 contains a SAM domain homologous to that of Scml2, and the two isolated domains were shown to form hetero-dimers (8, 9). Therefore, the C-terminal SAM domain of Scml2 likely functions as an anchor that attaches Scml2 to the PRC1 complex. Interestingly, several recent studies show that Scml2 can recruit the PRC1 complex to DNA via its SAM domain. Moreover, overexpression of the SAM domain disrupts PcG repression, suggesting a central role for Scm-like proteins in the recruitment of Polycomb complexes (19). However, the mechanism by which Scm-like proteins can recognize DNA in the first place remains unknown.
FIGURE 1.

SLED domain in human proteins. A, schematic representation of SLED-containing proteins in humans. SLED, MBT, and SAM domains are shown in red, cyan, and black, respectively. B, multiple sequence alignment of SLED domains from human proteins performed using ClustalW (29). Invariant residues are shown in red. The predicted secondary structure elements of the domain are shown above the Scml2 sequence. Scml2 cysteine residues are marked with an asterisk.
In this work, I present the solution NMR structure of a previously unidentified DNA-binding domain within human Scml2 (residues 354–468). This domain is located between the N-terminal MBT domains and the C-terminal SAM domain of Scml2. From an evolutionary perspective, the domain emerges in multicellular organisms and is conserved from the simplest multicellular eukaryotes to humans. It is exclusively found within Scm-like proteins, and therefore, I named it Scm-like embedded domain, or SLED. The SLED domain adopts a novel protein fold with no close structural homologues in the Protein Data Bank (PDB). Interestingly, the two previously identified lethal point mutations of the Scm gene in Drosophila map onto the SLED domain (5), suggesting that it plays a key functional role during development. The results presented in this work suggest that, in addition to its ability to bind methylated histones, Scml2 can also directly recognize double-stranded DNA via its SLED domain. The direct interaction of Scml2 with DNA involving this newly discovered domain may provide an important step in PRC1 recognition of its target genes.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The nucleotide sequence encoding the SLED domain of human Scml2 (amino acids 354–468) was codon-optimized for bacterial expression and synthesized in vitro by GenScript USA Inc. with NdeI and NotI restriction sites added at the 5′ and 3′ ends of the gene, respectively. The NdeI/NotI sites were then used to subclone the gene into a pET28b(+) vector (Novagen). Two point mutations of the SLED gene, N440A and C453Y, were created using site-directed mutagenesis. The accuracy of all resulting DNA constructs was confirmed by DNA sequencing (GENEWIZ).
The resulting plasmids were used to express the His6-tagged WT SLED and its N440A and C453Y mutants in Escherichia coli using the protocol described below. The bacteria were transformed with a SLED plasmid and grown in 50 ml of LB medium supplemented with kanamycin at 37 °C overnight. Cells were then transferred into the M9 minimal medium. 15N-labeled NH4Cl (1 g/liter, CIL) was used as the sole source of nitrogen, and glucose (3 g/liter) was used as the source of carbon. The 15N/13C-double-labeled protein sample was prepared using 13C-labeled glucose (CIL). Cells were grown at 37 °C to A600 of 1.0 absorbance units, induced with 1.0 mm isopropyl-1-thio-β-d-galactopyranoside for 10 h at 20 °C, and harvested by centrifugation. The bacterial pellet was resuspended in cold lysis buffer (50 mm sodium phosphate, 300 mm NaCl, 10 mm imidazole, pH 8.0) and lysed by sonication. HisPur beads (Thermo) were used to purify His6-tagged protein from the clarified lysate followed by thrombin (GE Healthcare) cleavage for 3 h at 37 °C and size-exclusion chromatography (Superdex 75, GE Healthcare). The final NMR samples contained 0.16–1.0 mm protein, 50 mm HEPES, pH 7.2, 100 mm NaCl, 2 mm DTT, and 10% D2O.
NMR Structure Determination
NMR spectra for the Scml2 SLED domain were collected at 15 °C on Agilent VNMRS 600 and 800-MHz spectrometers equipped with cold probes. The backbone 15N, 13C, and 1H resonances of the Scml2 SLED domain were assigned using 1H-15N HSQC, HNCA, HNCACB, HNCO, CBCA(CO)NH, and HBHA(CO)NH experiments. The aliphatic side-chain resonances were assigned using 1H-13C HSQC, HC(C)H-TOCSY and (H)CCH-TOCSY experiments. Aromatic side chains were assigned using aromatic 1H-13C HSQC and aromatic 13C NOESY experiments. 15N NOESY as well as aliphatic and aromatic 13C NOESY experiments (20) were used to obtain interproton distance restraints yielding 3421 and 146 restraints, respectively (see Table 1). NMR spectra were processed with NMRPipe (21) and analyzed with the program SPARKY (22). Nearly complete backbone (99%) and side-chain (93%) NMR resonance assignments were obtained.
TABLE 1.
Structural statistics of the Scml2 SLED domain
| Data collection | |
| NOE-based distance constraintsa | |
| Total | 3725 |
| Aliphatic | 3421 |
| Aromatic | 146 |
| Intraresidue [i = j] | 565 |
| Sequential [| i − j | = 1] | 963 |
| Medium range [1 < | i − j | < 5] | 703 |
| Long range [| i − j | ≥5] | 1190 |
| NOE constraints per restrained residueb | 29.2 |
| Dihedral angle constraints (TALOS) | 174 |
| Completeness of chemical shift assignmenta | 92.6% |
| Refinement | |
| Deviation from experimental restraints | |
| NOE (Å) | 0.023 ± 0.002 |
| Dihedral angles (°) | 0.72 ± 0.11 |
| r.m.s. deviations from standard covalent geometry: | |
| Bond length (Å) | 0.0139 ± 0.0002 |
| Bond Angles (°) | 0.96 ± 0.01 |
| Ramachandran plot statisticsa | |
| Core regions | 87.9% |
| Allowed regions | 12.1% |
| Generously allowed regions | 0% |
| Disallowed regions | 0% |
| Global quality scores | |
| r.m.s. deviations to mean (Å)c | |
| Backbone (residues 354–465) | 1.39 ± 0.33 (0.62 ± 0.08) |
| Heavy atoms (residues 354–465) | 1.70 ± 0.26 (1.11 ± 0.08) |
| F-score | 0.92 |
| Recall | 0.91 |
| Precision | 0.92 |
a Residues 350–468 were analyzed.
b There are 117 residues with conformationally restricting constraints.
c Residues 350–468 were analyzed; r.m.s. deviations calculated for SLED domain excluding disordered N- and C-terminal residues (residues 354–465) are shown in parentheses.
Structure calculation for the Scml2 SLED domain was performed using CYANA (23). The restraints for the backbone dihedral φ and ψ angles were derived from the backbone 1H, 15N, and 13C chemical shifts using the TALOS+ program (24). Intramolecular NOE correlations were assigned automatically in CYANA. Hydrogen bond restraints were added on the basis of NOE analysis. A total of 100 structures of the Scml2 SLED domain were generated followed by water refinement of the lowest energy 20 structures using CNS (25, 26).
Protein Sequence Conservation Analysis
SLED domain-containing proteins were identified within nonredundant reference sequences in the National Center for Biotechnology Information (NCBI) and Simple Modular Architecture Research Tool (SMART) (27, 28) protein databases. Multiple sequence alignments and visualization of the sequence conservation were performed using CLUSTAL W (29). The phylogenic tree of the SLED domains was created using the Interactive Tree of Life (iTOL) server (30).
DNA Binding Experiments
Chemical shift perturbation analysis was used to characterize Scml2 SLED-DNA binding. Specifically, 15N-labeled SLED domain was gradually titrated with unlabeled 11-bp double-stranded (ds) DNA. Complimentary oligonucleotides 5′-AGGAGCGGGAG-3′ and 5′-CTCCCGCTCCT-3′ were chemically synthesized (Integrated DNA Technologies (IDT)), heated to 98 °C for 10 min, and then annealed at room temperature to form the dsDNA. In a similar manner, two more double-stranded oligonucleotides were prepared (dsGGGCGCGCCC and dsTTTATATAAA), and three resulting double-stranded oligonucleotides were tested in separate NMR titration series for their ability to bind SLED domain. 15N-1H HSQC spectra of the SLED domain were collected at each point of the titration using the 800-MHz NMR spectrometer to monitor changes in 1H and 15N resonance frequencies of the domain induced by dsDNA binding. The observed changes in 15N and 1H frequencies relative to peak position in the free state, Δωi = (ΔωiN2 + ΔωiH2)½, plotted versus dsDNA/SLED ratio, yielded NMR titration profiles for each amino acid residue. Cumulative changes in peak positions (calculated as a sum of Δωi over all amide resonances) as a function of dsDNA concentration were used to calculate the dissociation constants (Kd) for the complexes using a two-state binding model. The resulting curves were least squares-fit to the following equation
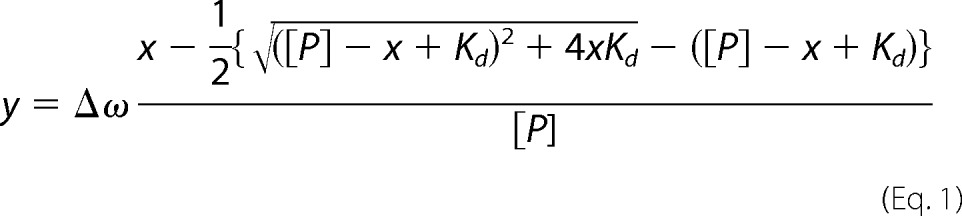 |
where [P] is the total protein concentration; x is DNA concentration; y is the corresponding change in peak position; Kd is the dissociation constant; and Δω is the frequency difference between free and DNA-bound protein. The per-residue Δωi frequency changes were used to map the DNA-binding site on the SLED domain structure. The titration data analysis was performed using SciDAVis.
RESULTS
The SLED Domain Is Evolutionarily Conserved in Metazoa
Detailed amino acid sequence analysis of the human Scml2 together with secondary structure prediction using the Jpred server (31) revealed the presence of a folded domain embedded between the N-terminal MBT repeats and the C-terminal SAM domain. The domain spans residues 354–468 and has no sequence homology to any other domain with known spatial structure in the PDB. Sequence BLAST against human proteins identified five proteins containing this domain, Scml2, Scmh1, SFMBT1, SFMBT2, and SCML4. The domain architecture of these proteins is shown in Fig. 1A, where newly identified domains are aligned and shown in red, MBT domains are shown in blue, and SAM domains are in black. Notably, all five identified proteins are sex comb on midleg-like with the number of N-terminal MBT domains varying from four (for SFMBT1 and SFMBT2) to zero (for SCML4). Because the domain is found exclusively within the Scm-like family, typically located between MBT and SAM domains, I designate it Scm-like embedded domain, or SLED. A multiple sequence alignment of the five members of SLED-containing proteins is shown in Fig. 1B where absolutely conserved residues are shown in red, and cysteine residues are marked with an asterisk. The predicted secondary structure elements are shown above the Scml2 sequence.
After identifying SLED-containing proteins in humans, I have searched for proteins with SLED domains in other species and have compared their domain architecture using SMART (27, 28, 32). The resulting phylogenetic tree with a representation of the domain architecture for each homologue is illustrated in Fig. 2, where SLED domains are shown as red squares. It is clear from Fig. 2 that SLED domains are present almost exclusively in the context of the C-terminal SAM domain (black circles) and the N-terminal MBT repeats (blue squares) across all multicellular species, which makes it a unique Scm-specific domain. Remarkably, the emergence of the SLED domain in evolution coincides with the development of multicellular eukaryotes because SLED-containing proteins are found exclusively in Metazoa (Fig. 2), consistent with the essential role of Scm-like proteins in cell differentiation. Note that, unlike SAM domains, MBT domains emerge simultaneously with SLEDs and are often found together, likely suggesting that the function of the SLED is closely related to that of the MBT domain.
FIGURE 2.

SLED domain conservation in multicellular organisms. A phylogenetic tree of SLED domain containing proteins is shown. Each species is represented by an individual color (right). The domain architecture for each homolog is shown following the name of the protein. SLED, MBT, and SAM domains are shown in red and cyan squares and black circles, respectively. The tree was generated using the Interactive Tree of Life server iTOL (30).
Solution Structure of the Human Scml2 SLED Domain
The solution structure of the SLED domain from human Scml2 (residues 354–468) was determined using NMR spectroscopy. A total of 100 structures were generated based on 3491 NOE-derived proton-proton distance restrains and 182 TALOS+-predicted dihedral angle restraints using CYANA (23, 33). The ensemble of the 20 lowest energy structures was further refined in explicit solvent using CNS (25, 26) to an r.m.s. deviation of 0.62 ± 0.08 and 1.11 ± 0.08 Å for the backbone and all heavy atoms, respectively. The full NMR structural statistics, including overall structure quality scores, are presented in Table 1.
The ensemble of the 20 lowest energy structures of the SLED domain is shown in Fig. 3A (backbone only). Overall, the structure is well defined with the exception of a few residues at the flexible N and C termini marked in blue and red, respectively. The domain adopts a novel fold with β1-α1-β2-α2-α3-β3-β4-α4-β5 topology (Fig. 3B). The core of the domain is formed by two 15-amino acid α-helices, α2 (amino acids 388–402) and α4 (amino acids 442–456), aligned parallel to each other, flanked by a three-stranded antiparallel β-sheet on one side and a long twisted β-hairpin on the other side packed against α helix 4 (Fig. 3C). The long twisted hairpin is formed by strands β3 and β4 (amino acids 422–427 and 430–435, respectively). The three-stranded β-sheet consists of strands β1 (356–361) and β2 (382–387) stabilized by four hydrogen bonds and a short C-terminal strand β5 (amino acids 462–463) stabilized by two hydrogen bonds, connecting Ser-463 to Tyr-360 of the β1 strand.
FIGURE 3.

Solution NMR structure of the SLED domain from human Scml2 (residues 354–467). A, NMR ensemble of the 20 lowest energy structures of the human Scml2 SLED domain (backbone representation only). The protein chain is rainbow-colored from blue to red; N and C termini are labeled. B, schematic representation of the SLED domain topology. C, ribbon representation of the SLED domain shown in two orientations; secondary structure elements are labeled. D, charge distribution on the surface of the SLED domain (shown in the same orientation as C).
The charge distribution on the surface of the Scml2 SLED domain is shown in Fig. 3D where the molecule is shown in two orientations. One side of the molecule (right panel) contains a remarkable hydrophobic cavity surrounded by positively charged residues Lys-414, Arg-418, and Arg-447.
Comparison of the human Scml2 SLED sequence to the sequences of its counterparts in other species revealed a set of conserved residues throughout the domain. A multiple sequence alignment of the Scml2 SLED domains from representative species (from fish to human) is shown in Fig. 4A, where residues with conservation of 90% and higher are highlighted and buried residues are annotated with an asterisk. Conserved hydrophobic residues correlate well with amino acid residues buried in the SLED structure forming the hydrophobic core. These residues are responsible for maintaining the overall SLED domain fold. Surface-exposed evolutionarily conserved residues are of special interest because they often correspond to functionally important regions on a protein surface, such as binding sites for interaction partners.
FIGURE 4.

Amino acid residue conservation in SLED domains. A, multiple sequence alignment of the Scml2 SLED domain across the species. Default ClustalW (29) colors are used for each residue. Residues with conservation of 90% or higher are highlighted, and residues that form the hydrophobic core of the domain are annotated with an asterisk. B, SLED surface conservation. Conserved residues (pink) are mapped on the surface of the human Scml2 SLED. Two orientations are shown to illustrate that conserved residues form a continuous patch on the SLED domain surface. The molecule on the right is rotated by 180° relative to the molecule on the left.
The surface conservation of the domain was analyzed, and the highly conserved amino acid residues were mapped on the Scml2 SLED structure (Fig. 4B). Remarkably, conserved surface-exposed residues are arranged in a continuous cluster on one side of the molecule (shown in pink on Fig. 4B). The majority of this site is formed by residues from the three-stranded β-sheet, including strands β1, β2, and β5. Such a conservation of the domain surface suggests that this side of the molecule is functionally important and may serve either as a binding site for other regions within Scml2 or as an interaction interface for other yet unidentified substrates.
Scml2 SLED Can Bind dsDNA
Although the SLED domain is evolutionarily linked to the MBT domains and is conserved across eukaryotes, the function of this domain remains unknown. In an effort to predict a possible function for the Scml2 SLED, an extensive search within the PDB for proteins with thee-dimensional structures similar to the SLED fold was performed using the DALI server (34–37). Remarkably, the only protein found to be structurally related to SLED (Z-score of 5.0, 6% sequence identity) is a 181-amino acid dsDNA-binding E. coli protein SeqA, which functions as a negative regulator of replication initiation in bacteria (PDB ID 1LRR) (38–40). Similar to SLED, SeqA includes two 15-amino acid α-helices in its core and forms a helical bundle similar in arrangement to the α1/α2/α3/α4 bundle of SLED. SeqA, however, lacks the three-stranded β-sheet and has two α-helices instead. The long β-hairpin characteristic of the SLED fold is much shorter in SeqA and is incorporated into an alternative arrangement of a three-stranded β-sheet and an α helix (Fig. 5).
FIGURE 5.
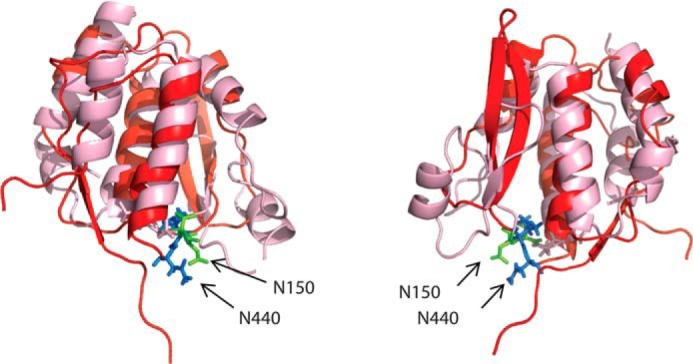
Structural alignment of the Scml2 SLED and SeqA. The SLED domain is shown as a red ribbon, and the SeqA (PDB ID: 1LRR) is shown in pink. The side chains of the SeqA loop residues involved in DNA binding are shown as green sticks (Asn-150, Thr-151), and the side chains of the SLED residues displaying the largest chemical shift changes upon DNA binding are shown as blue sticks (Asn-440, Ser-441).
Because the only DALI hit (albeit with a very limited structural similarity (Z-score of 5.0)) turned out to be a DNA-binding protein, I decided to test SLED for its ability to bind dsDNA. The human PRC1 complex has multiple known target genes, including HMX2, ASCL-1, and others (41–43). It is thought to recognize CpG-rich regions in the promoter regions upstream of these genes. Therefore, for my in vitro DNA binding assays, I used part of a CpG island within the promoter region of the ASCL-1 gene located in chromosome 12 (from 103,351,246 to 103,351,256). It is important to note that the ASCL-1 promoter contains hundreds of base pairs and I picked a stretch of only 11 nucleotides containing a single CpG sequence in the middle for the binding experiments. ASCL-1 is a known PRC1 target gene whose promoter is highly methylated in small-cell lung cancer and colon cancer, resulting in higher levels of gene expression when compared with normal cells.
To test the hypothesis that the Scml2 SLED domain can bind DNA, NMR chemical shift perturbation analysis was used. NMR chemical shifts are very sensitive to changes in protein structure. Therefore, their variations upon titration of a protein with its binding partner allow accurate mapping of the binding site and measurement of the binding affinity. A dsDNA 11-mer from ASCL-1 promoter region (ASCL1 hereafter) obtained by annealing two complimentary 5′-AGGAGCGGGAG-3′ and 5′-CTCCCGCTCCT-3′ oligonucleotides was gradually titrated into a 0.16 mm 15N-labeled SLED sample to a final 7:1 ratio of ASCL1 to SLED. The binding was monitored by collecting 15N-1H HSQC spectra of the SLED domain at each point of the titration. The overlay of eight spectra, one for each titration point in the series, is shown in Fig. 6A, where the free SLED domain spectrum is shown in blue, and the SLED spectrum in the presence of 7× molar excess of dsDNA is shown in red. The two insets on the right depict two selected spectral regions and clearly show large chemical shift changes (color gradient from blue to red) upon the addition of ASCL1 oligonucleotide. As seen in Fig. 6, A and B, only a subset of peaks changed position upon DNA addition, indicating a specific interaction between SLED and the ASCL1 oligonucleotide.
FIGURE 6.

Scml2 SLED domain and dsDNA binding. A, overlay of 15N/1H HSQC spectra of the free (blue) and DNA-bound SLED domain. The two insets show gradual changes in peak positions (from blue to red) in selected spectral regions upon the addition of ASCL1 dsDNA. B, NMR frequency difference Δω = (ΔωN2 + ΔωH2)½ (at 800 MHz for 1H) between the first and the last points of DNA titration for each amino acid residue of SLED are shown as a histogram. Δω values for backbone HN groups are shown in blue, and those for the HN groups of Gln and Asn side chains are shown in magenta. C, SLED amino acid residues that form the DNA-binding site (with Δω values > 30 Hz) are shown in red on a surface representation (left) and ribbon representation (right) of the SLED structure. Selected DNA-binding residues are labeled. Proline residues within the DNA-binding site are shown in purple on the SLED surface and labeled as P. Titration data for proline residues are not available because they are not present in the 15N/1H HSQC spectrum due to a missing HN group in their chemical structure.
Residues displaying the largest frequency changes (Δω > 30 Hz) were mapped onto the spatial structure of the SLED domain (shown in red in Fig. 6C), revealing the SLED-binding site for ASCL1 oligonucleotide. A distinct binding interface is clearly located on the same face of the molecule (red patch). Specifically, it includes the N terminus (residues 353–357) as well as residues forming the hydrophobic cavity (Leu-392, Val-388, Phe-448, and Phe-444), positively charged residues flanking the cavity (Lys-414 and Arg-447), and the loop region connecting β4 and α4 (residues 439–444). The residues Asn-440 and Ser-441 from this loop exhibit the most pronounced chemical shift changes (Fig. 6B). Interestingly, the corresponding loop of the SeqA protein is also involved in DNA recognition. It is remarkable that all residues mentioned above are highly conserved (Fig. 4A) and the identified DNA-binding site constitutes a portion of the larger conserved surface patch shown in Fig. 4B, suggesting that residues involved in SLED-DNA binding were preserved during evolution and are important for Scml2 function.
To confirm that the protein surface identified in Fig. 6 is indeed a DNA-binding site, Asn-440 located on this surface has been mutated into alanine, and the NMR titrations with ASCL1 oligonucleotide were repeated. This mutation is expected to compromise the DNA binding affinity of SLED. The result is shown in Fig. 7A, where the HSQC spectra of the free N440A mutant (blue) and N440A in the presence of 7 m excess of the oligonucleotide (red) are overlaid. As can be seen, binding was completely abrogated in the N440A mutant, confirming the relevance of this region in DNA binding.
FIGURE 7.

Mutational analysis of Scml2 SLED. A, overlay of 15N/1H HSQC spectra of the free (blue) N440A SLED domain and N440A in the presence of 7 m excess of ASCL1 oligonucleotide (red). B, DNA titration curves derived from the NMR chemical shift perturbation experiments. Cumulative chemical shift changes for all amino acid residues in the domain are plotted as a function of DNA to SLED ratio. Experimental data and fitting curves for three different double-stranded oligonucleotides (dsAGGAGCGGGAG, dsGGGCGCGCCC, and dsTTTATATAAA) are shown in red, purple, and blue, respectively. The resulting Kd values are shown at the bottom of the graph. C, the model of the C453Y mutant of the SLED domain. The packing of the Cys-453 (top) versus Tyr-453 (bottom) side chains within the hydrophobic core is shown. A portion of the protein is shown as a ribbon, and the side chains of the hydrophobic amino acid residues surrounding Cys-453 are shown as sticks. The side-chain atoms of the Cys-453 and Tyr-453 are shown as spheres. D, 15N/1H HSQC spectrum of the C453Y of the Scml2 SLED domain reveals that it is unfolded in solution.
Scml2 SLED DNA Binding Is Sequence-specific
Although I showed that the SLED domain of the Scml2 is capable of binding dsDNA, the question remains whether it has a preference toward specific DNA sequences. To answer this question, I tested whether SLED can preferentially bind CG-rich versus AT-rich DNA oligonucleotides. To this end, 0.25 mm 15N SLED was titrated with an excess of either CG-rich (dsGGGCGCGCCC) or AT-rich (dsTTTATATAAA) double-stranded oligonucleotides. Their binding was monitored using NMR, and the binding affinities were compared.
SLED global chemical shift perturbations as a function of dsDNA concentration are shown in Fig. 7B for ASCL1, GC-, and AT-rich oligonucleotides. The dissociation constant (Kd) for each complex was determined as described under “Experimental Procedures.” The best fits are shown as lines, and the resulting Kd values are listed at the bottom of the graph. The ASCL1 DNA binds to SLED the tightest among the three tested dsDNA sequences with Kd of 560.4 ± 27.4 μm. The CG-rich dsDNA binds with Kd of 631.7 ± 165.9 μm, and AT-rich sequence binds the weakest with Kd of 973.3 ± 288.9 μm. The ASCL1 dsDNA causes significantly larger changes in the SLED spectrum when compared with either AT-rich or CG-rich oligonucleotides.
Taken together these results suggest that SLED can bind dsDNA in a sequence-specific manner and that CpG-containing DNA sequences (ASCL1 and CG-rich) are preferred ligands when compared with AT-rich DNA. A more global and systematic search for DNA motifs recognized by the SLED domain is needed to reveal the optimal DNA sequence recognized in vivo.
DISCUSSION
I have characterized a new domain within human Scml2 conserved among multicellular organisms, referred to as SLED (Figs. 1 and 2). The solution NMR structure of the Scml2 SLED domain revealed a novel α/β protein fold (Fig. 3). In vitro binding experiments have shown that the domain can interact with dsDNA and has a preference toward CpG-rich motifs (Figs. 6 and 7B). The highly conserved residues Ser-441 and Asn-440, displaying the largest NMR chemical shift changes upon DNA binding, are located in the 438PVNS441 loop, connecting β4 strand and α4 helix (Figs. 4A and 6C). High amino acid conservation in the DNA-binding region of SLED suggests that DNA binding is likely a common property of SLED domains across Metazoa.
Interestingly, three-dimensional structural alignment using DALI revealed that the DNA-binding loop of the SLED (438PVNS441) corresponds to the DNA-binding loop of SeqA (145TNNN148). Moreover, mutation of Asn-440 into alanine (N440A) completely abolishes DNA binding by SLED (Fig. 7A). The amino acid composition and conformation of the two loops, however, are not identical between the two proteins, suggesting different modes of DNA interaction and specificity.
The SLED domain structure determined here can explain the devastating effect of the previously reported C511Y mutation in Drosophila Scm that maps onto its SLED domain. This mutation results in severe developmental malformations and is lethal at embryonic stage (5), highlighting the functional importance of the Scm SLED domain. Residue Cys-511 in Drosophila Scm corresponds to a highly conserved Cys-453 in human Scml2 SLED, which is a part of the hydrophobic core of the domain. Hence, a C511Y mutation in Scm likely results in destabilization of the domain, impairing its ability to recognize DNA (Fig. 7C). Indeed, mutation of Cys-453 of Scml2 to tyrosine leads to severe SLED domain destabilization and unfolding as seen in Fig. 7D, showing the HSQC spectrum of the mutant with very limited dispersion of the peaks in the spectrum typical for an unfolded protein. As seen in Fig. 7C, the side chain of Cys-453 is very tightly packed within the SLED hydrophobic core, and its replacement with a large bulky tyrosine side chain with its hydroxyl group not only introduces a polar group into a hydrophobic environment but also causes significant steric clashes, resulting in SLED domain destabilization.
Another lethal mutation in the Drosophila Scm gene, C425Y, maps onto the SLED domain and involves a cysteine residue that is not conserved between flies and humans. The corresponding residue in human Scml2, Gly-365, is located in the loop connecting the β1 strand and α1 helix of the SLED domain. Mutations in this loop are unlikely to affect the domain stability. It is conceivable, however, that this loop may be involved in SLED domain recognition of yet unidentified binding partners. Interestingly, despite the lack of amino acid conservation between Drosophila and human, this loop is conserved in vertebrates (Fig. 4A). It is located on the opposite face of the SLED domain relative to the DNA-binding site, suggesting that DNA binding is likely not the only function of the SLED domain and that its large conserved surface might contain binding sites for additional ligands.
Although the binding between the Scml2 SLED domain and a DNA fragment derived from a CpG island within the promoter region of the ASCL-1 gene examined in this work is rather weak, the affinity of this interaction is of the same order of magnitude as that of the N-terminal Scml2 MBT domains toward methylated histones. Therefore, one can expect that multiple interactions that mediate association of the full-length Scml2 with the nucleosome cumulatively increase the affinity of Scml2 and, subsequently, PRC1 complex to chromatin in vivo. I also showed that Scml2 SLED-DNA binding is sequence-dependent and that the DNA sequence used in this work is not necessarily the optimal one. More rigorous in vivo studies are necessary to determine the optimal DNA sequences recognized by Scml2 in the context of chromatin.
Remarkably, previous studies have shown that the full-length Scml2 can effectively bind DNA in a manner independent of its MBT domains, which is in agreement with results shown here (18). It was also suggested that Scml2 may contain a DNA-binding motif similar to an AT-hook that is responsible for DNA binding (18). The proposed motif spans the residues 285–310 in close proximity to the SLED domain (354–468), which may additionally enhance DNA binding by Scml2.
A list of potential targets of SLED may include a few other proteins that were shown to directly interact with Scml2, such as modified histones and several PRC1 subunits (2). Further systematic studies are necessary to identify additional Scml2 interaction partners.
This work presents the first experimental evidence that Scml2 harbors a conserved DNA-binding domain that may be responsible for PRC1 targeting to chromatin, providing a starting point for further structural and functional studies of Scml2-DNA interactions.
CONCLUSION
NMR spatial structure determination of the SLED domain, a previously unidentified conserved interaction module within the Scml2 transcription factor, is an important step toward uncovering Scml2 function. DNA binding by Scml2 SLED may prove important for PRC1 recognition of its target genes.
Acknowledgment
I thank Dr. D. M. Korzhnev for many helpful discussions and suggestions on how to improve the manuscript.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (code 2MEM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The 1H, 15N, 13C NMR chemical shift assignments and restraints used for structure calculation in this paper were deposited to the Biological Magnetic Resonance Bank database under BMRB ID 19526.
- PcG
- Polycomb group
- SLED
- Scm-like embedded domain
- MBT
- malignant brain tumor domain
- SAM
- sterile α motif
- PHC
- polyhomeotic-like protein
- HSQC
- heteronuclear single quantum correlation
- r.m.s.
- root mean square
- TOCSY
- total correlation spectroscopy.
REFERENCES
- 1. Aloia L., Di Stefano B., Di Croce L. (2013) Polycomb complexes in stem cells and embryonic development. Development 140, 2525–2534 [DOI] [PubMed] [Google Scholar]
- 2. Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D. (2012) PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simon J. A., Kingston R. E. (2013) Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang L., Jahren N., Miller E. L., Ketel C. S., Mallin D. R., Simon J. A. (2010) Comparative analysis of chromatin binding by Sex Comb on Midleg (SCM) and other Polycomb group repressors at a Drosophila Hox gene. Mol. Cell. Biol. 30, 2584–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bornemann D., Miller E., Simon J. (1998) Expression and properties of wild-type and mutant forms of the Drosophila sex comb on midleg (SCM) repressor protein. Genetics 150, 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montini E., Buchner G., Spalluto C., Andolfi G., Caruso A., den Dunnen J. T., Trump D., Rocchi M., Ballabio A., Franco B. (1999) Identification of SCML2, a second human gene homologous to the Drosophila Sex comb on midleg (Scm): a new gene cluster on Xp22. Genomics 58, 65–72 [DOI] [PubMed] [Google Scholar]
- 7. Shao Z., Raible F., Mollaaghababa R., Guyon J. R., Wu C. T., Bender W., Kingston R. E. (1999) Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98, 37–46 [DOI] [PubMed] [Google Scholar]
- 8. Kyba M., Brock H. W. (1998) The SAM domain of polyhomeotic, RAE28, and Scm mediates specific interactions through conserved residues. Dev. Genet. 22, 74–84 [DOI] [PubMed] [Google Scholar]
- 9. Kim C. A., Sawaya M. R., Cascio D., Kim W., Bowie J. U. (2005) Structural organization of a Sex-comb-on-midleg/polyhomeotic copolymer. J. Biol. Chem. 280, 27769–27775 [DOI] [PubMed] [Google Scholar]
- 10. Maurer-Stroh S., Dickens N. J., Hughes-Davies L., Kouzarides T., Eisenhaber F., Ponting C. P. (2003) The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 28, 69–74 [DOI] [PubMed] [Google Scholar]
- 11. Grimm C., Matos R., Ly-Hartig N., Steuerwald U., Lindner D., Rybin V., Müller J., Müller C. W. (2009) Molecular recognition of histone lysine methylation by the Polycomb group repressor dSfmbt. EMBO J. 28, 1965–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo Y., Nady N., Qi C., Allali-Hassani A., Zhu H., Pan P., Adams-Cioaba M. A., Amaya M. F., Dong A., Vedadi M., Schapira M., Read R. J., Arrowsmith C. H., Min J. (2009) Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 37, 2204–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Santiveri C. M., Lechtenberg B. C., Allen M. D., Sathyamurthy A., Jaulent A. M., Freund S. M., Bycroft M. (2008) The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine. J. Mol. Biol. 382, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 14. Li H., Fischle W., Wang W., Duncan E. M., Liang L., Murakami-Ishibe S., Allis C. D., Patel D. J. (2007) Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell 28, 677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grimm C., de Ayala Alonso A. G., Rybin V., Steuerwald U., Ly-Hartig N., Fischle W., Müller J., Müller C. W. (2007) Structural and functional analyses of methyl-lysine binding by the malignant brain tumour repeat protein Sex comb on midleg. EMBO Rep. 8, 1031–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Min J., Allali-Hassani A., Nady N., Qi C., Ouyang H., Liu Y., MacKenzie F., Vedadi M., Arrowsmith C. H. (2007) L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 14, 1229–1230 [DOI] [PubMed] [Google Scholar]
- 17. Wang W. K., Tereshko V., Boccuni P., MacGrogan D., Nimer S. D., Patel D. J. (2003) Malignant brain tumor repeats: a three-leaved propeller architecture with ligand/peptide binding pockets. Structure 11, 775–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nady N., Krichevsky L., Zhong N., Duan S., Tempel W., Amaya M. F., Ravichandran M., Arrowsmith C. H. (2012) Histone recognition by human malignant brain tumor domains. J. Mol. Biol. 423, 702–718 [DOI] [PubMed] [Google Scholar]
- 19. Kassis J. A., Kennison J. A. (2010) Recruitment of Polycomb complexes: a role for SCM. Mol. Cell. Biol. 30, 2581–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sattler M., Schleucher J., Griesinger C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34, 93–158 [Google Scholar]
- 21. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 22. Goddard T. D, Kneller D. G. (2008) SPARKY 3, University of California, San Francisco [Google Scholar]
- 23. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 24. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 26. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 27. Schultz J., Milpetz F., Bork P., Ponting C. P. (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U.S.A. 95, 5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Letunic I., Doerks T., Bork P. (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 30. Letunic I., Bork P. (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Letunic I., Doerks T., Bork P. (2009) SMART 6: recent updates and new developments. Nucleic Acids Res. 37, D229–D232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ikeya T., Terauchi T., Guntert P., Kainosho M. (2006) Evaluation of stereo-array isotope labeling (SAIL) patterns for automated structural analysis of proteins with CYANA. Magn. Reson. Chem. 44, Spec. No S152–S157 [DOI] [PubMed] [Google Scholar]
- 34. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holm L., Kääriäinen S., Wilton C., Plewczynski D. (2006) Using Dali for structural comparison of proteins. in Current Protocols in Bioinformatics, Chapter 5, Unit 5 5, 10.1002/0471250953.bi0505s14 [DOI] [PubMed] [Google Scholar]
- 36. Holm L., Sander C. (1997) Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 25, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holm L., Sander C. (1995) Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 [DOI] [PubMed] [Google Scholar]
- 38. Chung Y. S., Brendler T., Austin S., Guarné A. (2009) Structural insights into the cooperative binding of SeqA to a tandem GATC repeat. Nucleic Acids Res. 37, 3143–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujikawa N., Kurumizaka H., Nureki O., Tanaka Y., Yamazoe M., Hiraga S., Yokoyama S. (2004) Structural and biochemical analyses of hemimethylated DNA binding by the SeqA protein. Nucleic Acids Res. 32, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guarné A., Zhao Q., Ghirlando R., Yang W. (2002) Insights into negative modulation of E. coli replication initiation from the structure of SeqA-hemimethylated DNA complex. Nat. Struct. Biol. 9, 839–843 [DOI] [PubMed] [Google Scholar]
- 41. Ringrose L. (2007) Polycomb comes of age: genome-wide profiling of target sites. Curr. Opin. Cell Biol. 19, 290–297 [DOI] [PubMed] [Google Scholar]
- 42. Ringrose L., Paro R. (2007) Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134, 223–232 [DOI] [PubMed] [Google Scholar]
- 43. Schuettengruber B., Cavalli G. (2009) Recruitment of Polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136, 3531–3542 [DOI] [PubMed] [Google Scholar]