

Abstract
Background
Three major processes, constrictive vessel remodeling, intimal hyperplasia and retarded re-endothelialization, contribute to restenosis after vascular reconstructions. Clinically used drugs inhibit intimal hyperplasia but delay re-endothelialization and also cause constrictive remodeling. Here we have examined halofuginone (HF), a herbal derivative, for its beneficial effects on vessel remodeling and differential inhibition of intimal hyperplasia versus re-endothelialization.
Methods and Results
Two weeks after perivascular application to balloon-injured rat common carotid arteries, HF versus vehicle (n=6 animals) enlarged luminal area 2.14 fold by increasing vessel size (adaptive remodeling, 123%), reducing intimal hyperplasia (74.3%) without inhibiting re-endothelialization. Consistent with its positive effect on vessel expansion, HF reduced collagen type-1 (but not type-3) production in injured arteries as well as that from adventitial fibroblasts in vitro. In support of its differential effects on intimal hyperplasia versus re-endothelialization, HF produced greater inhibition of vascular smooth muscle cell versus endothelial cell proliferation at concentrations around 50 nM. Furthermore, HF at 50 nM effectively blocked Smad3 phosphorylation in smooth muscle cells which is known to promote smooth muscle cell proliferation, migration, and intimal hyperplasia, but HF had no effect on phospho-Smad3 in endothelial cells.
Conclusions
Periadventitial delivery of HF dramatically increased lumen patency via adaptive remodeling and selective inhibition of intimal hyperplasia without affecting endothelium recovery. HF is the first reported small molecule that has favorable effects on all three major processes involved in restenosis.
Keywords: halofuginone, adaptive remodeling, re-endothelialization, collagen subtypes, phospho-Smad3
Cardiovascular disease is the most common cause of death in the developed world. Atherosclerosis is the primary pathology underlying the great majority of cardiovascular morbidity including coronary artery and peripheral vascular disease1. Vascular interventions such as angioplasty and bypass are commonly performed to restore circulation to vital organs. However, such treatments inflict injury to the native vessel wall causing vessel re-narrowing (or restenosis) in a great number of patients. The injury, including denudation of the endothelial layer and mechanical stretching of the vessel, evokes an excessive wound healing response, including the release of various cytokines and growth factors at the injury site from monocytes and macrophages2. There are at least three major processes contributing to restenosis: 1) constrictive remodeling or overall constriction of the vessel wall which has been proposed to be attributable to excessive matrix protein production (e.g. collagen type -1) in the adventitia3, 2) intimal hyperplasia (IH) which is overgrowth of cells and matrix in the subintimal layer1, and 3) delayed recovery of the protective endothelial inner lining leading to thrombosis and also exacerbation of IH and constrictive remodeling2.
In contrast to our knowledge about IH, the mechanisms of arterial remodeling are much less understood3. Arterial remodeling results in a permanent change in vessel diameter; this might be through vessel expansion (termed adaptive remodeling) or shrinkage (termed constrictive remodeling)3. As has been observed in patients and in animal models of restenosis, constrictive remodeling accounts for a considerable portion of the lumen loss, and in many cases can be a greater contributor to restenosis than IH. Either prevention of constrictive remodeling or alternatively, induction of adaptive remodeling has been shown to attenuate post-angioplasty lumen loss (see review)3. It is thought that deposition of extracellular matrix (ECM) in the adventitia might be contributory to constrictive remodeling following angioplasty. In particular, an increase in collagen type 1 (Col-1) is often associated with constrictive remodeling; as is the case with other fibrotic diseases (e.g. in the liver, pancreas, and skin)3. Inhibition of Col-1 production has been used as a general strategy for treating fibrosis. Nevertheless, arterial remodeling is a complex process. It is likely that not only collagen content, but also crosslinking and collagen subtype, play important roles3. We have recently shown that an increase in the ratio of Col-3 (an elastic subtype of collagen) to Col-1 (a more rigid subtype) is associated with adaptive remodeling4.
While both IH and constrictive remodeling result in narrowing of the vessel lumen, rapid re-endothelialization following arterial injury can both prevent thrombosis as well as prevent IH2. In normal arteries, endothelial cells (ECs) form a protective inner lining that function as a barrier between the circulating blood and the vessel wall2. However, after interventions for atherosclerosis, ECs are denuded and SMCs are left exposed to cytokines and growth factors and thus transformed into active contributors to intimal growth and/or constrictive remodeling, resulting in restenosis. Therefore, the more rapidly the endothelial layer can be recovered, the greater the inhibitory effect on restenosis. Thus, a drug that selectively inhibits SMC proliferation with less impairment of EC growth provides an optimal recipe to prevent restenosis. ECs in general have been known to be more sensitive to growth inhibitors than SMCs. Probably for this reason only very few agents have been reported to selectively inhibit the growth of SMCs versus ECs5.
Currently the only clinically used anti-restenosis drugs are Rapamycin and Paclitaxel. While these two widely used drugs are effective in reducing IH, there is evidence that they cause constrictive remodeling6. Moreover, Rapamycin and Paclitaxel have been reported to inhibit re-endothelialization, causing late-stent thrombosis and sudden death2, 7. Therefore, a therapeutic agent that can manipulate in a favorable manner, each of the three processes that lead to restenosis would theoretically be an extremely effective tool for preventing restenosis versus a simple inhibitor of IH. To the best of our knowledge, no such agent has yet been reported.
Halofuginone (HF) is a derivative of febrifugine, one of the fifty fundamental herbs of traditional Chinese medicine. This ancient drug is attracting tremendous interest for its potent ECM modulating, anti-fibrotic, anti-metastatic, and anti-stress effects8. In a recently completed phase II clinical trial, HF was efficacious in reducing the lesions associated with Kaposi sarcoma. This drug has also entered phase I clinical trials for cancer9. A positive effect of HF on injury-induced IH was observed in two prior studies using less relevant models10, 11. However, whether HF affects post-injury arterial remodeling and re-endothelialization has not been previous reported. Of particular relevance to the process of vascular remodeling, HF is a specific inhibitor of Col-1 synthesis and thus has an anti-fibrotic effect in various tissues8. Based on the previous observations that excessive Col-1 deposition leads to constrictive remodeling3, we therefore hypothesized that HF may promote adaptive remodeling. Indeed, we found that HF not only reduced IH but importantly, also produced adaptive remodeling. Moreover, HF had the added benefit of preserving luminal re-endothelialization. Our findings suggest that when administered locally HF is a favorable anti-restenosis agent with significant potential clinic utility.
Methods
An expanded Methods section is available in Supplemental Material.
Rat carotid artery balloon angioplasty model, perivascular HF delivery, and morphometry
All animal studies conform to the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, 1996 revision) and protocols approved by the Institutional Animal Care and Use Committee at the University of Wisconsin. IRB approval has been obtained for use of cells derived from human samples. Carotid artery balloon angioplasty was performed in male Sprague-Dawley rats (Charles River, 250–300g) as previously described12. HF was then applied to the outside of the injured segment of the carotid artery by using F-127 pluronic gel (Sigma-Aldrich)12. This perivascular approach proved to be effective in the delivery of small molecule drugs (such as rapamycin) or even nanoparticles into the injured rat carotid arterial wall13. Prior to administration, 30 μg of HF from a DMSO stock was first dissolved in 30 μl of 10% DMSO and then mixed with 270 μl of 25% pluronic gel that was maintained on ice. We chose a concentration in vivo that was substantially higher than what has been shown to be effective in vitro because of the potential for HF to diffuse away from the arterial wall. In the control group, equal volume of vehicle (30 μl of 20% DMSO) mixed with pluronic gel was applied. Animals were sacrificed at 14 days post angioplasty, a time point at which significant remodeling occurred in our previous study12. Adaptive remodeling was defined as an increase of external elastic lamina (EEL) area. IH was quantified as a ratio of intima area versus media area. Re-endothelialization was assessed using Evans Blue assay as well as CD31 immunostaining5.
Cell culture and in vitro HF treatment
Rat aortic vascular smooth muscle cells (SMCs) and fibroblasts were isolated from the thoracoabdominal aorta of male Sprague-Dawley rats (anesthetized under 2.5% isoflurane) based on a protocol using an enzymatic dissociation method14. Rat aortic endothelial cells (ECs) were purchased from Genlantis. For HF treatment, cells were first starved for 12 or 24h in DMEM containing 0.5% FBS, the medium was then replaced with fresh DMEM supplemented with 10% FBS in which HF was added to a desired concentration. For control, equal volume of DMSO was added to a final concentration of 0.1 % (v/v versus medium). Proliferation of cultured SMCs was determined using cell number as a surrogate, for which Alamar Blue (Invitrogen) assay was performed as previously described5.
Statistical analysis
Data are presented as mean ± standard error (SEM). Statistical analysis was conducted using two-tailed unpaired Student’s t-test and ANOVA. Statistical significance was validated by P < 0.05.
Results
Halofuginone produces adaptive remodeling and reduces intimal hyperplasia in balloon-injured rat carotid arteries
Excessive Col-1 deposition has been suggested to be a cause of constrictive remodeling following arterial injury3, and HF is a specific inhibitor of Col-1 synthesis, in cell lines including fibroblasts8. These reports prompted us to test whether HF, due to its Col-1 inhibiting properties, might prevent constrictive arterial remodeling after injury.
We performed rat carotid balloon angioplasty, which is a widely accepted model to mimic angioplasty in humans, and then administered HF locally, by dissolving HF in pluronic gel and then applying the gel around the injured artery. The results of morphometric analyses using carotid sections collected on day 14 post angioplasty are shown in Figure 1. We found that compared to vehicle (DMSO) control, HF treatment produced a 123% increase in EEL area (Figure 1C), indicating that HF induced adaptive remodeling following balloon injury of rat carotid arteries. Further analysis revealed that the intima/media ratio, a measure of intimal hyperplasia (IH), decreased by 74.3% (Figure 1D), and luminal area increased by 2.14 fold (Figure 1E).
Figure 1. Effect of HF treatment on vessel size, neointima formation, and lumen size in rat carotid arteries following angioplasty.

Morphometric analysis was performed on the H&E-stained sections of rat carotid arteries that were collected on day 14 after angioplasty. A and B are representative sections from arteries treated with vehicle and HF, respectively. Arrow indicates external elastic lamina (EEL); arrowheads mark internal elastic lamina (IEL). C, D, and E are quantified data of arterial remodeling (EEL area), intimal hyperplasia (ratio of intima/media area), and lumen area, respectively. Each bar represents a mean (±SEM) of sections from 6 animals, *P< 0.05 compared to DMSO control.
Halofuginone inhibits the production of type 1 but not type 3 collagen from adventitial fibroblasts in vivo and in vitro
An increase in the ratio of Col-3/Col-1, the critical adventitial collagen subtypes, is believed to contribute to favorable arterial remodeling4. However, the effect of HF on Col-1 and Col-3 protein levels in arteries treated with angioplasty has not been previously evaluated. In order to assess whether HF might facilitate adaptive remodeling by affecting collagen homeostasis, we examined the effect of HF on the production of Col-1 and Col-3 in vivo and in vitro. Through immunostaining of carotid sections collected 14 days after angioplasty, we found that Col-1 was significantly reduced in the adventitia in HF-treated balloon-injured arteries compared to vehicle control (Figure 2C). In contrast, Col-3 levels were not significantly changed although there was a numerical increase (Figure 2F). We then further tested the HF effects on these two collagen subtypes in vitro using primary rat aortic adventitial fibroblasts. We found that while the Col-1 secretion, measured by Western blotting, from fibroblasts decreased by 30% (relative to control) in the presence of 50 nM HF, Col-3 secretion did not change significantly (Figure 2, G and H). The combined evidence from these in vivo and in vitro experiments indicates that HF specifically inhibits the production of Col-1 versus Col-3 by adventitial fibroblasts, thus providing a potential mechanism for this drug’s favorable effect on adaptive remodeling.
Figure 2. HF treatment inhibits the production of Col-1 but not Col-3 in the adventitia of rat carotid arteries after angioplasty as well as in adventitial fibroblasts in vitro.

Shown in A, B, D, and E are representative rat carotid artery sections immunostained for Col-1 (A and B) and Col-3 (D and E). Arteries treated with either vehicle (DMSO, A and D) or HF (B and E) were collected on day 14 following angioplasty. Arrowhead (red) marks IEL. Quantification of Col-1 (C) and Col-3 (F) in the adventitia was performed using Image J. HPF: high power field. Each bar represents a mean (±SEM) of all the sections taken from 4 animals (9 sections per animal), *P< 0.05 compared to DMSO control.
The effect of HF on secretion of Col-1 and Col-3 from rat aortic adventitial fibroblasts in vitro was also assessed (G and H). Fibroblasts were starved for 24h in DMEM containing 0.5% FBS. The medium was then replaced with DMEM containing 10% FBS added with either vehicle (DMSO) or HF (50 nM). After incubation for 48h, the medium was collected and concentrated and subjected to Western blotting analysis. G shows quantification of Col-1 and Col-3 levels normalized to β-actin. Each bar represents a mean (±SEM) of three separate experiments, *P< 0.05 compared to DMSO. H shows a representative Western blot.
Halofuginone inhibits rat SMC proliferation in balloon-injured arteries as well as in cell culture
SMC proliferation and migration are critical contributors to IH1. We investigated whether HF inhibited intimal growth by altering these SMC behaviors. We first addressed this question in vivo by immunostaining rat carotid sections for PCNA, a marker for proliferating cells. Indeed, PCNA-positive cells were greatly reduced (by 66.2%) in the HF-treated arteries relative to that of vehicle-treated controls (Figure 3A), indicating a strong inhibitory effect of HF on SMC proliferation in injured arteries. We then confirmed the inhibitory effect of HF on proliferation of cultured SMCs. As shown in Figure 3B, 10% serum produced a 2.2 fold increase in rat aortic medial SMC proliferation. While HF at 1 nM had no effect on SMC proliferation, 50 nM HF reduced serum-stimulated proliferation by 70.6% and 100 nM HF reduced by 82.3%.
Figure 3. HF treatment reduces PCNA-positive cells in vivo and SMC proliferation and migration in vitro.

A. PCNA immunostaining of carotid arteries treated periadventitially with either vehicle (DMSO) or HF. Sections were collected 14 days after angioplasty. Shown are representative cross sections with quantification of PCNA positive cells. HPF: high power field. Arrowhead (red) marks IEL. Each bar represents a mean (±SEM) of 36 sections from 4 animals, *P< 0.05.
B. Effect of HF on the proliferation of cultured rat SMCs. Cells were starved for 24h in DMEM containing 0.5% FBS. Medium was then changed to 10% FBS with either vehicle (DMSO) or different concentrations of HF. After incubation for 72h, Alamar Blue dye was added and fluorescence was measured 24h later. Each bar represents a mean (±SEM) of three separate experiments, *P< 0.05 compared to the condition of stimulation with 10% serum without HF treatment.
C. Effect of HF on the migration of cultured SMCs. Cells were starved for 24h and then stimulated with 10% FBS in the presence of vehicle or HF (50nM). A scratch was made prior to the FBS stimulation. Quantification of migration is expressed as a relative rate of recovery of the scratch wound. Each bar represents a mean (±SEM) of three separate experiments, *P< 0.05.
Moreover, using an in vitro scratch model of migration, we found that 50 nM HF inhibited rat aortic SMC migration by 73.6% 48h after treatment (Figure 3C). Taken together, our data indicate that HF potently inhibits proliferation as well as migration of vascular SMCs.
Halofuginone preserves re-endothelialization in balloon-injured arteries
Encouraged by the dramatic reduction of IH by HF and its favorable effect on adaptive remodeling, we next determined whether HF affects re-endothelialization, another important factor involved in the development of restenosis. Two weeks after angioplasty, we incised longitudinally the carotid arteries that were stained in the lumen with Evans Blue, a widely used method to measure re-endothelialization. Our data reveal that the ratio of re-endothelialized area (not stained by Evans Blue) versus total area was greater in HF-treated arteries versus vehicle-treated controls (Figure 4A). In order to confirm this finding, we performed another commonly used assay for re-endothelialization, immunostaining of CD31 (a marker of ECs). Similarly, we found that re-endothelialization was not reduced but rather, significantly increased in HF-treated vessels compared to vehicle-treated vessels subjected to angioplasty (Figure 4B). Thus, our data indicate that HF applied periadventitially preserved re-endothelialization of rat carotid arteries following balloon angioplasty.
Figure 4. HF treatment preserves re-endothelialization in rat carotid arteries after angioplasty.

A, Evans Blue staining. Arteries were treated at the time of balloon angioplasty with either vehicle (DMSO) or HF and stained with Evans Blue on day 14 after angioplasty. Shown are representative stained arteries; uninjured arteries were used as positive control. The areas denuded of endothelium are blue; re-endothelialized areas are unstained. Re-endothelialization was quantified and expressed as the ratio of unstained versus total area. Each bar represents a mean (±SEM) of 6 animals.
B. Immunostaining for CD31. Arteries were treated at the time of balloon angioplasty with either vehicle (a) or HF (b) and collected on day 14 after angioplasty for CD31 immunostaining. Inset in (a) shows the distinction of CD31 staining (brown) and nuclei counter-staining with hematoxylin (blue). Uninjured artery serves as a positive control for CD31 staining (c). Arrow (blue) indicates EEL; arrow head (red) marks IEL. Quantification (d) was performed as described in Methods. Each bar represents a mean (±SEM) of 4 animals.
Halofuginone preferentially inhibits SMC versus EC proliferation in vitro
A highly desirable property of drugs designed to inhibit restenosis is selective inhibition of SMC versus EC proliferation, leading to suppression of intimal growth but not re-endothelialization in arteries injured by vascular reconstruction. Very few agents have been shown to possess this beneficial feature5. The observed in vivo inhibitory effect of HF on IH but not re-endothelialization prompted us to examine differential effects of HF on the in vitro proliferation of primary aortic SMCs and aortic ECs both at passage 5. To observe the effect of HF over a range of concentrations, we created dose-response curves of both cell types. At high concentrations (>100 nM) HF equally inhibited the growth of both cell types. However, at some lower concentrations around 50 nM, HF preferentially inhibited SMC versus EC proliferation (Figure 5). These results indicate that HF is a preferential inhibitor of SMC versus EC proliferation in a certain range of concentrations.
Figure 5. Differential inhibitory effect of HF on the proliferation of SMCs versus ECs.
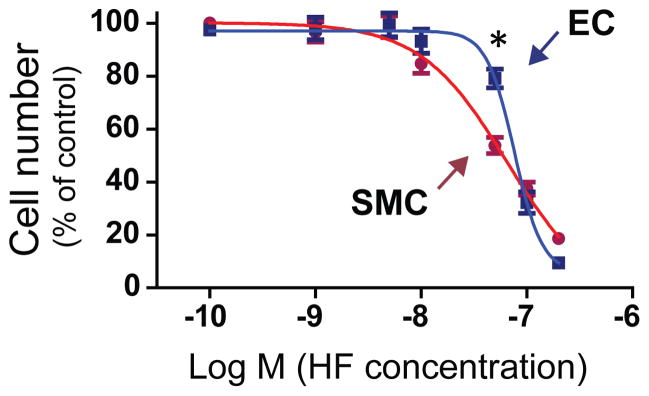
Dose response experiments using human aortic SMCs and human aortic ECs are described in detail in Methods (see supplementary materials). Shown is a representative of three similar experiments. Each data point is a mean (±SD) of triplicates, *P< 0.05 compared to SMCs at the same concentration (50 nM).
Halofuginone inhibits phosphorylation of Smad3 in balloon-injured arteries as well as in cultured rat aortic SMCs but not aortic ECs
We further explored the molecular mechanism by which HF differentially inhibits SMC versus EC proliferation by assessing its effect on the canonical TGFβ/Smad3 pathway. Our group has previously shown that TGFβ and its signaling effector Smad3 are up-regulated following arterial injury and contribute to neointima formation by stimulating proliferation of vascular SMCs12, 14. Knocking down Smad3 inhibits SMC proliferation and IH14, 15. Interestingly, an inhibitory effect of HF on Smad3 activation (phosphorylation) was observed in some other cell types16, 17 but has not been examined in vascular SMCs. We therefore investigated whether HF might inhibit Smad3 signaling differentially in SMCs versus ECs.
We first evaluated the effect of HF treatment on phospo-Smad3 in the injured arterial wall, providing a mechanism for its inhibitory effect on SMC proliferation and IH. We found that pSmad3 (Figure S1) were greatly increased in rat carotid arteries 14 days after angioplasty compared to uninjured arteries. Cells stained positively for pSmad3/Smad3 were located in the neointimal and medial layers (Figure 6, A and D). In particular, these Smad3-expressing cells were enriched in the region adjacent to the lumen. Remarkably, treatment of injured arteries with HF substantially reduced pSmad3 (by 54.9%, Figure 6C).
Figure 6. Differential inhibitory effect of HF on phospho-Smad3 in SMCs versus ECs.

A–F. Immunostaining of pSmad3. A, B, D, and E are representative rat carotid artery sections immunostained for pSmad3 (A and B) or Smad3 (D and E). Arteries treated with either vehicle (A and D) or HF (B and E) were collected on day 14 after angioplasty. Arrowhead (red) marks IEL. Quantifications of pSmad3 and Smad3 are presented in C and F, respectively. HPF: high power field. Each bar represents a mean (±SEM) of 36 sections from 4 animals, *P< 0.05.
G and H. Western blotting of pSmad3. In vitro experiments were performed to determine the effect of HF on Smad3 phosphorylation in rat aortic SMCs and rat aortic ECs. Cells were starved for 12h in DMEM containing 0.5% FBS and incubated with vehicle (DMSO) or HF (50nM) for 2h. The cells were then treated with TGFβ (5 ng/ml) for 30 min or with 10% FBS for 6h followed by cell lysate preparation and Western blotting. pSmad3 levels were quantified and normalized to β-actin. Each bar represents a mean (±SEM) of three independent experiments, *P< 0.05.
We then compared in vitro the effect of HF on Smad3 activation in SMCs and ECs, both primary cells from rat aorta (Figure 6, G and H). The Western blot data demonstrate that whereas in SMCs HF greatly reduced serum-stimulated Smad3 phosphorylation, pSmad3 levels in ECs were not changed by HF treatment. We then further determined if the HF effect on pSmad3 was mediated through the canonical TGFβ/Smad3 signaling pathway. Indeed, we found that in SMCs HF inhibited TGFβ-activated Smad3 signaling but did not affect pSmad3 in ECs. Thus, the combined in vivo and in vitro data indicate that HF effectively blocked phosphorylation of Smad3 in SMCs without altering Smad3 activation in ECs. These results suggest that HF may preferably inhibit SMC versus EC proliferation through a mechanism that involves differential inhibition of Smad3 signaling in these two cell types.
Discussion
In this study we have made a novel finding that when locally administered to balloon-injured rat carotid arteries, halofuginone (HF) effectively induces adaptive remodeling and selectively inhibits neointima growth without impeding re-endothelialization. The sum of HF’s effects is a dramatic twofold increase in lumen patency in treated arteries compared to controls. These favorable features of HF suggest that this small molecule might have an important clinical role in the treatment of stenotic diseases.
An inhibitory effect of HF on intimal hyperplasia has been examined in two prior studies10, 11. However, there are several distinct and novel differences between our findings and those of the previous studies. First, in the prior studies, the models used were less relevant to restenosis (ear crush in one and carotid anastomosis in the other). Moreover, the effect of HF in our study was substantial and much greater than what has been previously observed10, 11; we found a dramatic two-fold increase of luminal area in arteries treated with HF. Our findings show that the effect of HF is largely related to its ability to influence adaptive remodeling. Lastly, we found that HF has a selective effect on SMC proliferation and IH but spares re-endothelialization. Thus we believe that halofuginone is the first small molecule that has been shown to produce a beneficial effect on all three major processes critical to restenosis (intimal hyperplasia, adaptive remodeling and re-endothelialization).
While the effect of HF on IH has been explored10, 11, how HF affects arterial remodeling and endothelial recovery was not known. We tackled this question through rat carotid balloon angioplasty, a widely accepted model recapitulating restenosis despite the absence of an atherosclerotic plaque18. The critical role of adaptive remodeling in prevention of restenosis has been highlighted in several recent reports (for review, see Goel et al3). Studies in the clinical setting and in animal models have shown that constrictive remodeling can be an even more important contributor to restenosis than IH. Geometrically, relatively small changes in overall vessel diameter produced by adaptive remodeling can result in major changes in luminal area. For example, a 10% increase in vessel diameter without a change in plaque area can result in a 150% increase in luminal area19. In a similar manner but conversely, constrictive remodeling can cancel the gain of luminal area that is made by effective inhibition of IH. Our own studies in the rat carotid injury model reveal that rapamycin, which has a substantial inhibitory effect on IH, is also associated with constrictive remodeling (Kent et al, unpublished data).
Although an intraluminally implanted stent can prevent constrictive remodeling, this approach is not applicable to open vascular reconstructions (>300,000 cases per year in the US) including bypass and endarterectomy13. Moreover, stents are costly; they have the ability to incite in-stent IH2 and stent-edge constrictive remodeling6, making re-intervention much more challenging. Thus, stent-free approaches are becoming highly desirable for treating stenotic arteries and veins. Manipulating arterial remodeling from constrictive to adaptive, appears to be a promising strategy that can profoundly attenuate restenosis following open surgery, or angioplasty without the need to implant a stent. There are only a limited number of drugs or molecular interventions that have been shown to produce adaptive remodeling. As well, the mechanisms that influence arterial remodeling remain poorly understood3.
Adventitial extracellular matrix proteins, especially collagen, are thought to contribute to the mechanism of arterial remodeling3. Constrictive remodeling resembles the fibrotic process that contributes to multiple diseases19; excessive Col-1 accumulation leads to scarring and arterial constriction. As a specific inhibitor of Col-1 synthesis11, HF has shown efficacy in several conditions that have fibrosis as their cause8. Thus in a similar manner, by inhibiting Col-1, HF may prevent constrictive remodeling. There are at least two mechanisms that may contribute to HF-induced adaptive remodeling. First, inhibition of Col-1 production by HF likely prevents excessive Col-1 deposition in the adventitia thus mitigating the constrictive band that produces arterial narrowing. Second, the decrease of Col-1 content may alter the Col-1 to Col-3 ratio in the adventitia. Col-1 and Col-3 are the two major subtypes in the ECM of vasculature. While Col-1 confers rigidity and tensile strength, Col-3 provides elasticity to the vessel wall3. Studies in non-human primates as well as humans reveal that an increased ratio of Col-3/Col-1 is beneficial for normal vascular function and adaptive remodeling20. Moreover, we have recently found in balloon-injured rat carotid arteries, that adaptive remodeling induced by perivascular application of CTGF is associated with enhanced production of Col-3 versus Col-14. In the current study, a decrease in Col-1 (but not Col-3) content in HF-treated arteries favorably increased the Col-3/Col-1 ratio, and thus may have contributed to adaptive remodeling.
It is well recognized that both SMC proliferation and migration play critical roles in the formation of IH1. While an inhibitory effect of HF on bovine arterial SMC proliferation has been previously observed10, we provide herewith the first in vivo evidence of HF inhibition of cell proliferation in the injured arterial wall. Moreover, we found that migration of rat SMCs is impeded by HF as well. An effect of HF on SMC migration has not been previously reported. This impairment of proliferation as well as migration of SMCs may to a great extent account for the mitigation of IH in HF-treated arteries.
One of the major drawbacks of anti-restenotic drugs that inhibit SMC proliferation is their similar negative impairment of EC proliferation2. In fact, ECs are usually more sensitive to inhibitors than SMCs5. Vessels undergoing reconstruction or angioplasty are denuded of the endothelial layer and these drugs impair the process of re-endothelialization. The consequence is the potential for acute thrombosis since ECs provide the protective anti-thrombotic luminal lining2. Well-known examples are the two currently clinically applied drugs, Rapamycin and Paclitaxel. In addition to their therapeutic effect of inhibiting SMC proliferation and migration, these drugs also suppress the growth, mobility, and survival of ECs thus causing retarded re-endothelialization, thrombosis and enhanced restenosis2, 5. Obviously, the high likelihood that drugs will inhibit both SMC and EC proliferation imposes a major challenge for current and future interventions to treat restenosis. Drugs that selectively inhibit SMC proliferation but not EC growth are therefore highly desirable. Our data show that within a relatively large window of concentration (~5–100 nM), HF preferentially inhibits SMC versus EC proliferation. Indeed, consistent with these in vitro findings, HF treatment greatly inhibited injury-induced SMC neointimal proliferation but did not impair re-endothelialization. In contrast to our findings with HF, periadventitial administration of Rapamycin has been shown to significantly inhibit re-endothelialization7. Only a very few agents have been reported to differentially inhibit SMC versus EC proliferation5, and the molecular mechanisms for the selectivity are poorly understood. HF may provide a valuable tool for uncovering the mechanisms that differentially control SMC versus EC proliferation.
We investigated the mechanism underlying differential HF inhibition of SMC versus EC proliferation through the TGFβ/Smad3 pathway, which is a critical player in both cell types. Both TGFβ and Smad3 are up-regulated following arterial injury1. Studies from our lab and others have demonstrated that expression of Smad3 in injured arteries exacerbates IH whereas inhibition of Smad3 blocks SMC proliferation and IH14, 21. While HF has been previously shown to inhibit phosphorylation of Smad3 in pancreatic stellate cells16 and fibroblasts17, our data show that HF attenuates Smad3 phosphorylation in rat aortic SMCs as well. However in contrast, an inhibitory effect of HF on Smad3 phosphorylation was not observed in rat aortic ECs. This differential inhibitory effect of HF on the activation of Smad3 signaling in SMCs versus ECs is consonant with our observation that HF effectively attenuated SMC proliferation and IH but did not inhibit re-growth of the endothelium after arterial injury. Thus, this novel information of HF influence on Smad3 signaling provides interesting mechanistic insight into a preferential inhibition of SMC versus EC proliferation.
Study Limitations
The effect of HF in healthy rat carotid arteries may be different from that in diseased human vessels, which may limit the ability to extrapolate the current findings to outcomes in humans. It should also be noted that we have limited ability to translate the concentrations that were effective in vitro to concentrations in vivo. Thus, in the in vivo studies, we applied higher concentrations of HF assuming there would be substantial loss of the drug to the surrounding tissues due to diffusion. We were unable to measure the precise concentration of HF present within the arterial wall. Nonetheless, we observed substantial inhibition of cell proliferation and intimal hyperplasia by HF in balloon-injured arterial wall, suggesting that the concentration we used in vivo recapitulated HF’s in vitro effects.
Conclusions
We have identified HF as a drug that beneficially influences all three of the major processes that contribute to restenosis in a rat angioplasty model. HF stimulates adaptive remodeling while suppressing intimal growth and has no inhibitory effect on the recovery of the endothelial layer. There are no other small molecules reported that possess all three of these favorable properties.
Supplementary Material
Acknowledgments
We thank Dr. Bo Liu for helpful discussion and Sarah Franco for proof reading.
Sources of Funding
This work was supported by a National Heart, Lung, Blood Institute R01 Grant (HL-068673, to K. C. K), a T32 training Grant (HL-110853, to K.C.K.) and a Wisconsin Partnership Program New Investigator Award (ID 2832) (to L.-W. G.).
Footnotes
Disclosures
None.
References
- 1.Suwanabol PA, Kent KC, Liu B. Tgf-beta and restenosis revisited: A smad link. J Surg Res. 2011;167:287–297. doi: 10.1016/j.jss.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI. Vascular inflammation and repair: Implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc Interv. 2011;4:1057–1066. doi: 10.1016/j.jcin.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goel SA, Guo LW, Liu B, Kent KC. Mechanisms of post-intervention arterial remodelling. Cardiovasc Res. 2012;96:363–371. doi: 10.1093/cvr/cvs276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goel SA, Guo LW, Shi XD, Kundi R, Sovinski G, Seedial S, Liu B, Kent KC. Preferential secretion of collagen type 3 versus type 1 from adventitial fibroblasts stimulated by tgf-beta/smad3-treated medial smooth muscle cells. Cell Signal. 2012;25:955–960. doi: 10.1016/j.cellsig.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel SA, Guo LW, Wang B, Guo S, Roenneburg D, Ananiev GE, Hoffmann FM, Kent KC. High-throughput screening identifies idarubicin as a preferential inhibitor of smooth muscle versus endothelial cell proliferation. PloS one. 2014;9:e89349. doi: 10.1371/journal.pone.0089349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichimoto E, Fujimoto Y, Kubo K, Miyayama T, Iwata Y, Kitahara H, Kobayashi Y. Mechanism of edge restenosis after sirolimus-eluting stent implantation. The Journal of invasive cardiology. 2012;24:55–57. [PubMed] [Google Scholar]
- 7.Fukuda D, Enomoto S, Shirakawa I, Nagai R, Sata M. Fluvastatin accelerates re-endothelialization impaired by local sirolimus treatment. Eur J Pharmacol. 2009;612:87–92. doi: 10.1016/j.ejphar.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo CY, Mazitschek R, Whitman M. Halofuginone and other febrifugine derivatives inhibit prolyl-trna synthetase. Nat Chem Biol. 2012;8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng W, Robertson L, Gallinetti J, Mejia P, Vose S, Charlip A, Chu T, Mitchell JR. Surgical stress resistance induced by single amino acid deprivation requires gcn2 in mice. Sci Transl Med. 2012;4:118ra111. doi: 10.1126/scitranslmed.3002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagler A, Miao HQ, Aingorn H, Pines M, Genina O, Vlodavsky I. Inhibition of collagen synthesis, smooth muscle cell proliferation, and injury-induced intimal hyperplasia by halofuginone. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:194–202. doi: 10.1161/01.atv.17.1.194. [DOI] [PubMed] [Google Scholar]
- 11.Choi ET, Callow AD, Sehgal NL, Brown DM, Ryan US. Halofuginone, a specific collagen type i inhibitor, reduces anastomotic intimal hyperplasia. Arch Surg. 1995;130:257–261. doi: 10.1001/archsurg.1995.01430030027004. [DOI] [PubMed] [Google Scholar]
- 12.Kundi R, Hollenbeck ST, Yamanouchi D, Herman BC, Edlin R, Ryer EJ, Wang C, Tsai S, Liu B, Kent KC. Arterial gene transfer of the tgf-beta signalling protein smad3 induces adaptive remodelling following angioplasty: A role for ctgf. Cardiovasc Res. 2009;84:326–335. doi: 10.1093/cvr/cvp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi X, Chen G, Guo LW, Si Y, Zhu M, Pilla S, Liu B, Gong S, Kent KC. Periadventitial application of rapamycin-loaded nanoparticles produces sustained inhibition of vascular restenosis. PloS one. 2014;9:e89227. doi: 10.1371/journal.pone.0089227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC. Tgf-beta through smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol. 2009;297:H540–549. doi: 10.1152/ajpheart.91478.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edlin RS, Tsai S, Yamanouchi D, Wang C, Liu B, Kent KC. Characterization of primary and restenotic atherosclerotic plaque from the superficial femoral artery: Potential role of smad3 in regulation of smc proliferation. J Vasc Surg. 2009;49:1289–1295. doi: 10.1016/j.jvs.2008.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zion O, Genin O, Kawada N, Yoshizato K, Roffe S, Nagler A, Iovanna JL, Halevy O, Pines M. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas. 2009;38:427–435. doi: 10.1097/MPA.0b013e3181967670. [DOI] [PubMed] [Google Scholar]
- 17.McGaha TL, Phelps RG, Spiera H, Bona C. Halofuginone, an inhibitor of type-i collagen synthesis and skin sclerosis, blocks transforming-growth-factor-beta-mediated smad3 activation in fibroblasts. J Invest Dermatol. 2002;118:461–470. doi: 10.1046/j.0022-202x.2001.01690.x. [DOI] [PubMed] [Google Scholar]
- 18.Ali ZA, Alp NJ, Lupton H, Arnold N, Bannister T, Hu Y, Mussa S, Wheatcroft M, Greaves DR, Gunn J, Channon KM. Increased in-stent stenosis in apoe knockout mice: Insights from a novel mouse model of balloon angioplasty and stenting. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:833–840. doi: 10.1161/01.ATV.0000257135.39571.5b. [DOI] [PubMed] [Google Scholar]
- 19.Kakuta T, Currier JW, Haudenschild CC, Ryan TJ, Faxon DP. Differences in compensatory vessel enlargement, not intimal formation, account for restenosis after angioplasty in the hypercholesterolemic rabbit model. Circulation. 1994;89:2809–2815. doi: 10.1161/01.cir.89.6.2809. [DOI] [PubMed] [Google Scholar]
- 20.Qiu H, Depre C, Ghosh K, Resuello RG, Natividad FF, Rossi F, Peppas A, Shen YT, Vatner DE, Vatner SF. Mechanism of gender-specific differences in aortic stiffness with aging in nonhuman primates. Circulation. 2007;116:669–676. doi: 10.1161/CIRCULATIONAHA.107.689208. [DOI] [PubMed] [Google Scholar]
- 21.Lu P, Wang S, Cai W, Sheng J. Role of tgf-beta1/smad3 signaling pathway in secretion of type i and iii collagen by vascular smooth muscle cells of rats undergoing balloon injury. J Biomed Biotechnol. 2012;2012:965953. doi: 10.1155/2012/965953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.