

Abstract
Background/Aims
Few studies of gene variants that affect estrogen activity investigate their association with age at onset of Alzheimer's disease (AD) in women of different ethnicities. We investigated the influence of ESR1 polymorphisms on age at onset of AD in a multiethnic cohort of women.
Methods
Among 1,436 women participating in the Washington Heights Inwood Columbia Aging Project (WHICAP), association with age at AD onset was assessed for 41 single-nucleotide polymorphisms (SNPs) on the ESR1 gene using Cox proportional hazard models, adjusting for presence of an APOE ε4 allele, years of education, and body mass index (BMI).
Results
Six SNPs in self-identified White women were protectively associated with delayed age of AD onset in this self-identified group, including the two restriction fragment length polymorphisms (RFLPs) PvuII (rs2234693) and XbaI (rs9340799) (HR range 0.420 – 0.483). Two separate SNPs were found to affect age of AD onset in self-identified Black women.
Conclusions
ESR1 polymorphisms affect age of onset for AD in women, and risk alleles vary by ethnicity. These effects are possibly due to different linkage disequilibrium patterns or differences in comorbid environmental or cultural risk factors mediating SNP effect on risk for AD.
Keywords: AD, age of onset, Alzheimer disease and Hispanics, estrogen receptors, gene polymorphism, genetic association studies in subjects with dementia, female, estrogen receptor alpha, late-onset AD
Introduction
Estrogens are important in maintaining brain function in regions typically affected by Alzheimer's disease (AD) and variations in estrogen exposure over the lifetime may affect cognitive decline associated with AD [1,2]. Numerous papers have established that estrogen may have beneficial effects on multiple pathways that affect risk for AD. Estrogens promotes the growth and survival of cholinergic neurons[3,4] increase cholinergic activity[5], have antioxidant properties[6], and promote the nonamyloidogenic metabolism of the amyloid precursor protein[7]. Estrogens also play an important role in regulation of the vascular endothelium where they activate rapid vasodilatation, exert anti-inflammatory effects, stimulate endothelial growth and migration, and protect the vessels from atherosclerotic degeneration by elevating nitric oxide and prostaglandin levels[8,9]. However, evaluating the role of hormones and enzymes in aging and cognition is difficult since many hormone levels decline with age. It is likely that polymorphisms in genes encoding the estrogen pathway contribute to variations in lifetime hormone exposure, including age-related changes in hormone levels. Estrogen exerts its action through at least two receptors, estrogen receptor α (ERα), encoded by ESR1 on chromosome 6q25.1, and estrogen receptor β (ERβ)[2]. The association of polymorphisms in ESR1 with risk of AD has been investigated in a number of studies, but findings have been inconsistent [10-23]. However, most studies have been conducted in relatively homogeneous ethnic groups, and few polymorphisms have been assessed in a multiethnic cohort. Examination of SNPs in multiracial groups which are evaluated without taking ethnicity into account may have several limitations, including a loss of significant association due to different allele frequencies, different linkage disequilibrium patterns between ethnicities, or differences in the distribution of comorbid conditions and risk factors for AD by ethnic group. In this study, we examined the relationship between ESR1 SNPs and the risk of AD in a multiethnic by self-identified ethnicity as well as by genetic population ancestry markers [24]. The aims of this study were to confirm previous findings of ESR1 polymorphisms which were found to be significantly associated with risk for AD; to identify additional SNPs which confer risk for AD using a denser set of SNPs than in previous studies; and to examine whether ESR1 variants would affect risk for AD differently in groups of women with different self-identified ethnicity. We hypothesized that genetic variants would demonstrate different patterns of association between groups of different ethnicities due to different allele frequencies or linkage disequilibrium patterns between ethnic groups, as well as varying environmental risk factors.
Materials and Methods
Subjects
The initial cohort included 1,686 women participating in the Washington Heights Inwood Columbia Aging Project (WHICAP), a prospective study of aging and dementia among Medicare recipients age 65 years and older, residing in northern Manhattan. Each subject underwent an in - person interview of health and functional ability followed by a standardized medical assessment and neuropsychological battery [25]. Assessments were conducted at 18 - 24 month intervals over a mean of 6.1 years of follow-up. The population from which participants were drawn was comprised of individuals from several different countries of origin representing three broadly self - identified ethnicities (Caribbean Hispanic, n=400; African - American, n= 485; and non - Hispanic White of European ancestry, n=551). The sampling strategies and recruitment of these two cohorts have been described in detail elsewhere [24].
AD diagnosis was based on NINCDS - ADRDA criteria. Participants were classified as non-demented if they remained without cognitive or functional decline through their last study assessment (n=1107). Participants were classified as having incident AD if they were non-demented at baseline visit and then were classified as having probable or possible AD by NINCDS - ADRDA criteria at any later study visit (n=329). Participants with incident AD had no other medical or psychiatric conditions that might mimic AD including other neurologic conditions such as Parkinson's disease or stroke. Age at initial diagnosis of AD was used to estimate age at onset of dementia.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was reviewed and approved by the Columbia University institutional review board, and written informed consent was previously obtained from all subjects.
DNA Isolation, SNP selection and Genotyping
Genomic DNA was extracted from total peripheral blood leukocytes using standard methods. We used a multistep selection process to identify candidate SNPs for genotyping. We first selected SNPs within ESR1 that were previously reported to be associated with an increased incidence or earlier age at onset of AD in any population. We then referenced the International HapMap Project (www.hapmap.org) to select tagging SNPs in both Caucasian and African populations. To provide sufficient coverage of the gene, we selected SNPs to maintain a pairwise r2 threshold of 0.8 in SNPs with a minimum minor allele frequency of 0.2. We obtained an average intermarker distance of approximately 6.2 kilobase pairs between SNPs, which provided good coverage of the gene as viewed on linkage disequilibrium maps (Supplementary Figures 1-3).
Forty-one ESR1 SNPs as well as 100 ancestry informative markers (AIMs) were genotyped in a total of 1,436 samples using Illumina GoldenGate custom panels and the Illumina IScan platform. Genotyping was performed according to standard protocols (www.Illumina.com). The complete list of ESR1 SNPs that were genotyped, along with their minor allele frequencies (MAF) by self-identified ethnicity, is presented in Table 3. Duplicate genotyping was performed on ten percent of samples to verify accuracy, and the concordance rate was greater than 97 percent.
Table 3. ESR1 SNPs investigated.
| SNP # | SNP | Position | Distance (bp) | Minor Allele | MAF Self-identified White |
MAF Self-identified Hispanic |
MAF Self-identified Black |
|---|---|---|---|---|---|---|---|
| 1 | rs2077647 | 152171020 | C | 0.5 | 0.5 | 0.5 | |
| 2 | rs17847065 | 152171427 | 407 | A | 0.0 | 0.0 | 0.0 |
| 3 | rs11155814 | 152193127 | 21700 | G | 0.1 | 0.3 | 0.5 |
| 4 | rs7761133 | 152193806 | 679 | G | 0.2 | 0.4 | 0.6 |
| 5 | rs17081749 | 152193868 | 62 | C | 0.1 | 0.3 | 0.4 |
| 6 | rs6903763 | 152195099 | 679 | A | 0.1 | 0.1 | 0.2 |
| 7 | rs6909023 | 152195640 | 541 | A | 0.1 | 0.1 | 0.2 |
| 8 | rs6937568 | 152195907 | 267 | G | 0.0 | 0.1 | 0.2 |
| 9 | rs827421 | 152199065 | 3158 | A | 0.5 | 0.5 | 0.4 |
| 10 | rs17081777 | 152199272 | 207 | G | 0.0 | 0.1 | 0.1 |
| 11 | rs6902771 | 152199824 | 552 | A | 0.5 | 0.5 | 0.5 |
| 12 | rs3853250 | 152201843 | 2019 | C | 0.5 | 0.5 | 0.5 |
| 13 | rs4870056 | 152204170 | 2327 | A | 0.5 | 0.5 | 0.5 |
| 14 | rs2234693 | 152205278 | 1108 | C | 0.5 | 0.5 | 0.5 |
| 15 | rs9340799 | 152205324 | 46 | G | 0.4 | 0.3 | 0.3 |
| 16 | rs9322332 | 152208744 | 3420 | A | 0.5 | 0.4 | 0.4 |
| 17 | rs3936674 | 152209254 | 510 | A | 0.3 | 0.2 | 0.1 |
| 18 | rs712221 | 152222184 | 12930 | T | 0.5 | 0.5 | 0.4 |
| 19 | rs1709183 | 152235939 | 13755 | G | 0.2 | 0.3 | 0.4 |
| 20 | rs11155819 | 152241302 | 5363 | G | 0.3 | 0.3 | 0.1 |
| 21 | rs9340835 | 152241874 | 572 | A | 0.3 | 0.3 | 0.3 |
| 22 | rs9322335 | 152242072 | 198 | A | 0.2 | 0.3 | 0.3 |
| 23 | rs9322338 | 152242692 | 620 | A | 0.1 | 0.2 | 0.5 |
| 24 | rs6557170 | 152245047 | 2355 | A | 0.2 | 0.2 | 0.1 |
| 25 | rs9478251 | 152245222 | 175 | C | 0.0 | 0.2 | 0.3 |
| 26 | rs2347923 | 152269364 | 24142 | C | 0.3 | 0.4 | 0.4 |
| 27 | rs2347867 | 152271793 | 2429 | G | 0.3 | 0.5 | 0.6 |
| 28 | rs6557171 | 152276536 | 4743 | A | 0.3 | 0.4 | 0.4 |
| 29 | rs4870062 | 152279561 | 3025 | C | 0.3 | 0.4 | 0.5 |
| 30 | rs4583998 | 152302611 | 23050 | A | 0.3 | 0.5 | 0.5 |
| 31 | rs9371564 | 152304056 | 1445 | A | 0.3 | 0.5 | 0.6 |
| 32 | rs3020377 | 152314341 | 10285 | A | 0.6 | 0.5 | 0.3 |
| 33 | rs1884051 | 152325222 | 10881 | G | 0.4 | 0.4 | 0.5 |
| 34 | rs985695 | 152328648 | 3426 | A | 0.2 | 0.3 | 0.4 |
| 35 | rs726281 | 152344521 | 15873 | G | 0.2 | 0.5 | 0.7 |
| 36 | rs2982684 | 152348147 | 3626 | A | 0.1 | 0.1 | 0.2 |
| 37 | rs12212176 | 152351950 | 3803 | A | 0.2 | 0.1 | 0.1 |
| 38 | rs7757956 | 152359083 | 7133 | A | 0.1 | 0.2 | 0.2 |
| 39 | rs9340954 | 152362115 | 3032 | C | 0.3 | 0.4 | 0.6 |
| 40 | rs722208 | 152364828 | 2713 | G | 0.3 | 0.4 | 0.6 |
| 41 | rs2207396 | 152424325 | 59497 | A | 0.2 | 0.2 | 0.2 |
Bolded and italicized SNPs are those found to be significant in any self-identified ethnicity or AIMs-defined genetic ancestry
Assessment of ethnicity and genetic population ancestry
Participants were first categorized by self-identified ethnicity as given at time of study enrollment (with self-identified categories including Black of non-Hispanic ancestry, White of non-Hispanic ancestry, and Hispanic), as most previous studies on ESR1 polymorphisms and AD have used this means of classification. Participants were then re-categorized by genetic population ancestry. To control for potential confounding that can be generated by population stratification, we used a set of 100 unlinked ancestry informative markers (AIMs) from a panel of 650Y Illumina SNPs to assess population structure. The AIMs were selected because they have allele frequencies that are significantly different among three ethno - racial groups: non-Hispanic Whites, non-Hispanic African, and individuals of Mexican/Central American ancestry. To assess population stratification, we performed population structure analysis as implemented in the STRUCTURE program[26,27]. To anchor ancestry, we included data from Caucasians (CEPH), Yorubans (YRI) and Mexican/Central Americans from the HapMap project (Figure 1). Our self - identified White population closely aligned with the Caucasian (CEPH) samples in the HapMap dataset and our self - identified Black population clustered around the Yoruban (YRI) samples. As expected, Caribbean Hispanics clearly showed admixture of Caucasian (CEPH) and Yoruban (YRI) genetic population ancestry, and the range of admixture varied widely. We then classified participants into groups who were of predominant Caucasian ancestry as defined by the AIMs index (defined as ≥ 0.6 AIMs markers consistent with CEPH profile, n= 551) versus those who were of predominant African ancestry (defined as ≥ 0.6 AIMs markers consistent with YRI profile, n= 485). In doing so, individuals previously self-identified as Hispanic were reclassified as being of predominantly Caucasian or predominantly African AIMs-defined ancestry (if their AIMs index scores were ≥ 0.6 CEPH or YRI, respectively), or admixed/Hispanic if they did not have one predominant genetic ancestry (n= 400).
Figure 1. Plot of WHICAP participants by AIMs-defined ancestry versus HapMap populations.
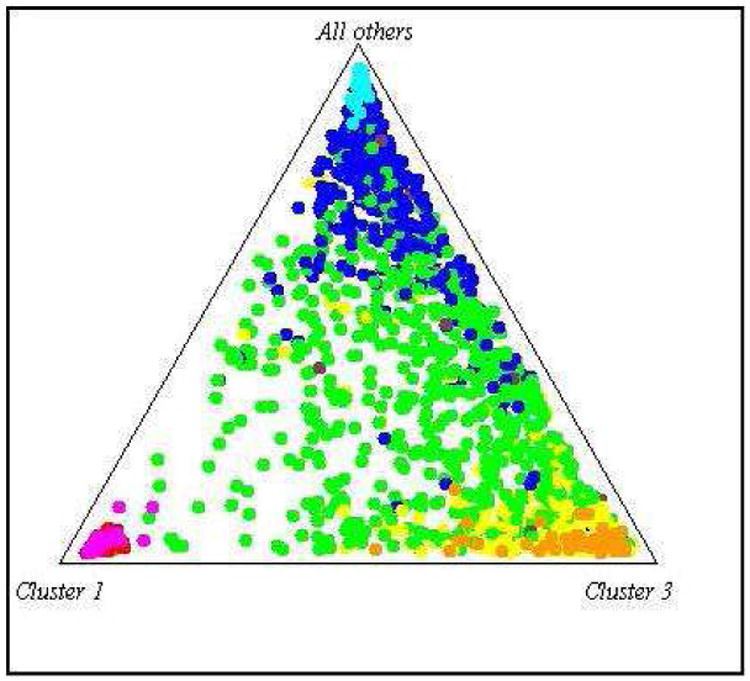
WHICAP participants
Yellow: Predominantly Caucasian AIMs-defined ancestry
Green: Admixed/Hispanic AIMs-defined ancestry
Blue: Predominantly African AIMs-defined ancestry
HapMap populations
Light Brown: Ancestrally homogenous Caucasian population (CEPH)
Light Blue:Ancestrally homogenous Yoruban Black population (YRI)
Red: Ancestrally homogenous East Asian population (CHJA)
Potential Confounders
Potential confounders included years of education, which was found to be independently associated with risk for AD in our group (Table 1); the presence of at least one APOE ε4 allele, which was included as a potential confounder because of its strong association with risk for AD in previous studies and because the allele frequencies of the ε4 allele vary substantially by race and ethnicity[28]; and body mass index (BMI), included because of the association of adipose tissue with higher estrogen levels [29-31]. Participants were classified according to the presence or absence of at least one APOE ε4 allele. Height and weight were measured at the initial evaluation to compute BMI. Other potential vascular risk factor confounders, including smoking and history of diabetes mellitus, were not included in the analyses because they were not found to be associated with risk for AD in our group (Table 1).
Table 1. Population Characteristics.
| Characteristic | Non Demented | Incident Alzheimer's Disease |
|---|---|---|
| Sample Size | 1107 | 329 |
| Age at time of enrollment (mean ± S.D.)* | 75.1 (5.8) | 77.4 (6.3) |
| Body Mass Index (mean ± S.D.) | 28.1 (5.7) | 27.8 (5.8) |
| Years of education (mean ± S.D.)* | 10.5 (4.6) | 7.5 (4.7) |
| Ever diagnosed with diabetes mellitus (n, %) | 174 (15.9) | 60 (18.2) |
| Current smokers (n, %) | 96 (8.6) | 31 (9.4) |
| At least one copy APOE ε4 (n, %) | 285 (26.0) | 91 (28.9) |
| Self-identified Ethnicity (n, %)* | ||
| White | 340 (89.5) | 40 (10.5) |
| Hispanic | 392 (68.4) | 181 (31.6) |
| Black | 375 (77.6) | 108 (22.4) |
p≤ 0.05
Statistical Analyses
Prior to association analysis, we assessed whether each SNP was in Hardy Weinberg equilibrium. This analysis was performed separately within each self – identified ethnicity as well as within each AIMs – defined group of unaffected individuals using the χ2 goodness-of-fit test in Haploview [32]. We used Cox proportional hazards modeling to assess the relationship between ESR1 genotypes and age at onset of AD, adjusting for presence of at least one APOE ε4 allele, years of education and BMI. To minimize the risk of a false-positive finding from rare variants and multiple testings, we computed empirical p-values by generating the null distribution on the basis of 1000 replicates of analyses. The use of empirical p-values corrects for the fact that multiple analyses were done. Cox modeling was done by stratifying first by self -reported ethnicity and then by AIMs - defined ancestry. We hypothesized that differences in associations between these two sets of analyses might reflect culturally - associated environmental risk factors for AD within groups defined by self-report. Conversely, similarities in significant SNPs between the two analyses would suggest a more direct genetic effect of ESR1 polymorphisms on risk for AD. The time to event variable was age at onset for participants who developed AD and age at last assessment for participants who remained non-demented throughout the follow-up period.
To provide the most robust model for observing an effect of the minor allele, SNPs were analyzed using a dominant model, in which participants homozygous for the common allele were used as the reference group and the risk group included participants who were heterozygous or homozygous for the minor allele.
Results
Demographic Characteristics
Table 1 presents the demographic characteristics of our cohort. The mean age of the participants at baseline was 77.0 (± 6.7) years, and ranged from 65 to 95 years. Mean length of follow-up was 6.1 (±4.3) years. The majority of women were self - identified as Hispanic (n= 573, 39.9 %) and Black (n=483, 33.6%), while 380 women were self - identified White (26.4%). Among all participants, 329 were classified as having incident AD (22.9%) and 1107 as non-demented. The frequency of incident AD was greater in self – identified Blacks and Hispanics than in Whites (Blacks: 22.4%; Hispanics: 31.6%; Whites: 10.5%). BMI, history of diabetes mellitus, and current smoking status did not differ significantly between individuals with or without AD when all ethnicities were analyzed as a whole; however, there was a significant level of variability (Table 2) among all covariates (BMI, presence of an APOE ε4 allele, and years of education) between the three self-identified ethnicities.
Table 2. Characteristics by self-identified ethnicity.
| Characteristic | Self-identified White | Self-identified Hispanic | Self-identified Black |
|---|---|---|---|
| Sample size (n) | 380 | 573 | 483 |
| Age at time of enrollment (mean ± S.D.)* | 77.9 (7.2) | 75.3 (6.9) | 77.3 (6.8) |
| Body Mass Index (mean ± S.D.)* | 26.2 (5.1) | 28.2 (6.3) | 28.3 (5.1) |
| Years of Education (mean ± S.D.)* | 12.4 (3.8) | 6.0 (4.3) | 11.0 (3.8) |
| At least one copy of APOE ε4 (n, %)* | 83 (21.8) | 137 (23.9) | 186 (32.3) |
p≤ 0.05
Analysis of ESR1 SNPs by self-defined ethnicity
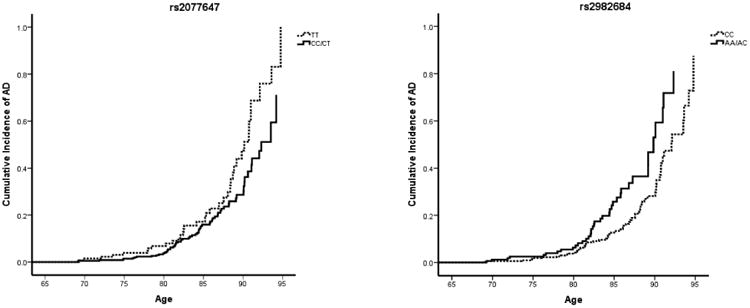
Table 4 presents the locations and minor allele frequencies (MAFs) of ESR1 SNPs that were found to be associated with age at onset of AD by self-defined ethnicity. It also reports the association between these ESR1 genotypes and their hazard ratios (HR) for AD, stratified by self-reported ethnicity. After adjusting for BMI, years of education, and the presence of an APOE ε4 allele, women who identified themselves as White and who were heterozygous or homozygous (i.e. carried one or two copies) for the A allele at rs6902771, for the C allele at rs2853250, for the A allele at rs4870056, for the C allele at rs2234693 (also known as the PvuII restriction site), for the G allele at rs9340799 (also known as the XbaI restriction site), and the A allele at rs9322332 had a roughly two-fold decrease in the hazard ratio for AD (Table 4; HR range 0.420 – 0.483) compared with women carrying no copies of these alleles (Table 4; HR for rs6902771 (AA/AG) = 0.437, empirical p-value = 0.015; HR for rs3853250 (CC/AC) = 0.442, empirical p-value = 0.017; HR for rs4870056 (AA/AG) = 0.420, empirical p-value = 0.011; HR for rs2234693 (CC/CT) = 0.443, empirical p-value = 0.014; HR for rs9340799 (GG/AG) = 0.441, empirical p-value = 0.017; HR for rs9322332 (AA/AC) = 0.483, empirical p-value = 0.033). Additionally, women who identified themselves as Black and who were heterozygous or homozygous (i.e. carried one or two copies) of the C allele at rs2077647 and for the A allele at rs2982684 had an increase and decrease in the hazard ratio for AD, respectively, compared with women carrying no copies of these alleles (HR for rs2077647 (CC/CT) = 0.629, 95% CI, 0.414 – 0.955, empirical p-value = 0.0.30; HR for rs2982684 (AA/AC) = 1.745, 95% CI, 1.152 – 2.644, empirical p-value = 0.009) (Table 4). Survival curves for these SNPs in self-identified Whites and Blacks are shown in Figures 2 and 3. These SNPs were not significantly associated with increased or decreased hazard rates for AD among women identifying themselves as Hispanic (Table 4).
Table 4. ESR1 SNPs associated with incident AD, by self-identified ethnicity.
| Self-identified White | Self-identified Hispanic | Self-identified Black | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP # | SNP | N | AD | HR (95% CI) | emp. p-value | SNP | N | AD | HR (95% CI) | emp. p-value | SNP | N | AD | HR (95% CI) | emp. p-value |
| 1 | rs2077647 (152171020); MAF (C): 0.5 | rs2077647 (152171020); MAF (C): 0.5 | rs2077647 (152171020); MAF (C): 0.5 | ||||||||||||
| CC/CT | 279 | 33 | 1.761 (0.680 - 4.556) | 0.244 | CC/CT | 146 | 282 | 1.183 (0.834 - 1.680) | 0.346 | CC/CT | 358 | 286 | 0.629 (0.414 - 0.955) | 0.030 | |
| TT | 97 | 6 | ref | TT | 418 | 103 | ref | TT | 124 | 88 | ref | ||||
| 11 | rs6902771 (152199824); MAF (A): 0.5 | rs6902771 (152199824); MAF (A): 0.5 | rs6902771 (152199824); MAF (A): 0.5 | ||||||||||||
| AA/AG | 272 | 21 | 0.437 (0.224 - 0.852) | 0.015 | AA/AG | 399 | 128 | 1.072 (0.763 - 1.505) | 0.690 | AA/AG | 352 | 85 | 1.361 (0.831 - 2.227) | 0.220 | |
| GG | 94 | 17 | ref | GG | 158 | 47 | ref | GG | 114 | 22 | ref | ||||
| 12 | rs3853250 (152201843); MAF (C): 0.5 | rs3853250 (152201843), MAF (C): 0.5 | rs3853250 (152201843); MAF (C): 0.5 | ||||||||||||
| CC/AC | 267 | 21 | 0.442 (0.226 - 0.862) | 0.017 | CC/AC | 411 | 131 | 1.076 (0.762 - 1.518) | 0.678 | CC/AC | 368 | 87 | 1.276 (0.762 - 2.137) | 0.354 | |
| AA | 99 | 17 | ref | AA | 146 | 45 | ref | AA | 97 | 20 | ref | ||||
| 13 | rs4870056 (152204170); MAF (A): 0.5 | rs4870056 (152204170); MAF (A): 0.5 | rs4870056 (152204170); MAF (A): 0.5 | ||||||||||||
| AA/AG | 262 | 20 | 0.420 (0.215 - 0.819) | 0.011 | AA/AG | 401 | 130 | 1.136 (0.807 - 1.599) | 0.464 | AA/AG | 340 | 82 | 1.156 (0.722 - 1.850) | 0.545 | |
| GG | 104 | 18 | ref | GG | 157 | 46 | ref | GG | 124 | 25 | ref | ||||
| 14 | rs2234693 (152205278); MAF (C):0.5 | rs2234693 (152205278); MAF (C): 0.5 | rs2234693 (152205278); MAF (C): 0.5 | ||||||||||||
| CC/CT | 275 | 23 | 0.443 (0.231 - 0.851) | 0.014 | CC/CT | 413 | 129 | 1.037 (0.740 - 1.452) | 0.834 | CC/CT | 381 | 88 | 1.275 (0.761 - 2.135) | 0.356 | |
| TT | 100 | 17 | ref | TT | 151 | 48 | ref | TT | 98 | 20 | ref | ||||
| 15 | rs9340799 (152205324); MAF (G): 0.4 | rs9340799 (152205324); MAF (G): 0.3 | rs9340799 (152205324); MAF (G): 0.3 | ||||||||||||
| GG/AG | 233 | 18 | 0.441 (0.226 - 0.863) | 0.017 | GG/AG | 309 | 98 | 0.919 (0.680 - 1.243) | 0.585 | GG/AG | 241 | 60 | 1.171 (0.786 - 1.745) | 0.437 | |
| AA | 143 | 21 | ref | AA | 254 | 79 | ref | AA | 239 | 48 | ref | ||||
| 16 | rs9322332 (152208744); MAF (A): 0.5 | rs9322332 (152208744); MAF (A): 0.4 | rs9322332 (152208744); MAF (A): 0.4 | ||||||||||||
| AA/AC | 257 | 21 | 0.483 (0.248 - 0.943) | 0.033 | AA/AC | 379 | 121 | 1.025 (0.737 - 1.427) | 0.881 | AA/AC | 146 | 77 | 1.399 (0.891 - 2.196) | 0.145 | |
| CC | 108 | 17 | ref | CC | 175 | 52 | ref | CC | 317 | 30 | ref | ||||
| 36 | rs2982684 (152348147); MAF (A): 0.1 | rs2982684 (152348147); MAF (A): 0.1 | rs2982684 (152348147); MAF (A): 0.2 | ||||||||||||
| AA/AC | 61 | 6 | 1.120 (0.461 - 2.725) | 0.802 | AA/AC | 134 | 38 | 0.748 (0.519 - 1.078) | 0.119 | AA/AC | 159 | 41 | 1.745 (1.152 - 2.644) | 0.009 | |
| CC | 305 | 32 | ref | CC | 424 | 138 | ref | CC | 306 | 66 | ref | ||||
Adjusted for years of education, body mass index (BMI), and presence of at least one APOE ε4 allele
MAF: Minor allele frequency
Emp. p-value: empirical p-value
Figure 2. Survival curves, ESR1 SNPs associated with incident AD in self-identified Whites.
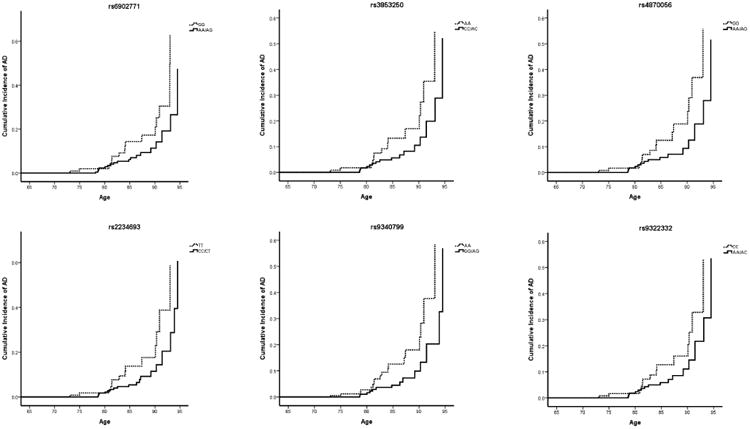
Figure 3. Survival curves, ESR1 SNPs associated with incident AD in self-identified Blacks.
Haplotype Analysis
Pairwise linkage disequilibrium (LD) was conducted between the six significant ESR1 SNPs in self-identified Whites, as implemented in the Haploview program using the D′ value[32] (Supplementary Table 1). The possibility of multi-locus association at adjacent variants was supported by the strong pairwise LD between SNP loci in this block, as well as the strong hazard rate findings at adjacent SNPs in this block (Supplementary Figure 1, also Table 4). In contrast, the two SNPS associated with increased or decreased likelihood of AD among African-Americans were not in strong LD (Supplementary Figure 3), indicating that they do not likely act by interaction with one another but perhaps in relation to separate, unrelated markers. We performed a “sliding window” haplotype analysis in the significant region in self-identified Whites, each analysis including two to four consecutive SNPs. While numerous haplotypes constructed from these ESR1 SNPs were found to be significantly associated with decreased risk for dementia (Supplementary Table 1), the most robust associations in self-identified Whites were haplotype C-A-C-G at SNPs 12 - 15 (rs3853250 through rs9340799, O.R. 0.656, p=0.009), which also contains the significant haplotypes A-C-G at SNPs 13 – 15 (rs4870056 through rs9340799, O.R. 0.656, p=0.009) and C-G at SNPs 14 – 15 (rs2234693 through rs9340799, O.R. 0.658, p=0.009).
Analysis of ESR1 SNPs by AIMs-defined Ancestry
We then repeated the analyses within strata defined by AIMs-defined predominant genetic ancestry. Using this categorization, one unique SNP was found to be associated with increased hazard ratio (decreased age of onset of AD) among women of predominantly Caucasian genetic ancestry (rs9322332, HR AA/AG = 1.862, 95% CI= 1.030 – 3.366, empirical p-value = 0.040) (Table 5). Additionally, rs2982684, which had previously been found to be associated with decreased age at onset of among self-identified Blacks, was also found to be associated with decreased age at onset among women of predominantly African genetic ancestry (rs2982684, HR AA/AC= 1.528, empirical p-value = 0.31) (Table 5). Following recategorization using AIMs-defined genetic ancestry, the six SNPs which previously had been found to be associated with delayed age at onset of AD among self-defined Whites (rs6902771, rs3853250, rs4870056, rs2234693, rs9340799, and rs9322332) were not found to be significant among women of predominantly Caucasian AIMs-defined ancestry (Supplementary Table 2). Likewise, rs2077647, which had been found to be associated with delayed age at onset among self-defined Blacks, was not associated with age at onset among women of predominantly African AIMs-defined ancestry (Supplementary Table 2).
Table 5. ESR1 SNPs associated with incident AD, by AIMs-defined predominant genetic ancestry.
| Predominantly Caucasian AIMs-defined ancestry | Admixed AIMs-defined ancestry | Predominantly African AIMs-defined ancestry | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP # | SNP | N | AD | HR (95% CI) | emp. p-value | SNP | N | AD | HR (95% CI) | emp. p-value | SNP | N | AD | HR (95% CI) | emp. p-value |
| 7 | rs6909023 (152195640); MAF (A): 0.1 | rs6909023 (152195640); MAF (A): 0.1 | rs6909023 (152195640); MAF (A): 0.2 | ||||||||||||
| AA/AG | 48 | 4 | 1.862 (1.030 – 3.366) | 0.040 | AA/AG | 166 | 36 | 0.264 (0.470 – 1.230) | 0.264 | AA/AG | 103 | 37 | 1.135 (0.760 – 1.696) | 0.536 | |
| GG | 318 | 34 | ref | GG | 300 | 71 | ref | GG | 453 | 137 | ref | ||||
| 36 | rs2982684 (152348147); MAF (A): 0.1 | rs2982684 (152348147); MAF (A): 0.2 | rs2982684 (152348147); MAF (A): 0.2 | ||||||||||||
| AA/AC | 159 | 17 | 0.720 (0.383 – 1.354) | 0.308 | AA/AC | 159 | 41 | 0.772 (0.491 – 1.211) | 0.260 | AA/AC | 134 | 38 | 1.528 (1.040 – 2.244) | 0.031 | |
| CC | 206 | 21 | ref | CC | 306 | 66 | ref | CC | 424 | 138 | ref | ||||
Adjusted for years of education, body mass index (BMI), and presence of at least one APOE ε4 allele
MAF: Minor allele frequency
Emp. p-value: empirical p-value
Discussion
Six SNPs in ESR1 were associated with an over two-fold decreased risk of AD among self-identified White women. Self-identified Whites with at least one minor allele at rs6902771, rs3853250, rs4870056, rs2234693, rs9340799, or rs9322332, had less than a one-half hazard ratio of AD compared with women without these risk alleles, indicating that these SNPs were associated with a delayed age of AD onset. These six intronic SNPs cover an 8.9 kb region with high linkage disequilibrium (minimum pairwise D′> 0.8), with rs2234693 (also known as restriction fragment length polymorphism (RFLP) PvuII) and rs9340799 (RFLP XbaI), located with 46 bp of one another. Haplotype analysis reveals that two- to four-SNP combinations of risk alleles among these six SNPs are all significantly associated with decreased risk for AD, with the most significant haplotype (C-A-C-G) including rs3853250, rs4870056, rs2234693, and rs9340799. When women were recategorized according predominant AIMs-defined genetic ancestry, these six SNPs were not associated with AD age at onset among women of predominantly Caucasian AIMs-defined ancestry; however one unique SNP, rs6909023, was associated with an increased hazard ratio (decreased age at onset) of AD (Table 5). The discrepancy between SNPs found to be significant by self-defined ethnicity versus AIMs-defined predominant genetic ancestry confirms our hypothesis that SNPs' effect on AD age at onset may be affected by environmental and/or cultural factors that affect women who define themselves as White, as opposed to women of predominant Caucasian genetic ancestry, as will be discussed in more detail below.
Among self-identified Blacks, women with at least one minor allele at rs2077647 had a significantly decreased hazard ratio (delayed age at onset) for AD; while women with at least one minor allele at rs2982684 had a significantly increased hazard ratio (earlier age at onset) for AD. When women were recategorized according to predominant AIMs-defined genetic ancestry, rs2982684 continued to be associated with decreased age at onset for AD among women of predominantly African AIMs-defined ancestry (Table 5), while rs2077647 was no longer significant. The persistent association of rs2982684 with decreased AD age at onset despite ethnic group recategorization indicates that this SNP may have a more direct genetic effect on AD among women of self-identified Black ethnicity or predominantly African genetic ancestry.
Among women of Hispanic self-defined ethnicity and women of Admixed AIMs-defined genetic ancestry, no significant findings were demonstrated. This may be due to the fact that women in these groups demonstrate significant heterogeneity in predominant genetic ancestry as well as environmental risk factors, including DM, smoking, and education.
Differential association of polymorphisms in a susceptibility gene for AD in groups of different self-identified ethnicities may occur for several reasons. First, differences in LD patterns between ethnic groups may contribute to discrepancies in genotype associations (Supplementary Figures 1-3). In our group, SNPs which were protective against AD in self – defined White women were located within a defined LD block (Supplementary Figure 1, Block 2). Notably, this block was less cohesive in self-defined Hispanics (Supplementary Figure 2, Blocks 2-3) and self-defined Blacks (Supplementary Figure 3, Blocks 3-4). This suggests that different LD patterns between ESR1 alleles and alleles of as yet unidentified loci for susceptibility to AD between populations of different genetic ancestries may contribute to the observed variability in genotypic association.
Second, it is also possible that differences in environmental or biological risk factors among women of different self-defined or genetic ancestries may play a significant role in phenotypic expression of the variants. This may explain why the six SNPs which were found to be protective in self-defined Whites were not found to be associated with age at AD onset among women of predominantly Caucasian genetic ancestry. As seen in Supplementary Table 3, when ethnic group stratification was changed from self-defined ethnicity to predominantly AIMs-defined genetic ancestry, the recategorization had the greatest effect on sample group when comparing self-identified Whites versus individuals of predominantly Caucasian AIMs-based ancestry, and self-identified Black versus women of predominantly African AIMS-based ancestry. Specifically, the shift in classification of race revealed that many individuals who identified themselves as Hispanic actually had genetic markers that were predominantly Caucasian or African. These individuals also had significantly higher rates of diabetes mellitus and smoking, and fewer years of education (Supplementary Table 3) than individuals of predominantly African AIMs who identified themselves as Black or participants of predominantly Caucasian AIMs who identified themselves as White. The potential mediation of SNP effect by vascular risk factors is reinforced by the different results in p-values for Cox analyses performed with and without the inclusion of vascular covariates (Supplementary Figure 4). For example, the inclusion of several vascular risk factors in the Cox analyses (including history of diabetes and current smoking) attenuated the significance of SNPs in self-identified White women (Supplementary Figure 4). These discrepancies indicate that environmental and biological covariate risk factors may affect the association of different alleles with age at onset for AD in this group.
While it is possible that discrepancies in SNPs found to be significantly associated with age at onset of AD may be due to different MAFs between ethnicities, this was not the case in our study group. Comparisons of SNPs found to be associated with age at AD onset demonstrate no significant difference in MAF between self-identified ethnicities (Tables 3 and 4) or between AIMs-defined predominant genetic ancestries (Table 5).
The association of polymorphisms in ESR1 with risk of AD has been investigated in a number of cohorts of Asian and European men and women, but findings have been inconsistent [10-18,20-23,33] ESR1 is more than 140 kb long and has eight exons and five domains[34]. The strongest associations within ESR1 have been found for the two RFLPs, PvuII (-397 T/C; rs2234693) and XbaI (-351 A/G; rs9340799), located 46 bp apart in the first intron, which have been associated with risk of developing cognitive impairment and AD [10,16,18,23]. These RFLPs have traditionally been recorded in PvuII as PP versus pp (where “P” is associated with the “T” allele, and “p” is associated with the “C” allele) and in XbaI as XX versus xx (where “X” is associated with the “A” allele and “x” is associated with the “G” allele). Capital “P” and “X” indicate the absence of the restriction site for each endonuclease, while lower case letters “p” and “x” indicate the presence of the restriction site. In our study, the C allele of rs2234693 (or “p”) and the G allele of rs9340799 (or “x”) were found to be protective, or associated with delayed age at AD onset; conversely the TT genotype of PvuII and the AA genotype of XbaI were found to be associated with earlier age at AD onset.
Several studies have similarly found an association between the T-allele of PvuII and A-allele of XbaI and increased risk for AD. One study of elderly Japanese men and women (205 cases versus 92 controls), found a marginal association between the “X” polymorphism (A-allele) of XbaI and risk for AD, (p=0.036) but no significant association between the PvuII “P” polymorphism (T-allele) and risk for AD (p=0.99)[15]. A second study of elderly Japanese found that frequencies of the “P” polymorphism (T-allele) of PvuII and “X” polymorphism (A-allele) of the XbaI RFLPs were significantly higher in the AD group than in the control group (49.4% P allele in AD cases v. 36.3% p allele in controls, p <0.01; 29.1% X allele in AD cases v. 16.7% X allele in controls, p <0.01) [33]. This finding was replicated by the same research group in two other studies of elderly Japanese which included both men and women [11,18]. A study of elderly Han Chinese men and women also found overrepresentation of “P” or “X” polymorphisms (T- and A-alleles, respectively) among AD patients compared with healthy controls (“P” allele: p=0.023, OR= 2.94, 95% CI 1.13- 7.1; “X” allele: p=0.046, OR = 2.28, 95% CI 1.003- 5.17)[14]. This trend was more distinct in elderly Han Chinese women than men (“P” allele: p= 0.016, OR=3.68, 95% CI 1.22- 11.08; “X” allele: p= 0.029, OR= 29.5, 95% CI 1.10- 7.94)[14]. A study of elderly Italian men and women found the ESR1 “PP” (TT) and “XX”(AA) genotypes to be associated with an increased risk for AD only in men (OR= 3.6, 95% CI 1.2- 10.9)[22]. The lowest APOE concentrations were observed in men carrying “PP” (TT) or “XX” (AA) genotypes (p=0.006), and in men carrying “PP” (TT) and/or “XX” (AA) genotypes together with the APOE ε4 allele (p=0.003). In women with AD, ESR1 “PP” (TT) and “XX” (AA) genotypes were also associated with lower MMSE values (p=0.0007)[22]. Finally, a separate study of elderly Italian men and women that combined the alleles of the PvuII and XbaI polymorphisms demonstrated that the “PPXX” (TTAA) haplotype was significantly more frequent in patients with AD than in controls[10]. The risk for AD increased seven-fold in individuals homozygous for the APOE ε4 allele with the “PPXX” (TTAA) ESR1 genotype[10].
Conversely, other studies have demonstrated an association between the “p” polymorphism (C-allele) of PvuII and “x” polymorphism (A-allele) of XbaI and increased risk for AD[16,17,20,23] or cognitive impairment[23], all in primarily homogenous Caucasian populations in Finland[16], Italy[17,20], and the United States[23]. Finally, other studies have found no association between XbaI I [12,21,35] or PvuII[12,21] genotypes and risk for AD. Conflicting results between our data and the data reported within previous studies may be due to several factors. First, study designs differ in sample size. Second, a difference in ethnicities between samples is a well-known confounding factor in genetic studies. It is also possible that the increased risk of AD of cognitive impairment with ERα polymorphisms is due to linkage disequilibrium with nearby genes that may in turn cause increased risk for developing AD. Finally, the age at onset of disease and clinical manifestations of disease may be modulated by environmental or cultural factors. It is possible that various risk factors lead to differential rates of progression.
A number of hypotheses for the functional significance of these polymorphisms have been reported in the literature. Given their location, 397 and 351 bp upstream from the start of exon 2, possible functional mechanisms include changed ESR1 expression via altered binding of transcription factors and influence on alternative splicing of the ESR1 gene. Both these mechanisms can be a result of either of these polymorphisms or through linkage disequilibrium with a truly functional, but so far unknown, sequence variation elsewhere in the ESR1 gene. To support the former hypothesis, Maruyama et al.[12] showed that the intronic region of the ESR1 gene carrying the PvuII and XbaI polymorphisms demonstrates a weak enhancer activity. Such activity appears to be influenced by genotype, as the enhancer activity of the G-allele appeared to higher than the A-allele. Additionally, according to Herrington et al. [36], the T-allele of the PvuII RFLP eliminates a functional binding site for the transcription factor B-myb. This implies that the presence of this allele may result in lower ESR1 transcription or may alter stability or structure of the ERα transcript and the subsequent ERα protein. This is supported by one study which demonstrated that the PvuII T-allele was associated with decreased plasma estradiol (E2) levels in an allele-dose-dependent manner in postmenopausal women [37], and findings in other studies that the PvuII polymorphism T-allele is associated with conditions affected by lower levels of E2 including increased risk of osteoporosis [38,39] and myocardial infarction [40]. The fact that the XbaI polymorphism A-allele is also associated with lower E2 levels may be due to linkage disequilibrium with the PvuII SNP or another functional polymorphism, or to functional significance of the XbaI polymorphism itself.
Overall, our findings confirm previous studies' findings of a strong association between ESR1 polymorphisms and age at onset for Alzheimer's disease among women. We also extend these studies through denser genotyping, rather than relying on imputation which can introduce false positives in multi-ethnic cohorts. From this effort, we identified additional SNPs that are associated with AD risk, and characterized how these SNPs vary among individuals of different AIMs-defined ancestries. This allowed us to perform haplotype analysis among the six most significant SNPs in self-identified White women, which identified haplotype C-A-C-G at SNPs 12 - 15 (rs3853250 through rs9340799) containing the RFLPs PvuII and XbaI as significantly decreasing risk for AD, (O.R. 0.656, p=0.009). Moreover, our studies clearly illustrates the importance of controlling for population stratification as well as for environmental risk factors in association studies, as SNPs which we found to be associated with AD vary significantly between the models within one ethnic group and among ethnic groups.
We note that most SNPs examined were intronic, and therefore may not be the critical location of the pathological variants, but may serve as markers for the critical region or may otherwise influence the expression of critical genetic markers. Further studies may characterize other genetic mechanisms that may contribute to AD, including methylation and copy number variations (CNVs). Future studies with denser genotyping to achieve high resolution in all ethnic groups, along with gene expression studies, may further provide biological insights. Additional insight may also be gained through future studies conducting similar analyses in men.
Supplementary Material
Supplementary Figure 1. ESR1 LD plot, self-identified White ethnicity
Supplementary Figure 2. ESR1 LD plot, self-identified Hispanic ethnicity
Supplementary Figure 3. ESR1 LD plot, self-identified Black ethnicity
Supplementary Figure 4. Significant SNPs in self-identified White women, with and without vascular covariates
Supplementary Table 1. Haplotype Analysis
Supplementary Table 2. Comparison of ESR1 SNPs associated with incident AD, by self-identified ethnicity versus AIMs-defined predominant genetic ancestry
Supplementary Table 3. Environmental and biological characteristics by self-identified ethnicity and AIMs-defined ancestry
Acknowledgments
Dr. Janicki, author, is funded by NIH grant 5-T32MH020004-1, the Charles and Ann Lee Saunders Brown Fellowship Fund, and the Louis V. Gerstner Jr. Scholars Foundation.
Ms. Park, author, is funded by 5P01 HD035897, R01 MH084995, and R01 NS060113.
Dr. Cheng, author, is funded by NIH grants U24 AG026395, R01 AG028786, R01 NS060113, R01 MH084995, R01 AG036469, and P50 AG008702.
Dr. Clark, author, is funded by NIH grants R01 NS060113, R01 NS073872, 5 P50 AG008702, 1RC2NS070276, 2P50NS038370-11, the NINDS Parkinson's Disease iPS Cell Line Research Consortium and P50 “Genetics of Down Syndrome”, and receives research support from the Alzheimer's Association, the Parkinson's Disease Foundation and the Michael J. Fox Foundation.
Dr. Lee, author, is funded by NIH grants R37 AG15473, U24 AG026395, 5P01 HD035897, R01 MH084995, P50 AG008702, U01 AG023749, R01 NS060113, R01 AG036469, and receives research support from the Alzheimer's Association.
Dr. Schupf, author, is funded by NIH grants 5R01AG014673-09, R01AG037212, U01 AG023749, R01 AG028786, P01 HD35897, P50 AG08702, R01 AG034189, 1 R21CA125461-01A2, 1 R01AG034189, 1 R01AG0306040-01, 5R01AG007370-17, and receives research support from the Alzheimer's Association. Dr. Schupf has consulted for Elan/Jannsen Pharmaceuticals
Footnotes
Financial Disclosures: Dr. Janicki reported no biomedical financial interests or potential conflicts of interest.
Ms. Park reported no biomedical financial interests or potential conflicts of interest.
Dr. Cheng reported no biomedical financial interests or potential conflicts of interest.
Dr. Clark reported no biomedical financial interests or potential conflicts of interest.
Dr. Lee reported no biomedical financial interests or potential conflicts of interest.
References
- 1.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. Journal of Comparative Neurology. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.McEwen BS. Invited review: Estrogens effects on the brain: Multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–2801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 3.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 4.Toran-Allerand CD, Miranda RC, Bentham WD, Sohrabji F, Brown TJ, Hochberg RB, MacLusky NJ. Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc Natl Acad Sci U S A. 1992;89:4668–4672. doi: 10.1073/pnas.89.10.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luine VN. Estradiol increases choline acetyltransferase activity in specific basal forebrain nuclei and projection areas of female rats. Exp Neurol. 1985;89:484–490. doi: 10.1016/0014-4886(85)90108-6. [DOI] [PubMed] [Google Scholar]
- 6.Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- 7.Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J Biol Chem. 1994;269:13065–13068. [PubMed] [Google Scholar]
- 8.Gerhard M, Ganz P. How do we explain the clinical benefits of estrogen? From bedside to bench. Circulation. 1995;92:5–8. doi: 10.1161/01.cir.92.1.5. [DOI] [PubMed] [Google Scholar]
- 9.Simoncini T, Mannella P, Genazzani AR. Rapid estrogen actions in the cardiovascular system. Ann N Y Acad Sci. 2006;1089:424–430. doi: 10.1196/annals.1386.001. [DOI] [PubMed] [Google Scholar]
- 10.Brandi ML, Becherini L, Gennari L, Racchi M, Bianchetti A, Nacmias B, Sorbi S, Mecocci P, Senin U, Govoni S. Association of the estrogen receptor alpha gene polymorphisms with sporadic Alzheimer's disease. Biochemical and biophysical research communications. 1999;265:335–338. doi: 10.1006/bbrc.1999.1665. [DOI] [PubMed] [Google Scholar]
- 11.Ji Y, Urakami K, Wada-Isoe K, Adachi Y, Nakashima K. Estrogen receptor gene polymorphisms in patients with Alzheimer's disease, vascular dementia and alcohol-associated dementia. Dementia and Geriatric Cognitive Disorders. 2000;11:119–122. doi: 10.1159/000017224. [DOI] [PubMed] [Google Scholar]
- 12.Maruyama H, Toji H, Harrington CR, Sasaki K, Izumi Y, Ohnuma T, Arai H, Yasuda M, Tanaka C, Emson PC, Nakamura S, Kawakami H. Lack of an association of estrogen receptor alpha gene polymorphisms and transcriptional activity with Alzheimer disease. Archives of Neurology. 2000;57:236–240. doi: 10.1001/archneur.57.2.236. [DOI] [PubMed] [Google Scholar]
- 13.Lambert JC, Harris JM, Mann D, Lemmon H, Coates J, Cumming A, St-Clair D, Lendon C. Are the estrogen receptors involved in Alzheimer's disease? Neurosci Lett. 2001;306:193–197. doi: 10.1016/s0304-3940(01)01806-7. [DOI] [PubMed] [Google Scholar]
- 14.Lin GF, Ma QW, Zhang DS, Zha YL, Lou KJ, Shen JH. Polymorphism of alpha-estrogen receptor and aryl hydrocarbon receptor genes in dementia patients in Shanghai suburb. Acta Pharmacologica Sinica. 2003;24:651–656. [PubMed] [Google Scholar]
- 15.Usui C, Shibata N, Ohnuma T, Higashi S, Ohkubo T, Ueki A, Nagao M, Arai H. No genetic association between the myeloperoxidase gene -463 polymorphism and estrogen receptor-alpha gene polymorphisms and Japanese sporadic Alzheimer's disease. Dementia and Geriatric Cognitive Disorders. 2006;21:296–299. doi: 10.1159/000091437. [DOI] [PubMed] [Google Scholar]
- 16.Mattila KM, Axelman K, Rinne JO, Blomberg M, Lehtimaki T, Laippala P, Roytta M, Viitanen M, Wahlund L, Winblad B, Lannfelt L. Interaction between estrogen receptor 1 and the epsilon 4 allele of apolipoprotein E increases the risk of familial Alzheimer's disease in women. Neuroscience Letters. 2000;282:45–48. doi: 10.1016/s0304-3940(00)00849-1. [DOI] [PubMed] [Google Scholar]
- 17.Porrello E, Monti MC, Sinforiani E, Cairati M, Guaita A, Montomoli C, Govoni S, Racchi M. Estrogen receptor alpha and APOE epsilon 4 polymorphisms interact to increase risk for sporadic AD in Italian females. European journal of Neurology : the Official Journal of the European Federation of Neurological Societies. 2006;13:639–644. doi: 10.1111/j.1468-1331.2006.01333.x. [DOI] [PubMed] [Google Scholar]
- 18.Isoe-Wada K, Maeda M, Yong J, Adachi Y, Harada H, Urakami K, Nakashima K. Positive association between an estrogen receptor gene polymorphism and Parkinson's disease with dementia. European Journal of Neurology : the Official Journal of the European Federation of Neurological Societies. 1999;6:431–435. doi: 10.1046/j.1468-1331.1999.640431.x. [DOI] [PubMed] [Google Scholar]
- 19.Isoe K, Urakami K, Adachi Y, Nakashima K. Genetic association of estrogen receptor gene polymorphisms with Alzheimer's disease. Alzheimer's Research. 1997;3:195–197. [Google Scholar]
- 20.Monastero R, Cefalu AB, Camarda C, Noto D, Camarda LK, Caldarella R, Imbornone E, Averna MR, Camarda R. Association of estrogen receptor alpha gene with Alzheimer's disease: A case-control study. J Alzheimers Dis. 2006;9:273–278. doi: 10.3233/jad-2006-9306. [DOI] [PubMed] [Google Scholar]
- 21.Ma SL, Tang NL, Tam CW, Lui VW, Lau ES, Zhang YP, Chiu HF, Lam LC. Polymorphisms of the estrogen receptor alpha (ESR1) gene and the risk of Alzheimer's disease in a southern Chinese community. Int Psychogeriatr. 2009;21:977–986. doi: 10.1017/S1041610209990068. [DOI] [PubMed] [Google Scholar]
- 22.Corbo RM, Gambina G, Ruggeri M, Scacchi R. Association of estrogen receptor alpha (ESR1) PvuII and XbaI polymorphisms with sporadic Alzheimer's disease and their effect on apolipoprotein E concentrations. Dementia and Geriatric Cognitive Disorders. 2006;22:67–72. doi: 10.1159/000093315. [DOI] [PubMed] [Google Scholar]
- 23.Yaffe K, Lui LY, Grady D, Stone K, Morin P. Estrogen receptor 1 polymorphisms and risk of cognitive impairment in older women. Biological Psychiatry. 2002;51:677–682. doi: 10.1016/s0006-3223(01)01289-6. [DOI] [PubMed] [Google Scholar]
- 24.Tang MX, Cross P, Andrews H, Jacobs DM, Small S, Bell K, Merchant C, Lantigua R, Costa R, Stern Y, Mayeux R. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001;56:49–56. doi: 10.1212/wnl.56.1.49. [DOI] [PubMed] [Google Scholar]
- 25.Stern Y, Andrews H, Pittman J, Sano M, Tatemichi T, Lantigua R, Mayeux R. Diagnosis of dementia in a heterogeneous population. Development of a neuropsychological paradigm-based diagnosis of dementia and quantified correction for the effects of education. Arch Neurol. 1992;49:453–460. doi: 10.1001/archneur.1992.00530290035009. [DOI] [PubMed] [Google Scholar]
- 26.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. ApoE and Alzheimer disease meta analysis consortium. JAMA : the Journal of the American Medical Association. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 29.Cauley JA, Gutai JP, Kuller LH, LeDonne D, Powell JG. The epidemiology of serum sex hormones in postmenopausal women. American Journal of Epidemiology. 1989;129:1120–1131. doi: 10.1093/oxfordjournals.aje.a115234. [DOI] [PubMed] [Google Scholar]
- 30.Meldrum DR, Davidson BJ, Tataryn IV, Judd HL. Changes in circulating steroids with aging in postmenopausal women. Obstetrics and Gynecology. 1981;57:624–628. [PubMed] [Google Scholar]
- 31.Judd HL, Shamonki IM, Frumar AM, Lagasse LD. Origin of serum estradiol in postmenopausal women. Obstetrics and Gynecology. 1982;59:680–686. [PubMed] [Google Scholar]
- 32.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 33.Isoe K, Urakami K, Wakutani Y, Yong J, Adachi Y, Nakashima K. Screening for mutation of the presenilin 2 gene in Alzheimer's disease. Dementia and Geriatric Cognitive Disorders. 1997;8:60. doi: 10.1159/000106602. [DOI] [PubMed] [Google Scholar]
- 34.Ponglikitmongkol M, Green S, Chambon P. Genomic organization of the human oestrogen receptor gene. The EMBO Journal. 1988;7:3385–3388. doi: 10.1002/j.1460-2075.1988.tb03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prince JA, Feuk L, Sawyer SL, Gottfries J, Ricksten A, Nagga K, Bogdanovic N, Blennow K, Brookes AJ. Lack of replication of association findings in complex disease: An analysis of 15 polymorphisms in prior candidate genes for sporadic Alzheimer's disease. European Journal of Human Genetics : EJHG. 2001;9:437–444. doi: 10.1038/sj.ejhg.5200651. [DOI] [PubMed] [Google Scholar]
- 36.Herrington DM, Howard TD, Brosnihan KB, McDonnell DP, Li X, Hawkins GA, Reboussin DM, Xu J, Zheng SL, Meyers DA, Bleecker ER. Common estrogen receptor polymorphism augments effects of hormone replacement therapy on E-selectin but not C-reactive protein. Circulation. 2002;105:1879–1882. doi: 10.1161/01.cir.0000016173.98826.88. [DOI] [PubMed] [Google Scholar]
- 37.Schuit SC, de Jong FH, Stolk L, Koek WN, van Meurs JB, Schoofs MW, Zillikens MC, Hofman A, van Leeuwen JP, Pols HA, Uitterlinden AG. Estrogen receptor alpha gene polymorphisms are associated with estradiol levels in postmenopausal women. Eur J Endocrinol. 2005;153:327–334. doi: 10.1530/eje.1.01973. [DOI] [PubMed] [Google Scholar]
- 38.van Meurs JB, Schuit SC, Weel AE, van der Klift M, Bergink AP, Arp PP, Colin EM, Fang Y, Hofman A, van Duijn CM, van Leeuwen JP, Pols HA, Uitterlinden AG. Association of 5′ estrogen receptor alpha gene polymorphisms with bone mineral density, vertebral bone area and fracture risk. Hum Mol Genet. 2003;12:1745–1754. doi: 10.1093/hmg/ddg176. [DOI] [PubMed] [Google Scholar]
- 39.Ioannidis JP, Ralston SH, Bennett ST, Brandi ML, Grinberg D, Karassa FB, Langdahl B, van Meurs JB, Mosekilde L, Scollen S, Albagha OM, Bustamante M, Carey AH, Dunning AM, Enjuanes A, van Leeuwen JP, Mavilia C, Masi L, McGuigan FE, Nogues X, Pols HA, Reid DM, Schuit SC, Sherlock RE, Uitterlinden AG. Differential genetic effects of ESR1 gene polymorphisms on osteoporosis outcomes. JAMA. 2004;292:2105–2114. doi: 10.1001/jama.292.17.2105. [DOI] [PubMed] [Google Scholar]
- 40.Schuit SC, Oei HH, Witteman JC, Geurts van Kessel CH, van Meurs JB, Nijhuis RL, van Leeuwen JP, de Jong FH, Zillikens MC, Hofman A, Pols HA, Uitterlinden AG. Estrogen receptor alpha gene polymorphisms and risk of myocardial infarction. JAMA. 2004;291:2969–2977. doi: 10.1001/jama.291.24.2969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. ESR1 LD plot, self-identified White ethnicity
Supplementary Figure 2. ESR1 LD plot, self-identified Hispanic ethnicity
Supplementary Figure 3. ESR1 LD plot, self-identified Black ethnicity
Supplementary Figure 4. Significant SNPs in self-identified White women, with and without vascular covariates
Supplementary Table 1. Haplotype Analysis
Supplementary Table 2. Comparison of ESR1 SNPs associated with incident AD, by self-identified ethnicity versus AIMs-defined predominant genetic ancestry
Supplementary Table 3. Environmental and biological characteristics by self-identified ethnicity and AIMs-defined ancestry