

Abstract
The recent discovery of several potentially pathogenic autoantibodies has helped identify patients with clinically distinctive central nervous system diseases that appear to benefit from immunotherapy. The associated autoantibodies are directed against the extracellular domains of cell-surface–expressed neuronal or glial proteins such as LGI1, N-methyl-D-aspartate receptor, and aquaporin-4. The original descriptions of the associated clinical syndromes were phenotypically well circumscribed. However, as availability of antibody testing has increased, the range of associated patient phenotypes and demographics has expanded. This in turn has led to the recognition of more immunotherapy-responsive syndromes in patients presenting with cognitive and behavioral problems, seizures, movement disorders, psychiatric features, and demyelinating disease. Although antibody detection remains diagnostically important, clinical recognition of these distinctive syndromes should ensure early and appropriate immunotherapy administration. We review the emerging paradigm of cell-surface–directed antibody–mediated neurological diseases, describe how the associated disease spectrums have broadened since the original descriptions, discuss some of the methodological issues regarding techniques for antibody detection and emphasize considerations surrounding immunotherapy administration. As these disorders continue to reach mainstream neurology and even psychiatry, more cell-surface–directed antibodies will be discovered, and their possible relevance to other more common disease presentations should become more clearly defined.
Autoantibody-associated diseases of the central nervous system (CNS) are among the most rapidly expanding fields in clinical neurology. The first such antibodies were detected by binding to brain tissue sections and targeted intracellular proteins (Hu, Yo, Ri, Tr, CV2/CRMP5); they were termed paraneoplastic, given their frequent association with an underlying malignancy.1–4 However, antibodies directed against these intracellular antigens are unlikely to access their target in vivo and are not, in general, considered to be pathogenic.2,3 Rather, it is thought that T-cell–mediated inflammatory responses are the primary mechanism of neuronal destruction, and these disorders will not be considered further here.5
By contrast, antibodies that target the extracellular domains of cell-surface antigens, usually integral membrane proteins, can modulate the number or function of the target protein and are potentially pathogenic (Table1).3,6,7 These neuronal- or glial-surface–directed antibodies8 are detected by demonstration of antibody binding to the surface of human embryonic kidney cells that have been made to express the specific target (Fig 1A–C), to the surface of cultured live neurons (Fig 1D), and to fixed brain sections (see Fig 1E; these techniques have different merits and will be discussed later).3,7,9 The clinical syndromes associated with neuroglial surface-directed antibodies (NGSAbs) frequently show a rapid onset, response to immunotherapies, good correlations between symptom course and antibody levels, and are less frequently associated with malignancies.2,7,10,11
Table 1.
Clinical Associations of the Most Common Central Nervous System Cell-Surface–Directed Antibodies
| NMDAR | LGI1 | CASPR2 | AMPAR | GlyR | GABAB | AQP4 | MOG | |
|---|---|---|---|---|---|---|---|---|
| Frequent clinical associations | Diffuse encephalitis with psychiatric features, cognitive impairment, seizures, movement disorder, dysautonomia, and reduction in consciousness | LE with faciobrachial dystonic seizures and serum hyponatremia | Morvan syndrome with psychiatric features, insomnia, dysautonomia, and neuromyotonia (often with LGI1 antibodies); less frequently LE | LE | PERM but also some SPS and related syndromes | LE | NMOSD | NMOSD |
| Tumor/infectious associations | Ovarian teratoma in about 30%45,46; Relapses post-HSV encephalitis with NMDAR antibodies75–78 | <10% (various tumors described)29,30,68,69 | Thymoma (∼30%)20,157 | Lung, breast, thymoma (∼50%)41,55 | Thymoma rarely (<10%)18,89,158 | Lung (∼50%)42 | Rare159; relapses often precipitated by various infections131 | None yet known |
| Expanding phenotypic spectrum | Few cases with purely psychotic features48,53,160; predominant movement disorder161; few with predominant cryptogenic epilepsy syndrome162 | Cryptogenic epilepsies60,62 | Cryptogenic epilepsies60,62; Guillain-Barre–like syndrome163 | Atypical psychosis55 | LE, brainstem encephalitis124; cryptogenic epilepsies60,164 | Encephalopathy165 | Encephalopathy, including pediatric ADEM166 | |
| Approximate number of reported cases since first description | >700 in 6 years | ∼250 in 3 years | ∼30 in 3 years | ∼25 in 4 years | ∼60 in 5 years | ∼30 in 3 years | >2,000 in 8 years | ∼30 in 1 year |
| Prevalence in clinically defined tested cohorts | 9/48 (19%) with unknown encephalitis25 | 6/62 (10%) with unknown encephalitis25 | 24/27 (89%) with Morvan syndrome20 | 15/410 (4%) with suspected autoimmune encephalitis41 | Mainly seen in PERM21,89,91,158; 10/81 (12%) with SPS18; 1/48 (2%) pediatric encephalopathies37 | 10/70 (14%) of LE cases42 | 70–80% of patients with NMOSD100,110 | 16/215 (7%) NMOSD110 |
| Prevalence (%) in healthy/disease cohorts136 a | 0–1.2/0–1.3 | 0–0.06/0–0.0432,33 | 0–0.29/0–0.32 | 0–0.23/0–0.0441 | 0.06–1.2/0.1218 | 0/0–0.0442 | 0/0–0.04100 | 0–0.06/0.08–2112 |
| Primary cell type/antigenic target | Neuron/NR1 subunit | Neuron | Neuron | Neuron/GluR1/2 | Neuron/α1 receptor | Neuron/B1 subunit | Astrocyte/M23 arrays | Oligodendrocyte |
For neuronal- or glial-surface–directed antibodies reported in >20 cases and in >1 publication. LE produces amnesia, confusion, and seizures (additional features noted above within each antibody specificity).
Data are from Dahm et al,136 plus other papers as referenced within the table.
ADEM = acute disseminated encephalomyelitis; AMPAR = α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; AQP4 = aquaporin-4; CASPR2 = contactin-associated protein 2; GABAB = γ-aminobutyric acidB; GlyR = glycine receptor; HSV = herpes simplex virus; LE = limbic encephalitis; LGI1 = leucine-rich glioma-inactivated 1; MOG = myelin oligodendrocyte glycoprotein; NMDAR = N-methyl-D-aspartate receptor; NMOSD = neuromyelitis optica spectrum disorder; PERM = progressive encephalomyelitis with rigidity and myoclonus; SPS = stiff-person syndrome.
Figure 1.
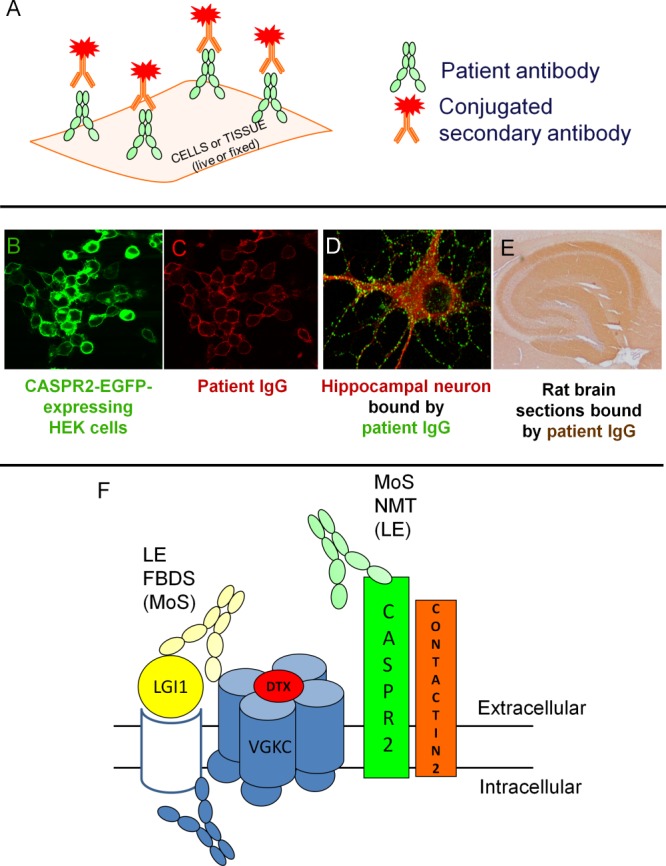
(A) Transfected cells or tissue (neuronal cultures or brain sections) may be labeled with patient antibody and then a secondary, dye/enzyme-conjugated antibody to allow visualization. Graphic courtesy of Dr T. Moloney. (B, C) Cell-based assay. Enhanced green fluorescent protein (EGFP)-tagged antigen (in this case contactin-associated protein 2 [CASPR2]; B; green) is bound by patient immunoglobulin (Ig) G (C; red). (D) Hippocampal neuronal cultures labeled with leucine-rich glioma-inactivated 1 (LGI1)-IgG (green) and intracellularly stained with microtubule-associated protein 2, a neuronal marker (red; image courtesy of Dr L. Zuliani). (E) Sagittal rat brain section showing hippocampal staining with patient serum N-methyl-D-aspartate receptor–IgG (image from Irani et al46). (F) Depiction of the voltage-gated potassium channel (VGKC) complex labeled with radioiodinated dendrotoxin (DTX). Antibodies bind the extracellular domains of LGI1 (in patients with limbic encephalitis, faciobrachial dystonic seizures [FBDS], and, less so, Morvan syndrome [MoS]) and CASPR2 (in patients with MoS more frequently than in neuromyotonia (NMT) or LE). Contactin-2 antibodies are rare. Some antibodies may bind the intracellular domains of some molecules within the VGKC complex (blue antibody): these antibodies may precipitate the VGKC-complex but only those directed against extracellular epitopes are likely to be pathogenic. HEK = human embryonic kidney.
Since 2001, a number of NGSAbs have been identified (see Table1). The most common are antibodies to the N-methyl-D-aspartate receptor (NMDAR) and components of the voltage-gated potassium channel (VGKC) complex (see Figs 1F and 2A), which includes leucine-rich glioma-inactivated 1 (LGI1), contactin-associated protein 2 (CASPR2), and contactin-2. In the United Kingdom, NMDAR, aquaporin-4 (AQP4) and LGI1 antibodies occur at a rate of approximately 2 new cases per million persons per year.12,13
Figure 2.
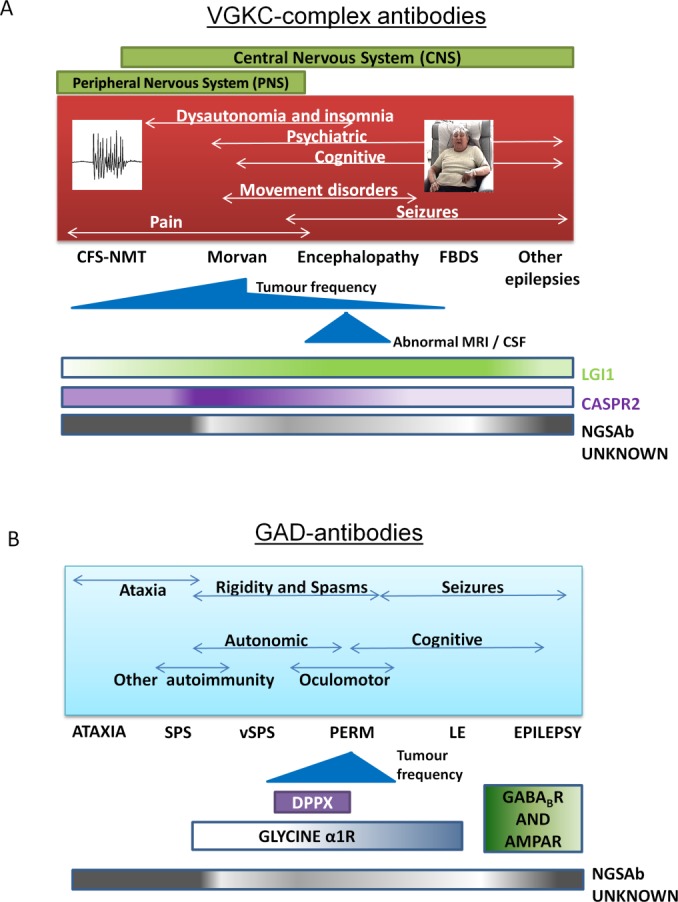
(A) The phenotype spread of voltage-gated potassium channel (VGKC) complex, leucine-rich glioma-inactivated 1 (LGI1), and contactin-associated protein 2 (CASPR2) antibodies. The relative proportions of patients with LGI1 and CASPR2 antibodies and those who remain without a known cell-surface antigenic target (Neuroglial cell-surface antibody [NGSAb] unknown) are depicted in the gradient bars. Movement disorders include ataxia, chorea, and parkinsonism.44,93,94 A number of patients, especially those with cramp fasciculation syndrome–neuromyotonia (CFS-NMT; high frequency discharges shown) and epilepsy (excluding faciobrachial dystonic seizures [FBDS]) currently have no defined antigenic target (NGSAb unknown), although their sera precipitate VGKC complexes in the radioimmunoassay. (B) High levels of glutamic acid decarboxylase (GAD) antibodies are associated with a variety of syndromes, including cerebellar ataxia, stiff-person syndrome (SPS), variant SPS (vSPS), progressive encephalomyelitis with rigidity and myoclonus (PERM), limbic encephalitis (LE), and epilepsy. Glycine-alpha1 receptor antibodies have been reported in patients with PERM, vSPS, and SPS in order of decreasing frequency; dipeptidyl peptidase IV–related protein (DPPX) antibodies in some patients with vSPS/PERM; and antibodies against γ-aminobutyric acidB receptor (GABABR) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR) in some patients with GAD antibodies and LE but not to date in cases with epilepsy alone. Their relative frequencies, and those of the NGSAb unknown patients, are depicted in the gradient bars, where a darker color implies a higher chance of finding that antibody.
Glutamic acid decarboxylase (GAD) antibodies lie in a hinterland between classical paraneoplastic and NGSAbs (see Fig 2B). GAD is an intracellular enzyme, GAD-antibody titers do not usually correlate with clinical outcomes, and yet GAD-antibody–associated syndromes (see Fig 2B) are infrequently seen with tumors.14,15 Furthermore, despite GAD's intracellular localization, some articles do highlight the pathogenic potential of GAD antibodies.16 Interestingly, there are recent reports of NGSAbs coexisting with GAD antibodies,17–19 and the coexistence of >1 potentially relevant NGSAb, or 1 NGSAb and 1 intracellularly directed antibody, is increasingly recognized.20–22
Many previous reviews2,3,7,10,23 have used the autoantibody specificity to categorize associated syndromes (see Table1). Here, we also outline the clinical features that should alert neurologists to the possibility of a NGSAb (Table2) and may assist in treating the patient before antibody results are available. One consequence of the increasing availability and commercial accessibility of NGSAb testing is the recognition of patients with “positive” antibodies that do not fit established disease phenotypes or are discovered in nonclassical clinical scenarios. Therefore, we also discuss the relative merits of antibody-detection methods and the diversification of recognized phenotypes or “phenotype spread” in NGSAb-associated syndromes (examples in Fig 2). Finally, we review the rationale and evidence for immunosuppressive therapy in these conditions.
Table 2.
Features That Should Raise Suspicion of Neuronal-Surface–Directed Antibodies in Patients Presenting with Cognitive Impairment, Psychiatric Features, Epilepsy, Movement Disorders, or Demyelinating Disease
| Clinical Syndrome | |||||
|---|---|---|---|---|---|
| Cognitive Impairment | Psychiatric | Epilepsy | Movement Disorders | Demyelinating Disease | |
| Onseta | Rapid | Rapid without a Prodrome of Social Withdrawal | Rapid, Especially When No Prior Seizure History | Rapid | Acute Relapses |
| Clinical Features | Memory as a conspicuously affected cognitive domain; often associated seizures including FBDS and hyponatremia (with VGKCc/LGI1-antibodies); underlying cancer possible (GABABR/AMPAR) | First lifetime episode of psychosis; associated seizure(s) and movement disorder (NMDAR/AMPAR); neuromyotonia, dysautonomia, and insomnia (Morvan: CASPR2 > LGI1) | Frequent, localization-related seizures; associated cognitive/psychiatric features in young females (often NMDAR/GAD) or middle-age (VGKCc/LGI1) | Variable phenomenologies including chorea, dystonia, and stereotypies; associated neuropsychiatric features such as anxiety, OCD, or depression (NMDAR/D2R); ataxia (CASPR2, GAD, mGluR1/5); preceding streptococcal infection; hyperekplexia and rigidity (glycine, DPPX) | LETM, severe/bilateral ON, intractable nausea with medullary lesion, drug-refractory pain, often with poor spontaneous recovery (AQP4) or overall better prognosis (MOG) |
| MRI and CSF (protein, cells, oligoclonal bands) findings | T2/FLAIR hyperintensity on brain MRI, particularly in medial temporal lobes; normal CSF usually | Various subcortical or cortical T2/FLAIR changes; some appearances suggestive of demyelination; CSF pleocytosis and oligoclonal bands | T2/FLAIR hyperintensity on brain MRI, particularly in medial temporal lobes; normal CSF usually | Usually normal, sometimes medial temporal lobe hyperintensities; often normal CSF | White matter lesions can look very similar to MS; brain may be normal; longitudinally extensive myelitis; CSF typically shows pleocytosis without bands |
| Associated cell-surface–directed antibodies | VGKCc (LGI1, CASPR2), AMPAR, GABABR, GAD, DPPX | NMDAR; LGI1 and CASPR2; D2R; AMPAR | VGKCc (especially LGI1), GAD, NMDAR | NMDAR, glycine, VGKCc, GAD, DPPX, D2R | AQP4, MOG |
One feature common to many of these presentations is the rapid onset, often within days to a few weeks. Routine CSF examination is normal in many cases, especially VGKC-complex antibodies, with variable presence of oligoclonal bands and raised immunoglobulin G index.
AMPAR = α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; AQP4 = aquaporin-4; CASPR2 = contactin-associated protein 2; CSF = cerebrospinal fluid; D2R = dopamine receptor D2; DPPX = dipeptidyl peptidase IV–related protein; FBDS = faciobrachial dystonic seizures; FLAIR = fluid-attenuated inversion recovery; GABABR = γ-aminobutyric acidB receptor; GAD = glutamic acid decarboxylase; LETM = longitudinally extensive transverse myelitis; LGI1 = leucine-rich glioma-inactivated 1; mGluR = metabotropic glutamatergic receptor; MOG = myelin oligodendrocyte glycoprotein; MRI = magnetic resonance imaging; MS = multiple sclerosis; NMDAR = N-methyl-D-aspartate receptor; OCD = obsessive–compulsive disorder; ON = optic neuritis; VGKCc = voltage-gated potassium channel complex.
Red Flags to the Presence of a Cell-Surface CNS-Directed Autoantibody When Seeing Patients With…
Cognitive Impairment: Encephalitis, Encephalopathy, and Dementia
The onset of amnesia, disorientation, and personality change over days or a few weeks should prompt consideration of a rapidly progressive dementia (see Table2).24 Antibody-associated conditions should be considered high within this differential diagnosis, particularly VGKC-complex antibodies in middle-aged and elderly patients (see Table1).25–28
VGKC-complex antibody–associated encephalopathy/limbic encephalitis (LE) was first described in 2 patients in 200129 and in 2 series in 2004.30,31 It subsequently became clear that these antibodies, measured by radioimmunoprecipitation from solubilized mammalian brain membranes, were directed against the extracellular domains of neuronal proteins that are tightly complexed with VGKCs in situ (see Fig 1F).32 In patients with LE, the commonest target is LGI1 (in 80–90%),32,33 in 5 to 10% of cases the target is CASPR2, very few have contactin-2 antibodies, and some patients have no defined target.32 Seizures are present in the majority of LGI1-antibody–positive cases, and serum hyponatremia is seen in about 60%.30–33 These 2 features—seizures and hyponatremia—coupled with a subacute onset, provide a strong indicator of an immunotherapy-responsive disease, distinct from other rapidly progressive dementias such as Creutzfeldt–Jakob disease (CJD).34 However, routine cerebrospinal fluid (CSF) analysis and brain magnetic resonance imaging (MRI) are normal in around 75% and 50% of cases with VGKC-complex antibodies respectively,30–33 so the key to confirming this diagnosis is antibody testing. Various epilepsies, movement disorders (discussed below), and neuromyotonia are also recognized in patients with VGKC-complex antibodies. Over time, the spread in the demographic associated with VGKC-complex antibodies has meant females are now recognized as often as males, and the associated tumor frequency appears to be decreasing (Fig 3A). Some recent reports have noted children with cognitive impairment, seizures, and VGKC-complex antibodies, although they lack the distinctive LGI1 or CASPR2 antibodies, hyponatremia, or faciobrachial dystonic seizures (FBDS; see below) often seen in adults (see Fig 3C).35–39
Figure 3.

(A) The epidemiology of patients with voltage-gated potassium channel (VGKC) complex antibodies in published series that reported >3 cases since the first publication in 2001 and 2013. There have been trends toward fewer males (p = 0.068) and tumors (p = 0.397). (B) The epidemiology of patients with N-methyl-D-aspartate (NMDA)-receptor antibodies in series that reported >3 cases since the first publication in 2007 and 2013. There have been significant reductions in tumors over time (p = 0.044) and a trend toward an increasing number of males, whereas the frequency of affected children has not altered. (C) Differences in the features of adults and children with VGKC-complex antibodies. Significant differences were seen in percentage of males (**p = 0.006) and presence of leucine-rich glioma-inactivated 1 (LGI1)/contactin-associated protein 2 (CASPR2) antibodies (Ab; *p = 0.036); there were nonsignificant differences for serum hyponatremia (↓Na+; p = 0.21) and tumors (p = 0.51; Mann–Whitney U tests). (D) Modified Rankin score in 64 patients with nonparaneoplastic LGI1-antibody–associated encephalopathy at peak of illness (pink) and latest follow-up (black). Data were modified from Irani et al.32 Patients were treated with corticosteroids (ST), with or without intravenous immunoglobulins (IVIG) and/or plasma exchange (PLEX). One patient died in the ST + IVIG + PLEX group. Median follow-up was for 48 months (range = 19–95) with no differences between groups (p = 0.77, Kruskal–Wallis test).
Other NGSAbs have been identified in a small number of patients with typical limbic encephalopathies (see Table1); γ-aminobutyric acidB receptor (GABABR) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR) antibodies have been described mostly in association with tumors,40–42 including small-cell lung, thymoma, and breast. Importantly, even in these patients with NGSAbs and paraneoplastic disease, the syndromes appear to be treatment-responsive, emphasizing the pathogenic importance of NGSAbs.
Psychiatric Features
Patients with VGKC-complex and GAD antibodies may present with psychiatric features, including delusions, hallucinations, depression, mania, and prominent emotionality, but usually these are overwhelmed by amnesia, disorientation, and executive dysfunction.15,32,43,44 By contrast, patients with NMDAR antibodies typically show a diffuse encephalitis with prominent and early psychiatric features,45,46 which may be the first and only recognized feature of the disease.47,48
NMDAR antibodies were first described in young women with ovarian teratomas, who typically developed encephalitis with CSF pleocytosis and abnormal magnetic resonance brain imaging.49 Since this description, the phenotype spread of NMDAR antibodies has evolved so that NMDAR-antibody encephalitis is now predominantly a disease of children and young adults without a tumor (see Fig 3B), usually with a normal brain MRI.46,50,51 After a brief prodrome, with fever and headache, the disease often evolves in 2 major stages: an early stage involving seizures and cognitive/psychiatric features, followed 10 to 20 days later by a movement disorder, dysautonomia, and a reduction in consciousness.46
Phenotype spread of NMDAR antibodies has led to the recognition of isolated psychiatric presentations.47,48,52,53 This has fueled renewed interest in the possibility of an autoimmune basis for some forms of schizophrenia. Prominent psychotic features are found in patients with many other NGSAbs including Morvan syndrome (see Fig 2A; see below),20 and basal ganglia encephalitis.54 Furthermore, atypical psychosis has been reported in patients with AMPAR antibodies.55 Patients with these illnesses often show sleep disturbances and movement disorders in conjunction with psychiatric features.20,54 Further studies are now required to understand the relevance of NGSAbs to the assessment and treatment of new onset psychosis.
Given that antigenic targets other than LGI1 and NMDAR are detected relatively infrequently in cognitive or psychiatric presentations (see Tables1 and 2),17,41 it may be that these 2 most common antibody targets represent the base, rather than the tip, of the rapidly progressive dementia iceberg.
Epilepsy
Many patients with adult onset focal epilepsies prove to be a diagnostic and therapeutic challenge. Around 40% do not show responsible imaging abnormalities (“cryptogenic”), and 25% are antiepileptic drug (AED)-refractory.56 Several studies have confirmed the presence of VGKC-complex antibodies (mainly low levels, <400pM, usually without LGI1 or CASPR2 reactivity) and GAD antibodies (often high levels) in around 10% of adults with longstanding epilepsies.57–59
More importantly, a small percentage of patients with cryptogenic epilepsies, often with frequent seizures and subtle neuropsychiatric features, have LGI1 (and occasionally CASPR2) antibodies at the onset of their disease, and exhibit a favorable response to immunotherapies (see Table2).60–62 NGSAbs may also be relevant to the etiology of other forms of epilepsy; for example, LE may precede adult onset temporal lobe epilepsy with mesial temporal sclerosis by several years in up to 25% of patients.63,64 Therefore, NGSAbs appear to have pathogenic potential in some forms of adult onset epilepsies.65
Another example of phenotype spread is the recent observation of a newly described epilepsy termed FBDS.66–69 FBDS are the clearest example of a clinically distinctive seizure semiology that appears almost pathognomonic for the presence of the cell-surface–directed LGI1 antibodies. FBDS are adult onset, brief (often <2 seconds), frequent (median 50 per day) events usually involving dystonic posturing of the arm and ipsilateral face grimacing. With increased recognition, this phenotype has spread to include examples of simultaneously bilateral posturing, events of up to 30 seconds in duration, some superimposed clonic movements, and an age range from 28 to 92 years.69
Importantly, FBDS occur before the onset of cognitive impairment in around 70% of cases, and they often show a striking preferential response to corticosteroids over AEDs.66–71 Their recognition gains further significance given that cognitive impairment might be prevented if FBDS are effectively treated early in the disease course.66–69 Similarly, ictal bradycardia preceding cognitive impairment in patients with VGKC-complex/LGI1 antibodies may be a conserved phenotype that permits earlier diagnosis and immunosuppressive treatment.72
Movement Disorders
Anti–basal ganglia antibodies (ABGAs) have been described in association with poststreptococcal neurological illnesses, but they show limited syndrome specificity, levels correlate poorly with disease course, and they do not appear to target the extracellular domain of neuronal proteins.73,74 However, patients with ABGAs can harbor other antibodies with pathogenic potential. In particular, NMDAR antibodies have been found in the serum and CSF of 50% of patients with a diagnosis of “encephalitis lethargica,”75 and dopamine-2–receptor antibodies have been reported in some patients with basal ganglia encephalitis, Sydenham chorea, and Tourette syndrome.54 In many of these patients, the movement disorder is the prominent feature, but there may be coexistent psychiatric symptoms, including obsessive–compulsive disorder, depression, and anxiety.
The movement disorder in patients with NMDAR antibodies frequently shows prominent orofacial involvement with additional complex bilateral movements of the limbs.46,50,76 However, the hyperkinetic movement phenomenology is heterogeneous and can include myoclonus, dystonia, chorea, and athetosis.77
Interestingly, in the past 2 years several studies have identified NMDAR antibodies in patients with proven herpes simplex virus encephalitis (HSVE).22 The strongest association appears to be in children developing choreoathetosis and cognitive impairment weeks to months after proven HSVE, suggesting that NGSAbs can arise as a result of virally induced neuronal damage.78–81 Given the marked similarity of this presentation to many typical patients with NMDAR antibodies, and the observed response to immunotherapy, these NMDAR antibodies are likely to be pathogenic.78,82
The spectrum of stiff-person–related disorders retain commonality in their subacute onset of otherwise unexplained spasms and stiffness and frequent association with high-titer GAD antibodies.83,84 The phenotypes can extend from axial-dominant stiff-person syndrome (SPS), or variant SPS with single limb involvement, to the progressive encephalomyelitis with rigidity and myoclonus (PERM) subtype with prominent encephalopathy and brainstem features. Some studies suggest that the variable degree of autonomic involvement, seizures, cerebellar ataxia, and cognitive impairment allows subcategorization of these disorders (see Fig 2B).83–86 In addition to these SPSs, the phenotype spread of GAD-antibody–associated syndromes now includes LE and some epilepsies (Fig 2B).15,17
Antibodies to GABAAR-associated protein and gephyrin, both intracellular proteins, in some SPS patients are unlikely to represent in vivo targets of pathogenic antibodies.87,88 By contrast, the recent discovery of glycine-alpha1–receptor antibodies in PERM and a few cases with SPS,89–91 the association of dipeptidyl peptidase IV–related protein antibodies in 3 patients with a distinctive form of SPS,92 and the recognition that some patients with GABABR-antibody and AMPAR-antibody LE also harbor GAD antibodies, suggest that potentially pathogenic NGSAbs may also be present in GAD-antibody–positive patients.17–19 Moreover, although classical SPS often responds adequately to benzodiazepines, antibodies to surface epitopes of glycine receptors are now found in a range of PERM and related syndromes,91 with excellent and preferential responses to immunotherapies. It is possible that NGSAbs against other proteins involved in inhibitory neurotransmission are awaiting discovery in several patients with GAD-antibody–associated disorders.19
Finally, movement disorders (see Fig 2A) are also now recognized within the phenotypes of VGKC-complex antibody–associated disorders with reports of ataxia,32,93 parkinsonism,44 and chorea,44,94 and often coexist with cognitive impairment and seizures.32,44
Demyelinating Disease
Neuromyelitis optica (NMO) is a disorder characterized by attacks of longitudinally extensive transverse myelitis (LETM) and optic neuritis. NMO was formerly classified as an aggressive variant of multiple sclerosis (MS), but is now known to fall within the spectrum of NGSAb-associated CNS disorders95,96 and may respond adversely to medications commonly used for treatment of MS.97,98 Unlike antibody disorders targeting neuronal cell-surface antigens, the pathogenic antibody in about 80% of NMO patients targets the AQP4 water channel on astrocytes.95,99,100 Here again, the recognition of the AQP4 antibody has led to identification of different presentations, such as early intractable vomiting,96,101 fulminant parenchymal cerebral lesions,102 white matter lesions on brain MRI that can look similar to those seen in MS,103 and non–longitudinally extensive myelitis.104,105 By extension, the identification of the AQP4 antibody has led to the related syndrome being labeled under the umbrella term of NMO spectrum disorder (NMOSD), which encompasses all associated phenotypes, including isolated LETM or optic neuritis, and has enabled earlier diagnosis and treatment after the first NMO attack.104–106
Importantly, pain is an increasingly recognized, disabling, and treatment-refractory feature of NMO. Neuropathic-type pain has also been associated with VGKC-complex antibodies, either in isolation107 or as part of neuromyotonia or Morvan syndrome.20 It is often immunotherapy-responsive, and there is an association with CASPR2 antibodies.20,107
As in all the diseases discussed here, some patients with typical symptoms of NMO do not have the recognized antibody. Although this may be due to technical limitations, the existence of other antibodies is likely in some of these currently seronegative patients. In NMOSD, for instance, a significant number of AQP4-antibody–negative patients have antibodies against myelin oligodendrocyte glycoprotein (MOG), which may herald a less aggressive prognosis.108–112 Most recently, patients with NMDAR antibodies have been described with marked demyelinating changes on imaging113 and clinical features overlapping with NMO and MS.114–116 Like AQP4 and MOG, NMDARs are also expressed on glia.117
Current and Novel Assays Using Serum- and CSF-Based Diagnostics and Related Insights into Disease Biology
Antibody Testing: Native, Fixed, and Solubilized Antigens
Classical paraneoplastic antibodies bind intracellular antigens that are exposed on paraformaldehyde-fixed rodent brain sections (see Fig 1E) and also after denaturation on Western blots. By contrast, although NGSAbs can often be detected on brain sections, they do not usually bind well to denatured antigens. The technique most likely to detect potentially pathogenic antibodies is the binding of serum or CSF immunoglobulin (Ig) G to live cells that express the native antigen on their surface (Cell-based assays (CBAs); see Fig 1A–C).3,8 Absence of binding to cells that express another antigen helps to demonstrate antibody specificity.3,32,46
CBAs employing live mammalian cells have the advantage of exposing the patients' antibodies only to the extracellular domains of native antigens.17,32,46,99 Fixed or permeabilized CBAs, however, may detect nonpathogenic antibodies to intracellular and fixation-exposed epitopes. Some groups prefer a combination of these CBA techniques, but only partly based on the antigen specificity (for example compare Boronat et al17 to Lancaster et al42), whereas others adhere to a single method for all antigens.32,46,100 In our view, the ideal would be for local diagnostic laboratories to have live neuronal cultures for fast detection of a range of potential NGSAbs and/or live CBAs for identifying the targets; but these live cell assays are time-consuming and costly. Currently, therefore, most diagnostic laboratories use commercial kits that provide fixed brain tissue and fixed antigen-expressing cells with the possible limitations discussed above.
The clinical relevance of NGSAbs against LGI1 and CASPR2 is widely accepted. Contactin-2 antibodies are less common and often coexist with LGI1 or CASPR2 antibodies (see Fig 1F).32 Antibodies that immunoprecipitate the VGKC complex from solubilized mammalian brain membranes, but do not show LGI1/CASPR2/contactin-2 reactivity or bind to the surface of hippocampal neurons, may be directed against epitopes that are intracellular in vivo (eg, Fig 1F) and therefore not be pathogenic. This is consistent with their appearance in patients with chronic epilepsies60 and a variety of conditions that are not immunotherapy-responsive, such as sporadic CJD, transient global amnesia, and MS.118–120 It is similarly of interest that VGKC-complex antibodies from children rarely show LGI1/CASPR2/contactin-2 reactivity.37–39 However, sera from some patients with VGKC-complex antibodies without LGI1/CASPR2/contactin-2 reactivity do bind hippocampal neurons and associate with immunotherapy-responsive syndromes (A. Vincent, unpublished observations); the identity of these target proteins is not yet known.
CSF and Serum Antibody Levels at Onset and after Treatment: Relationships to Intrathecal Synthesis
There has been some confusion regarding the relative roles of CSF and serum antibodies in these diseases. In all known NGSAb syndromes, absolute concentrations of NGSAbs in the serum are consistently higher than in CSF at diagnosis.30,31,41,42,46,75 These high serum levels of NGSAbs are especially unsurprising in patients with systemic tumors, including ovarian teratomas and small-cell lung cancers, which express the antigenic target and are highly likely to trigger the peripheral antibody production.45,121 But they are also consistently seen in nonparaneoplastic patients. In the minority of patients reported with NGSAbs in CSF and not in serum at disease onset,122 the apparent absence of peripheral antibody would necessitate invoking a strikingly different mechanism of autoimmunization and may be a consequence of the assay methodology (see below).
Absolute serum IgG levels are usually around 400× higher than those in CSF.3 With a normal blood–brain barrier (BBB), therefore, the CSF to serum ratios of any circulating antibody would be 1:400, indicating that the specific antibody within the CSF is not being produced intrathecally. However, for NMDAR antibodies, the ratio is often much higher, from only 1:4 to 1:3,204 (median ≈ 1:15),46,75,123 and similar ratios of 1:10 to 1:200 and 1:14 to 1:320 are reported for VGKC-complex antibodies30,31 and AMPAR antibodies, respectively.41 This indicates that there is more antigen-specific antibody in the CSF than expected by natural diffusion.
A comparison of the antigen-specific antibody to the amount of total IgG in the 2 compartments ([(CSF NGSAb)/(CSF total IgG)]/[(serum NGSAb)/(serum total IgG)]) indicates the “intrathecal synthesis” of the particular antibody. In the normal situation, it would be 1. Values >1 imply that clonal B cells producing antigen-specific IgG have traversed the BBB and expanded intrathecally to secrete the specific antibodies. Intrathecal synthesis is seen with most NGSAbs,45,46,75,123 but its extent varies considerably between different diseases. Whereas it is frequently high in patients with NMDAR,46,50 AMPAR,41 GABABR,42 and glycine-receptor antibodies,124 it is often lower in patients with VGKC-complex/LGI1 antibodies.30,31 Conversely, when NGSAbs are detected in serum but not in CSF, which can be the case at the onset of disease (A. Vincent, unpublished observations), it may be that the NGSAbs have traversed the BBB and have bound directly to their target within the brain parenchyma,125 and that intrathecal synthesis will follow as the disease progresses. Alternatively, their absence in the CSF may imply a low chance of pathogenicity.
Some of the confusion regarding serum versus CSF antibody levels, however, arises from different ways of presenting the data; in some studies the absolute levels of serum and CSF NGSAbs have first been normalized to total IgG levels in serum or CSF (eg, Dalmau et al45), whereas in others absolute concentrations are presented (eg, Irani et al,46 Suh-Lailam et al123). In addition, other interstudy differences include the dilutions used to test serum and CSF (compare Lancaster et al42 to Boronat et al17), and differences between live and fixed CBAs (discussed above).121 In Oxford, use of relatively concentrated serum (usually 1:20) and 1:1 diluted CSF, and exposure to purely extracellular targets (live CBA) has not identified any positive CSFs with negative paired sera. Nevertheless, there may be rare situations where the antibody generation occurs purely within the CNS, and for this reason clinicians should consider sending both serum and CSF for diagnosis of NGSAb-related disorders when possible.
These considerations are of relevance to treatments. Serum antibodies are often vulnerable to standard acute immunosuppressive therapies, such as corticosteroids, intravenous immunoglobulins (IVIG), and particularly plasma exchange (PLEX). Access to CNS antibodies and plasma cells, however, may be difficult with many chemotherapeutic agents.126 Therefore, it is unsurprising that we and others have observed cases where CSF antibodies postimmunotherapy appear more closely to track the disease course (Irani, Gelfand, and Geschwind, unpublished).122 However, there are examples where serum antibodies best correlate with the clinical course (A. Vincent, unpublished observations). So, as with testing at diagnosis, it may be that both serum and CSF provide complementary paraclinical information to guide ongoing immunotherapy and, in some cases, assessing for relapse activity. Given intrathecal synthesis of NGSAbs in most of these conditions, it may be that CNS-directed B-cell or plasma-cell depletion will be most effective. Nevertheless, the repeated observations that PLEX is clinically useful and tumor removal hastens recovery45,46,127–130 suggest that removal of a plasma factor—such as antibodies—and reduction in ongoing peripheral autoimmunization ameliorate the CNS disease.
“False-Negative” Autoantibodies, “False-Positive” Autoantibodies, and Autoantibodies with Unproven Clinical Relevance
As the antibody specificity is often the defining feature of a reported cohort, a major difficulty with interpretation of the results lies in the absence of an independent gold standard for any of these conditions. This is especially evident for NMOSD.131
Given several recent independent reports of serum and/or CSF NMDAR antibodies in asymptomatic individuals132 and patients with nonrelapsing HSVE,22 paroxysmal exercise-induced dyskinesias,133 CJD,134 myelitis,135 and Klebsiella pneumoniae meningitis (A. Tebo, personal communication),123 and the detection of serum NMDAR or VGKC-complex antibodies in a few patients with CJD and other nonimmune disorders (M. Rossi et al, submitted),118,119 the specificity and clinical relevance of some neuronal antibodies is in question.
When the assay methodologies include specificity controls, such as absent binding of antigen-specific sera to other antigens, results in healthy or disease controls do not necessarily represent false positives. Rather, these observation suggest that low levels of autoantibodies can exist in patients without recognized antibody-associated syndromes (see Tables1 and 2), and may represent secondary immunization after neuronal damage and either have no effect, or could alter the course of the disease. A recent study suggested a high, age-related rate of NGSAbs especially against NMDAR (10%) in disease and healthy controls. Also, around 1% of healthy controls had CASPR2 and MOG NGSAbs.136 Although these figures include IgA and IgM subclasses, and some titers are low, they do emphasize the need to exercise caution in the interpretation of the clinical relevance of a positive result.
At present, despite ongoing phenotype spread, diagnosis and treatment decisions for NGSAb-associated disorders must continue to be based on the relevance of such antibodies to the clinical syndrome, and clinicians need to strike a balance between appropriate early, aggressive treatment and exposing patients to unnecessary immunotherapies.
Treatments: Concepts and Syndrome-Specific Approaches
There are no published randomized–controlled clinical trials in NGSAb-associated CNS diseases. Available treatment evidence comes from case and cohort studies, which suggest that early and more aggressive immunotherapy improves outcomes.30,46,50,68,69,137 Therapeutic aims in NGSAb-associated conditions include symptomatic control of the acute episode, reduction of existing antibody levels, suppression of future antibody production, and timely withdrawal of potentially toxic medications. In addition, tumor removal should always be considered when relevant. Important questions to ask prior to planning treatment regimens include when and how disability is accrued, and what the natural history of the condition is. In addition, when addressing the question of worsening, relapsing, or persistent symptoms in a patient with an established NGSAb-related diagnosis, it is important to assess whether such symptomatology reflects ongoing CNS disease activity or is the consequence of prior antibody-induced injury. It is also important to provide symptomatic treatment for comorbid psychiatric symptoms, seizures, fatigue, sleep disturbances, and pain syndromes that can accompany these syndromes or the postencephalitic aftermath.
In conjunction with symptomatic management, the acute phase is usually managed with variable combinations of high-dose corticosteroids, IVIG, and PLEX. There is evidence for a more rapid return of function after early corticosteroids in VGKC-complex antibody–associated LE30 and FBDS.69 The efficacy of IVIG and PLEX in NGSAb-associated encephalitis is unproven, but supportive reports do exist, especially for PLEX.128–130 However, our open-label observational analysis suggests that the addition of IVIG, PLEX, or both to corticosteroids in 64 patients with VGKC-complex/LGI1 antibody–associated LE did not appear to alter outcomes at a median of 4 years of follow-up (see Fig 3D; data modified from Irani et al32). More robust data for PLEX are available for the treatment of NMO.104,106,138,139 Nevertheless, despite limited evidence IVIG and PLEX continue to be popular options because they have a rapid onset of action and are relatively accessible, and there remains an intuitive belief that their mechanisms of action and their efficacy in similar diseases should translate to clinical benefit. As IgGs are replaced with a half-life of 21 days,140 PLEX typically only serves as a bridge while more definitive immunotherapies are commenced to reduce antibody production. The use of high doses and long durations of corticosteroids in many of these conditions make steroid-sparing options important especially in diseases whose natural histories show the potential for a chronic or relapsing–remitting course, such as NMO and NMDAR-antibody encephalitis.
Observational studies have shown that NMO relapses are significantly reduced by chronic immunotherapy with glucocorticoids,141 azathioprine,142 methotrexate,143 mycophenolate mofetil (MMF),144 rituximab,145–147 mitoxantrone,148,149 toculizimab,150 and eculizimab.151 No head-to-head or placebo trial has been performed, but in single-arm analyses azathioprine does appear to associate with a higher annualized relapse rate than MMF or rituximab,152 and was also associated with a relatively high rate of lymphoma.142 In NMDAR-antibody encephalitis, there is some evidence that patients with limited or no improvement after first-line immunotherapies may show longer-term benefit from more aggressive regimens including addition of rituximab and/or cyclophosphamide.46,137 This is very different from the experience with VGKC-complex/LGI1–antibody encephalitis (see Fig 3D), where there has been little published use of second-line immunotherapy, as this is usually a monophasic illness that has traditionally been treated with steroids with or without addition of PLEX/IVIG.30,153,154 However, the toxicity of prolonged steroid regimes in the elderly, unanswered questions about relative efficacy of steroids versus other immunosuppressive options, and a significant minority of patients with poor cognitive and functional outcomes despite acute immunosuppression155 mean more aggressive or targeted immunotherapies such as rituximab156 merit future study.
Conclusions
Over the past decade, there has been a marked increase in the number of NGSAb targets identified that associate with CNS diseases, and a rapid expansion in the clinical features and demographics seen in conjunction with each antibody. This phenotype spread has highlighted the relevance of NGSAbs to neurologists with an interest in epilepsy, cognition and behavior, movement disorders, and demyelinating disease. This antibody awareness has also highlighted the increasing requirement to exercise clinical judgment in the context of an incongruent clinical–serological picture and has emphasized the importance of understanding assay methodologies and appreciating concepts surrounding rational immunotherapy administration. Vigilance for an underlying tumor is required in all these syndromes to varying degrees, but the distinction between cell-surface and intracellular antigenic targets may be more important than the presence of an underlying tumor in guiding prognosis of paraneoplastic syndromes.
Future studies will need to address the pathogenicity of the antibodies, which patients with serum/CSF antibodies develop disease, whether B cells or NGSAbs or both enter the CNS to initiate the disease, and whether antibody titers help guide immunotherapy escalation and withdrawal. Research should aim to identify the clinical epidemiology of positive test results in determining the best guidelines for screening NGSAbs and its cost-effectiveness in the neurology clinic, and to offer efficient methods for worldwide diagnosis.
Acknowledgments
S.R.I. was supported by Epilepsy Research UK (P1201), by the Fulbright UK-US commission and the MS society (962/12), and by the BMA Vera Down Grant. A.V. was supported by the Oxford Biomedical Centre and the National Institute for Health Research. J.M.G. was supported by the National Center for Advancing Translational Sciences of the NIH (award number KL2TR000143). The contents of this review are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
All coauthors have seen and agree with the contents of the article, the International Committee of Medical Journal Editors requirements for authorship have been met, and each author believes that the article represents honest work.
Authorship
The authors have all made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data, drafting the manuscript, or revising it critically for important intellectual content, and have given final approval of the version to be published.
Potential Conflicts of Interest
S.R.I.: coapplicant patent with Oxford University for VGKC complex antibodies licensed to Euroimmun; royalties, (licensee ISIS). J.M.G.: medical–legal consulting related to CNS inflammatory–demyelinating disease. A.V.: consultancy, Athena Diagnostics; employment, Oxford University; holds patent with Oxford University for VGKC complex antibodies licensed to Euroimmun; royalties, Euroimmun, Athena Diagnostics.
References
- Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135–1140. doi: 10.1136/jnnp.2003.034447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol. 2011;122:381–400. doi: 10.1007/s00401-011-0876-1. [DOI] [PubMed] [Google Scholar]
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10:759–772. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- Leypoldt F, Wandinger KP. Paraneoplastic neurological syndromes. Clin Exp Immunol. 2014;175:336–348. doi: 10.1111/cei.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Tanaka K, Shinozawa K, et al. Cytotoxic T cells react with recombinant Yo protein from a patient with paraneoplastic cerebellar degeneration and anti-Yo antibody. J Neurol Sci. 1998;161:88–90. doi: 10.1016/s0022-510x(98)00257-3. [DOI] [PubMed] [Google Scholar]
- Vincent A, Lily O, Palace J. Pathogenic autoantibodies to neuronal proteins in neurological disorders. J Neuroimmunol. 1999;100:169–180. doi: 10.1016/s0165-5728(99)00210-6. [DOI] [PubMed] [Google Scholar]
- Lancaster E, Dalmau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8:380–390. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani L, Graus F, Giometto B, et al. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry. 2012;83:638–645. doi: 10.1136/jnnp-2011-301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, et al. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Irani SR, Vincent A. Autoimmune encephalitis—new awareness, challenging questions. Discov Med. 2011;11:449–458. [PubMed] [Google Scholar]
- Irani SR, Vincent A. The expanding spectrum of clinically-distinctive, immunotherapy-responsive autoimmune encephalopathies. Arq Neuropsiquiatr. 2012;70:300–304. doi: 10.1590/s0004-282x2012000400015. [DOI] [PubMed] [Google Scholar]
- Granerod J, Cousens S, Davies NW, et al. New estimates of incidence of encephalitis in England. Emerg Infect Dis. 2013;19(9) doi: 10.3201/eid1909.130064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari N, Lillevang ST, Skejoe HP, et al. A population-based study of neuromyelitis optica in Caucasians. Neurology. 2011;76:1589–1595. doi: 10.1212/WNL.0b013e3182190f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131:2553–2563. doi: 10.1093/brain/awn183. [DOI] [PubMed] [Google Scholar]
- Malter MP, Helmstaedter C, Urbach H, et al. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol. 2010;67:470–478. doi: 10.1002/ana.21917. [DOI] [PubMed] [Google Scholar]
- Vincent A. Stiff, twitchy or wobbly: are GAD antibodies pathogenic? Brain. 2008;131:2536–2537. doi: 10.1093/brain/awn221. [DOI] [PubMed] [Google Scholar]
- Boronat A, Sabater L, Saiz A, et al. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology. 2011;76:795–800. doi: 10.1212/WNL.0b013e31820e7b8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013;70:44–50. doi: 10.1001/jamaneurol.2013.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T, Alexopoulos H, McMenamin M, et al. Neuronal surface and glutamic acid decarboxylase autoantibodies in nonparaneoplastic stiff person syndrome. JAMA Neurol. 2013;70:1140–1149. doi: 10.1001/jamaneurol.2013.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72:241–255. doi: 10.1002/ana.23577. [DOI] [PubMed] [Google Scholar]
- Turner MR, Irani SR, Leite MI, et al. Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology. 2011;77:439–443. doi: 10.1212/WNL.0b013e318227b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss H, Finke C, Holtje M, et al. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol. 2012;72:902–911. doi: 10.1002/ana.23689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado S, Pinto AN, Irani SR. What should you know about limbic encephalitis? Arq Neuropsiquiatr. 2012;70:817–822. doi: 10.1590/s0004-282x2012001000012. [DOI] [PubMed] [Google Scholar]
- Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:97–108. doi: 10.1002/ana.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granerod J, Ambrose HE, Davies NW, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10:835–844. doi: 10.1016/S1473-3099(10)70222-X. [DOI] [PubMed] [Google Scholar]
- Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011;70:437–444. doi: 10.1002/ana.22454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable MS, Sheriff H, Dalmau J, et al. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54:899–904. doi: 10.1093/cid/cir1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom MH, Smith S, Akdal G, Geschwind MD. Immunologically mediated dementias. Curr Neurol Neurosci Rep. 2009;9:359–367. doi: 10.1007/s11910-009-0053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50:73–78. doi: 10.1002/ana.1097. [DOI] [PubMed] [Google Scholar]
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701–712. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- Thieben MJ, Lennon VA, Boeve BF, et al. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004;62:1177–1182. doi: 10.1212/01.wnl.0000122648.19196.02. [DOI] [PubMed] [Google Scholar]
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain. 2010;133:2734–2748. doi: 10.1093/brain/awq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776–785. doi: 10.1016/S1474-4422(10)70137-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol. 2008;65:1341–1346. doi: 10.1001/archneur.65.10.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamija R, Renaud DL, Pittock SJ, et al. Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr Neurol. 2011;44:275–281. doi: 10.1016/j.pediatrneurol.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Haberlandt E, Bast T, Ebner A, et al. Limbic encephalitis in children and adolescents. Arch Dis Child. 2011;96:186–191. doi: 10.1136/adc.2010.183897. [DOI] [PubMed] [Google Scholar]
- Hacohen Y, Wright S, Waters P, et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry. 2013;84:748–755. doi: 10.1136/jnnp-2012-303807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman J, Brenner T, Gill D, et al. VGKC antibodies in pediatric encephalitis presenting with status epilepticus. Neurology. 2011;76:1252–1255. doi: 10.1212/WNL.0b013e3182143552. [DOI] [PubMed] [Google Scholar]
- Suleiman J, Brilot F, Lang B, et al. Autoimmune epilepsy in children: case series and proposed guidelines for identification. Epilepsia. 2013;54:1036–1045. doi: 10.1111/epi.12142. [DOI] [PubMed] [Google Scholar]
- Jeffery OJ, Lennon VA, Pittock SJ, et al. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology. 2013;81:882–887. doi: 10.1212/WNL.0b013e3182a35271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–434. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67–76. doi: 10.1016/S1474-4422(09)70324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers KJ, Lennon VA, Rundell JR, et al. Psychiatric manifestations of voltage-gated potassium-channel complex autoimmunity. J Neuropsychiatry Clin Neurosci. 2011;23:425–433. doi: 10.1176/jnp.23.4.jnp425. [DOI] [PubMed] [Google Scholar]
- Tan KM, Lennon VA, Klein CJ, et al. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70:1883–1890. doi: 10.1212/01.wnl.0000312275.04260.a0. [DOI] [PubMed] [Google Scholar]
- Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–1098. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010;133:1655–1667. doi: 10.1093/brain/awq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70:1133–1139. doi: 10.1001/jamaneurol.2013.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi MS, Irani SR, Lang B, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol. 2011;258:686–688. doi: 10.1007/s00415-010-5788-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10:63–74. doi: 10.1016/S1474-4422(10)70253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66:11–18. doi: 10.1002/ana.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer C, Stepniak B, Schneider A, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.110. Sep 3. doi: 10.1038/mp.2013.110. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Steiner J, Walter M, Glanz W, et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry. 2013;70:271–278. doi: 10.1001/2013.jamapsychiatry.86. [DOI] [PubMed] [Google Scholar]
- Dale RC, Merheb V, Pillai S, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain. 2012;135:3453–3468. doi: 10.1093/brain/aws256. [DOI] [PubMed] [Google Scholar]
- Graus F, Boronat A, Xifro X, et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology. 2010;74:857–859. doi: 10.1212/WNL.0b013e3181d3e404. [DOI] [PubMed] [Google Scholar]
- Shorvon SD. The etiologic classification of epilepsy. Epilepsia. 2011;52:1052–1057. doi: 10.1111/j.1528-1167.2011.03041.x. [DOI] [PubMed] [Google Scholar]
- Majoie HJ, de Baets M, Renier W, et al. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res. 2006;71:135–141. doi: 10.1016/j.eplepsyres.2006.06.003. [DOI] [PubMed] [Google Scholar]
- McKnight K, Jiang Y, Hart Y, et al. Serum antibodies in epilepsy and seizure-associated disorders. Neurology. 2005;65:1730–1736. doi: 10.1212/01.wnl.0000187129.66353.13. [DOI] [PubMed] [Google Scholar]
- Peltola J, Kulmala P, Isojarvi J, et al. Autoantibodies to glutamic acid decarboxylase in patients with therapy-resistant epilepsy. Neurology. 2000;55:46–50. doi: 10.1212/wnl.55.1.46. [DOI] [PubMed] [Google Scholar]
- Brenner T, Sills GJ, Hart Y, et al. Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia. 2013;54:1028–1035. doi: 10.1111/epi.12127. [DOI] [PubMed] [Google Scholar]
- Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure. 2013;22:776–779. doi: 10.1016/j.seizure.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Quek AM, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69:582–593. doi: 10.1001/archneurol.2011.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav. 2007;10:529–538. doi: 10.1016/j.yebeh.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Bien CG, Schulze-Bonhage A, Deckert M, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology. 2000;55:1823–1828. doi: 10.1212/wnl.55.12.1823. [DOI] [PubMed] [Google Scholar]
- Irani SR, Bien CG, Lang B. Autoimmune epilepsies. Curr Opin Neurol. 2011;24:146–153. doi: 10.1097/WCO.0b013e3283446f05. [DOI] [PubMed] [Google Scholar]
- Barajas RF, Collins DE, Cha S, Geschwind MD. Adult-onset drug-refractory seizure disorder associated with anti-voltage-gated potassium-channel antibody. Epilepsia. 2010;51:473–477. doi: 10.1111/j.1528-1167.2009.02287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Buckley C, Vincent A, et al. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology. 2008;71:1647–1648. doi: 10.1212/01.wnl.0000326572.93762.51. [DOI] [PubMed] [Google Scholar]
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900. doi: 10.1002/ana.22307. [DOI] [PubMed] [Google Scholar]
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013;136:3151–3162. doi: 10.1093/brain/awt212. [DOI] [PubMed] [Google Scholar]
- Yoo JY, Hirsch LJ. Limbic encephalitis associated with anti-voltage-gated potassium channel complex antibodies mimicking Creutzfeldt-Jakob disease. JAMA Neurol. 2014;71:79–82. doi: 10.1001/jamaneurol.2013.5179. [DOI] [PubMed] [Google Scholar]
- Plantone D, Renna R, Grossi D, et al. Teaching NeuroImages: basal ganglia involvement in facio-brachial dystonic seizures associated with LGI1 antibodies. Neurology. 2013;80:e183–e184. doi: 10.1212/WNL.0b013e31828f17fa. [DOI] [PubMed] [Google Scholar]
- Naasan G, Irani SR, Bettcher BM, et al. Episodic bradycardia: a neurocardiac prodrome to VGKC-complex/LGI1-antibody encephalitis. JAMA Neurol. doi: 10.1001/jamaneurol.2014.1234. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RC, Heyman I, Giovannoni G, Church AW. Incidence of anti-brain antibodies in children with obsessive-compulsive disorder. Br J Psychiatry. 2005;187:314–319. doi: 10.1192/bjp.187.4.314. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Trikouli E, Martino D, et al. Anti-basal ganglia antibodies in patients with atypical dystonia and tics: a prospective study. Neurology. 2004;63:156–158. doi: 10.1212/01.wnl.0000131900.71337.c7. [DOI] [PubMed] [Google Scholar]
- Dale RC, Irani SR, Brilot F, et al. N-methyl-D-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol. 2009;66:704–709. doi: 10.1002/ana.21807. [DOI] [PubMed] [Google Scholar]
- Stamelou M, Plazzi G, Lugaresi E, et al. The distinct movement disorder in anti-NMDA receptor encephalitis may be related to status dissociatus: a hypothesis. Mov Disord. 2012;27:1360–1363. doi: 10.1002/mds.25072. [DOI] [PubMed] [Google Scholar]
- Baizabal-Carvallo JF, Stocco A, Muscal E, Jankovic J. The spectrum of movement disorders in children with anti-NMDA receptor encephalitis. Mov Disord. 2013;28:543–547. doi: 10.1002/mds.25354. [DOI] [PubMed] [Google Scholar]
- Hacohen Y, Deiva K, Pettingill P, et al. N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord. 2014;29:90–96. doi: 10.1002/mds.25626. [DOI] [PubMed] [Google Scholar]
- Hacohen Y, Dlamini N, Hedderly T, et al. N-methyl-D-aspartate receptor antibody-associated movement disorder without encephalopathy. Dev Med Child Neurol. 2014;56:190–193. doi: 10.1111/dmcn.12321. [DOI] [PubMed] [Google Scholar]
- Mohammad SS, Sinclair K, Pillai S, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-Methyl-D-aspartate receptor or dopamine-2 receptor. Mov Disord. 2014;29:117–122. doi: 10.1002/mds.25623. [DOI] [PubMed] [Google Scholar]
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology. 2013;81:1637–1639. doi: 10.1212/WNL.0b013e3182a9f531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftberger R, Armangue T, Leypoldt F, et al. Clinical neuropathology practice guide 4–2013: post-herpes simplex encephalitis: N-methyl-Daspartate receptor antibodies are part of the problem. Clin Neuropathol. 2013;32:251–254. doi: 10.5414/NP300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker RA, Revesz T, Thom M, et al. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 1998;65:633–640. doi: 10.1136/jnnp.65.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinck HM, Thompson PD. Stiff man syndrome and related conditions. Mov Disord. 2002;17:853–866. doi: 10.1002/mds.10279. [DOI] [PubMed] [Google Scholar]
- McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69:230–238. doi: 10.1001/archneurol.2011.991. [DOI] [PubMed] [Google Scholar]
- Shaw PJ. Stiff-man syndrome and its variants. Lancet. 1999;353:86–87. doi: 10.1016/S0140-6736(05)76151-1. [DOI] [PubMed] [Google Scholar]
- Raju R, Rakocevic G, Chen Z, et al. Autoimmunity to GABAA-receptor-associated protein in stiff-person syndrome. Brain. 2006;129:3270–3276. doi: 10.1093/brain/awl245. [DOI] [PubMed] [Google Scholar]
- Butler MH, Hayashi A, Ohkoshi N, et al. Autoimmunity to gephyrin in stiff-man syndrome. Neuron. 2000;26:307–312. doi: 10.1016/s0896-6273(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71:1291–1292. doi: 10.1212/01.wnl.0000327606.50322.f0. [DOI] [PubMed] [Google Scholar]
- Mas N, Saiz A, Leite MI, et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 2011;82:1399–1401. doi: 10.1136/jnnp.2010.229104. [DOI] [PubMed] [Google Scholar]
- Stern WM, Howard R, Chalmers RM, et al. Glycine receptor antibody mediated progressive encephalomyelitis with rigidity and myoclonus (PERM): a rare but treatable neurological syndrome. Pract Neurol. 2014;14:123–127. doi: 10.1136/practneurol-2013-000511. [DOI] [PubMed] [Google Scholar]
- Balint B, Jarius S, Nagel S, et al. Progressive encephalomyelitis with rigidity and myoclonus: a new variant with DPPX antibodies. Neurology. 2014;82:1521–1528. doi: 10.1212/WNL.0000000000000372. [DOI] [PubMed] [Google Scholar]
- Becker EB, Zuliani L, Pettingill R, et al. Contactin-associated protein-2 antibodies in non-paraneoplastic cerebellar ataxia. J Neurol Neurosurg Psychiatry. 2012;83:437–440. doi: 10.1136/jnnp-2011-301506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Irani SR, Cheeran BJ, et al. Immunotherapy-responsive chorea as the presenting feature of LGI1-antibody encephalitis. Neurology. 2012;79:195–196. doi: 10.1212/WNL.0b013e31825f0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim W, Li XF, et al. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. 2012;18:1480–1483. doi: 10.1177/1352458512439439. [DOI] [PubMed] [Google Scholar]
- Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–245. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- Waters P, Vincent A. Detection of anti-aquaporin-4 antibodies in neuromyelitis optica: current status of the assays. Int MS J. 2008;15:99–105. [PubMed] [Google Scholar]
- Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–671; discussion 669. doi: 10.1212/WNL.0b013e318248dec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. 2011;76:1229–1237. doi: 10.1212/WNL.0b013e318214332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey CR, Bermel RA. Fulminant cerebral demyelination in neuromyelitis optica. Neurology. 2011;77:193. doi: 10.1212/WNL.0b013e3182242d6e. [DOI] [PubMed] [Google Scholar]
- Pittock SJ, Lennon VA, Krecke K, et al. Brain abnormalities in neuromyelitis optica. Arch Neurol. 2006;63:390–396. doi: 10.1001/archneur.63.3.390. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM. Neuromyelitis optica spectrum disorders. Continuum (Minneap Minn) 2010;16:105–121. doi: 10.1212/01.CON.0000389937.69413.15. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–1032. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Lennon VA, Aston PA, et al. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology. 2012;79:1136–1144. doi: 10.1212/WNL.0b013e3182698cab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–1277. doi: 10.1212/WNL.0b013e31826aac4e. [DOI] [PubMed] [Google Scholar]
- Rostasy K, Mader S, Hennes EM. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult Scler. 2013;19:1052–1059. doi: 10.1177/1352458512470310. [DOI] [PubMed] [Google Scholar]
- Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–481. doi: 10.1212/WNL.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. 2014;71:276–283. doi: 10.1001/jamaneurol.2013.5857. [DOI] [PubMed] [Google Scholar]
- Di Pauli F, Mader S, Rostasy K, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. 2011;138:247–254. doi: 10.1016/j.clim.2010.11.013. [DOI] [PubMed] [Google Scholar]
- Hacohen Y, Absoud M, Woodhall M, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry. 2014;85:456–461. doi: 10.1136/jnnp-2013-306411. [DOI] [PubMed] [Google Scholar]
- Zoccarato M, Saddi MV, Serra G, et al. Aquaporin-4 antibody neuromyelitis optica following anti-NMDA receptor encephalitis. J Neurol. 2013;260:3185–3187. doi: 10.1007/s00415-013-7182-x. [DOI] [PubMed] [Google Scholar]
- Kruer MC, Koch TK, Bourdette DN, et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology. 2010;74:1473–1475. doi: 10.1212/WNL.0b013e3181dc1a7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MJ, Hoftberger R, Iizuka T, et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;75:411–428. doi: 10.1002/ana.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olberg H, Haugen M, Storstein A, Vedeler CA. Neurological manifestations related to level of voltage-gated potassium channel antibodies. J Neurol Neurosurg Psychiatry. 2013;84:941–943. doi: 10.1136/jnnp-2013-305252. [DOI] [PubMed] [Google Scholar]
- Paterson RW, Zandi MS, Armstrong R, et al. Clinical relevance of positive voltage-gated potassium channel (VGKC)-complex antibodies: experience from a tertiary referral centre. J Neurol Neurosurg Psychiatry. 2014;85:625–630. doi: 10.1136/jnnp-2013-305218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus-Leppan H, Rudge P, Mead S, et al. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol. 2013;70:919–922. doi: 10.1001/jamaneurol.2013.2077. [DOI] [PubMed] [Google Scholar]
- Irani SR, Vincent A. NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;11:298–304. doi: 10.1007/s11910-011-0186-y. [DOI] [PubMed] [Google Scholar]
- Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–177. doi: 10.1016/S1474-4422(13)70282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh-Lailam BB, Haven TR, Copple SS, et al. Anti-NMDA-receptor antibody encephalitis: performance evaluation and laboratory experience with the anti-NMDA-receptor IgG assay. Clin Chim Acta. 2013;421:1–6. doi: 10.1016/j.cca.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Carvajal-González A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics clinical features and outcomes. Brain. 2014 doi: 10.1093/brain/awu142. Jun 20. pii: awu142. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T, Alexopoulos H, Pettingill P, et al. Immunization against GAD induces antibody binding to GAD-independent antigens and brainstem GABAergic neuronal loss. PLoS One. 2013;8:e72921. doi: 10.1371/journal.pone.0072921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon LL, Soussain C, Jahnke K, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007;25:2295–2305. doi: 10.1200/JCO.2006.09.9861. [DOI] [PubMed] [Google Scholar]
- Smith JH, Dhamija R, Moseley BD, et al. N-methyl-D-aspartate receptor autoimmune encephalitis presenting with opsoclonus-myoclonus: treatment response to plasmapheresis. Arch Neurol. 2011;68:1069–1072. doi: 10.1001/archneurol.2011.166. [DOI] [PubMed] [Google Scholar]
- Nunez-Enamorado N, Camacho-Salas A, Belda-Hofheinz S, et al. Fast and spectacular clinical response to plasmapheresis in a pediatric case of anti-NMDA encephalitis [in Spanish] Rev Neurol. 2012;54:420–424. [PubMed] [Google Scholar]
- Pham HP, Daniel-Johnson JA, Stotler BA, et al. Therapeutic plasma exchange for the treatment of anti-NMDA receptor encephalitis. J Clin Apher. 2011;26:320–325. doi: 10.1002/jca.20311. [DOI] [PubMed] [Google Scholar]
- Ehrlich S, Fassbender CM, Blaes C, et al. Therapeutic apheresis for autoimmune encephalitis: a nationwide data collection [in German] Nervenarzt. 2013;84:498–507. doi: 10.1007/s00115-012-3710-7. [DOI] [PubMed] [Google Scholar]
- Jacob A, McKeon A, Nakashima I, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–930. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- Hansen HC, Klingbeil C, Dalmau J, et al. Persistent intrathecal antibody synthesis 15 years after recovering from anti-N-methyl-D-aspartate receptor encephalitis. JAMA Neurol. 2013;70:117–119. doi: 10.1001/jamaneurol.2013.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labate A, Quattrone A, Dalmau J, Gambardella A. Anti-N-methyl-D-aspartate-glutamic-receptor encephalitis presenting as paroxysmal exercise-induced foot weakness. Mov Disord. 2013;28:820–822. doi: 10.1002/mds.25510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol. 2012;259:1979–1981. doi: 10.1007/s00415-012-6489-3. [DOI] [PubMed] [Google Scholar]
- Outteryck O, Baille G, Hodel J, et al. Extensive myelitis associated with anti-NMDA receptor antibodies. BMC Neurol. 2013;13:211. doi: 10.1186/1471-2377-13-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm L, Ott C, Steiner J, et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014 doi: 10.1002/ana.24189. May 23. doi: 10.1002/ana.24189. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–165. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato DK, Nakashima I, Takahashi T, et al. Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology. 2013;80:2210–2216. doi: 10.1212/WNL.0b013e318296ea08. [DOI] [PubMed] [Google Scholar]
- Lim YM, Pyun SY, Kang BH, et al. Factors associated with the effectiveness of plasma exchange for the treatment of NMO-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler. 2013;19:1216–1218. doi: 10.1177/1352458512471875. [DOI] [PubMed] [Google Scholar]
- Kaplan AA. Therapeutic plasma exchange: core curriculum 2008. Am J Kidney Dis. 2008;52:1180–1196. doi: 10.1053/j.ajkd.2008.02.360. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Misu T, Miyazawa I, et al. Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler. 2007;13:968–974. doi: 10.1177/1352458507077189. [DOI] [PubMed] [Google Scholar]
- Costanzi C, Matiello M, Lucchinetti CF, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–666. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- Kitley J, Elsone L, George J, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry. 2013;84:918–921. doi: 10.1136/jnnp-2012-304774. [DOI] [PubMed] [Google Scholar]
- Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. 2009;66:1128–1133. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- Cree BA, Lamb S, Morgan K, et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–1448. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology. 2011;76:1310–1315. doi: 10.1212/WNL.0b013e3182152881. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim W, Park MS, et al. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol. 2011;68:473–479. doi: 10.1001/archneurol.2010.322. [DOI] [PubMed] [Google Scholar]
- Weinstock-Guttman B, Ramanathan M, Lincoff N, et al. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease) Arch Neurol. 2006;63:957–963. doi: 10.1001/archneur.63.7.957. [DOI] [PubMed] [Google Scholar]
- Araki M, Aranami T, Matsuoka T, et al. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2013;23:827–831. doi: 10.1007/s10165-012-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- Mealy MA, Wingerchuk DM, Palace J, et al. Comparison of Relapse and Treatment Failure Rates Among Patients With Neuromyelitis Optica: Multicenter Study of Treatment Efficacy. JAMA Neurol. 2014;71:324–330. doi: 10.1001/jamaneurol.2013.5699. [DOI] [PubMed] [Google Scholar]
- Wong SH, Saunders MD, Larner AJ, et al. An effective immunotherapy regimen for VGKC antibody-positive limbic encephalitis. J Neurol Neurosurg Psychiatry. 2010;81:1167–1169. doi: 10.1136/jnnp.2009.178293. [DOI] [PubMed] [Google Scholar]
- Jaben EA, Winters JL. Plasma exchange as a therapeutic option in patients with neurologic symptoms due to antibodies to voltage-gated potassium channels: a report of five cases and review of the literature. J Clin Apher. 2012;27:267–273. doi: 10.1002/jca.21233. [DOI] [PubMed] [Google Scholar]
- Frisch C, Malter MP, Elger CE, Helmstaedter C. Neuropsychological course of voltage-gated potassium channel and glutamic acid decarboxylase antibody related limbic encephalitis. Eur J Neurol. 2013;20:1297–1304. doi: 10.1111/ene.12186. [DOI] [PubMed] [Google Scholar]
- Irani SR, Gelfand JM, Bettcher BM, et al. Effect of rituximab in patients with leucine-rich, glioma-inactivated 1 antibody-associated encephalopathy. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.463. May 19. doi: 10.1001/jamaneurol.2014.463. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A, Irani SR. Caspr2 antibodies in patients with thymomas. J Thorac Oncol. 2010;5:S277–S280. doi: 10.1097/JTO.0b013e3181f23f04. [DOI] [PubMed] [Google Scholar]
- Iizuka T, Leite MI, Lang B, et al. Glycine receptor antibodies are detected in progressive encephalomyelitis with rigidity and myoclonus (PERM) but not in saccadic oscillations. J Neurol. 2012;259:1566–1573. doi: 10.1007/s00415-011-6377-2. [DOI] [PubMed] [Google Scholar]
- Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol. 2008;65:629–632. doi: 10.1001/archneur.65.5.629. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70:1133–1139. doi: 10.1001/jamaneurol.2013.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Agusti I, Dalmau J, Sevilla T, et al. Isolated hemidystonia associated with NMDA receptor antibodies. Mov Disord. 2011;26:351–352. doi: 10.1002/mds.23315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehusmann P, Dalmau J, Rudlowski C, et al. Diagnostic value of N-methyl-D-aspartate receptor antibodies in women with new-onset epilepsy. Arch Neurol. 2009;66:458–464. doi: 10.1001/archneurol.2009.5. [DOI] [PubMed] [Google Scholar]
- Tuzun E, Kinay D, Hacohen Y, et al. Guillain-Barre-like syndrome associated with lung adenocarcinoma and CASPR2 antibodies. Muscle Nerve. 2013;48:836–837. doi: 10.1002/mus.23851. [DOI] [PubMed] [Google Scholar]
- Ekizoglu E, Tuzun E, Woodhall M, et al. Investigation of neuronal autoantibodies in two different focal epilepsy syndromes. Epilepsia. 2014;55:414–422. doi: 10.1111/epi.12528. [DOI] [PubMed] [Google Scholar]
- Magana SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology. 2009;72:712–717. doi: 10.1212/01.wnl.0000343001.36493.ae. [DOI] [PubMed] [Google Scholar]
- Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler. 2013;19:941–946. doi: 10.1177/1352458512466317. [DOI] [PubMed] [Google Scholar]