

Abstract
Myeloproliferative neoplasms (MPNs) are often characterized by JAK2 or calreticulin mutations, indicating aberrant trafficking in pathogenesis. This study focuses on Mpl trafficking and Jak2 association using two model systems: Human Erythroleukemia cells (HEL; JAK2V617F) and K562 myeloid leukemia cells (JAK2WT). Consistent with a putative chaperone role for Jak2, Mpl and Jak2 associate on both intracellular and plasma membranes (shown by proximity ligation assay) and siRNA-mediated knockdown of Jak2 led to Mpl trapping in the ER. Even in Jak2 sufficient cells, Mpl accumulates in punctate structures that partially co-localize with ER-Tracker, the ER exit site marker (ERES) Sec31a, the autophagy marker LC3 and LAMP1. Mpl was fused to miniSOG, a genetically-encoded tag for correlated light and electron microscopy. Results suggest that a fraction of Mpl is taken up into autophagic structures from the ER and routed to autolyososomes. Surface biotinylation shows that both immature and mature Mpl reach the cell surface; in K562 cells Mpl is also released in exosomes. Both forms rapidly internalize upon ligand addition, while recovery is primarily attributed to immature Mpl. Mpl appears to reach the plasma membrane via both conventional ER-Golgi and autolysosome secretory pathways, as well as recycling.
Keywords: Autophagy, JAK2, miniSOG, Mpl, MPNs
Introduction
Thrombopoietin (Tpo) and its receptor, Mpl, are the principal regulators of early and late thrombopoiesis (1). Mpl (myeloproliferative leukemia virus oncogene) is expressed in platelets, hematopoietic stem cells and early progenitors, where its gene product initiates critical cell survival and proliferation signals after Tpo stimulation (2, 3). When Mpl levels are analysed by SDS-PAGE and western blotting of normal platelet lysates, two distinct bands reflect the maturation state of the glycosylated receptor (4). The mature, 84 kDa form of Mpl bears N-linked carbohydrates that have been processed during transit through the Golgi and reaches the platelet surface through the conventional secretory pathway. The faster mobility, 74 kDa protein represents an immature form that has been modified only by core-glycosylation in the endoplasmic reticulum (ER). Tpo binding to Mpl results in lower circulating Tpo levels, providing a negative feedback pathway for platelet control of thrombopoiesis (5, 6). Conversely, suppression of Mpl protein expression in platelets led to elevated platelet counts in recent murine models, which was attributed at least in part to increased circulating Tpo (7, 8).
Mpl signalling is mediated through members of the Janus kinase family, principally Jak2 and Tyk2. Mpl surface levels are often abnormally low in platelets of MPN patients, which may be accompanied by accumulation of the immature, ER core-glycosylated form (9-11). Most MPN patients carry either an activating form of JAK2 (V617F and others) or mutations in the ER resident protein, calreticulin (12, 13). Although more rare, cases have also been described expressing mutant forms of Mpl (W515K/L). Thus, a possible scenario is that interactions between these proteins lead to disease because of aberrant Mpl synthesis, trafficking or degradation (11, 14, 15).
We focus here on the trafficking patterns of Mpl, using hematopoietic model systems that express either homozygous wild type or mutant JAK2. We explore the hypothesis that normal Jak2 is a chaperone for Mpl routing to the cell surface and recycling (4, 16, 17), a role that may be poorly mediated by Jak2V617. Consistent with the chaperone hypothesis, Tong et al (2006) showed that Jak2 binding to the Mpl Box domain contributes to both stability of the receptor and its exit from the ER (17). Here, we also employed sophisticated microscopy methods and transfection methods to gain insight into the fate of Mpl protein that fails to progress through the classical secretory pathway (ER to Golgi to plasma membrane).
Two interconnected and rapidly advancing cell biology fields provide a foundation for interpreting our results. The first is the unfolded protein response (UPR), which triggers several distinct pathways to sequester and dispose of misfolded proteins that accumulate in the ER (18). One fate for such proteins can be retrograde translocation from the ER into the cytosol and subsequent proteosomal destruction, through a group of processes referred to as ERAD (ER-associated degradation) (19-21). A second pathway, coined ERAD-II, stimulates autophagosome formation and routing of autophagocytosed material to the lysosomal pathway where it is potentially degraded (22). In yeast and mammalian cells, core-glycosylated ER proteins can also be routed to the cell surface through an unconventional pathway that is dependent upon GRASP (Golgi reassembly stacking proteins) and other machinery associated with autophagy (23-25).
In this report, we provide evidence that association of Mpl with wildtype Jak2 improves the likelihood for successful exit from the ER, Golgi processing and transit to the surface. When Jak2 is limiting or absent, Mpl accumulates in the ER. This pathway may be saturable since, even in the presence of endogenous Jak2, immature Mpl can be routed into early autophagosomal structures that are physically connected to the ER and resemble isolation membranes (IM) described by others (26). Through the autophagy pathway, Mpl reaches organelles with morphology characteristic of autophagosomes and secretory autolysosomes, including presence of multivesicular bodies (MVB). This provides a plausible route for limited amounts of ER-core glycosylated Mpl receptor to reach the cell surface and, in some cases, to be released from cells in exosomes. The emerging field of autophagy-based unconventional secretion is associated with such diverse processes as inflammation and tissue remodelling (27, 28). This novel pathway may now be applicable to both the causation and potential treatment of myeloproliferative diseases.
Results
Jak2WT protein shifts the ratio of mature: immature Mpl glycoprotein
The thrombopoietin receptor, Mpl, is critical for normal platelet development and function. Figure 1A shows western blotting results of Mpl expression in platelet lysates prepared from healthy human donors. Note that Mpl migrates in SDS-PAGE as 2 distinct bands. The top band (84 kDa) represents the mature, fully glycosylated form of the receptor. The faster migrating band (74 kDa) represents the ER core-glycosylated form of the glycoprotein. The ratio of these two forms is approximately equal, despite the lack of an extensive endoplasmic reticulum in platelets.
Figure 1.
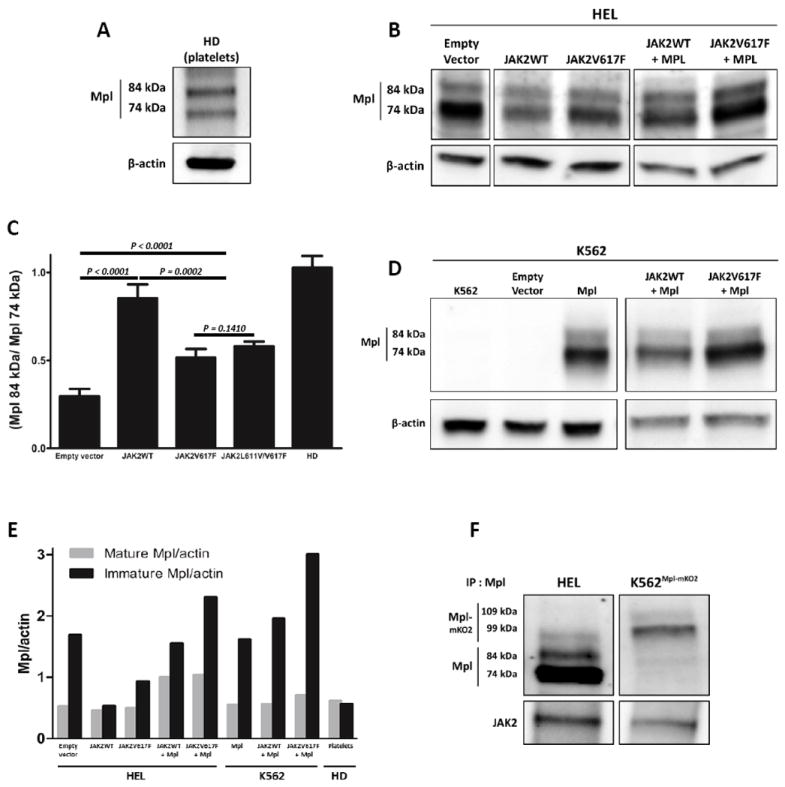
Ratios of mature:immature Mpl in normal platelets and cell lines expressing wildtype or mutant JAK2. A) Western blot analysis of Mpl in healthy donors’ (HD) platelets shows 2 distinct bands representing the mature glycoprotein (top band) and the ER-core glycosylated form (bottom band). The ratio is ~1. B) Western blot analysis of HEL whole cell lysate after transfection with cDNA coding for JAK2WT or JAK2V617F, alone or co-transfected with cDNA coding for Mpl. C). Summary of the ratios of mature:immature Mpl for at least 3 independent (2 for the JAK2V617F transfection) blotting experiments under the protocol shown in (B). Based on two-tailed student t-test statistical analysis, there is a significant difference (p=0.0001) in the ratio of Mpl isoforms in cells transfected with JAK2WT by comparison with control cells. Differences were also statistically significant in HEL cells transfected with either JAK2V617F or JAK2L611V/V617F by comparison to controls (p=0.0159 and p=0.0009, respectively). The mature:immature ratio is also significantly higher in cells transfected with JAK2WT than in cells transfected with JAK2V617F or JAK2L611V/V617F (p<0.01). D) Western blot analysis of K562 lysates after transfection with cDNA coding for Mpl, alone or co-transfected with cDNA coding for JAK2WT or JAK2V617F. E) Summary of data in (A), (B) and (D), showing the relative band intensity of mature and immature Mpl and normalized for levels of β-actin in each lane. F) Immunoprecipitation of the Mpl-Jak2 complex in different cell lines analyzed by immunoblotting anti-Mpl and anti-JAK2.
Previous work has connected expression levels of mRNA for JAK2WT with Mpl maturation and trafficking (29). We chose to evaluate the role for Jak2 proteins in this process using hematopoietic cell lines that express either exclusively mutant or wildtype JAK2. For the mutant JAK2 case, Human Erythroid Leukemia (HEL) cells were chosen based upon homozygous expression and amplification of JAK2V617F. As shown in Figure 1B (lane 1), the ER core-glycosylated form comprises over 75% of the total Mpl protein in lysates from HEL cells. Lane 2 shows results in cells transiently transfected with a vector for expression of JAK2WT. The key result in this figure is the observation that production of wildtype Jak2 results in a lower fraction of Mpl in the immature form.
Additional lanes in Figure 1B show conditions after transient transfection with vectors for expressing mutant JAK2 (V617F, L611V/V617F), with or without a vector for exogenous expression of Mpl (as a control for possible transcriptional feedback regulation).Figure 1C reports the quantitation of the entire data set from Fig 1A-B, expressed as a ratio of mature:immature glycoprotein. Changes in the ratios for cells producing wildtype Jak2 versus control cells are highly significant (P<0.0001). Results are also significantly different between cells transfected with wildtype JAK2 and mutant forms of JAK2.
To represent a hematopoietic cell line expressing only endogenous JAK2WT, we focused on K562 cells (30). K562 leukemia cells express the Bcr-Abl fusion protein typical of chronic myelogenous leukemia (CML), share characteristics of both early granulocyte and erythroid lineages, and do not express Mpl. Figure 1D reports western blotting results in K562 cells after transient transfection of MPL, with and without co-transfection with JAK2WT or JAK2V617F. In the background of endogenous JAK2WT, elevated production of normal Jak2 does not appreciably shift the ratio of mature: immature Mpl (compare lanes 3,4 in Figure 1D).
Figure 1E summarizes the data from normal platelets and the two cell lines. In this plot, levels of Mpl are normalized based on beta actin expression in the same samples. The most notable results are 1) introduction of JAK2WT into HEL cells markedly lowers the levels of ER core-glycosylated Mpl and 2) immature Mpl accumulates when the relative expression of wildtype and mutant Jak2 protein is markedly shifted in favour of mutant Jak2. With less than 25% of Mpl in the mature form, HEL cells resemble the poor maturation seen in platelets from MPN patients (29). When wildtype JAK2 is introduced into HEL cells, there is a marked shift to a higher ratio of mature:immature Mpl and a pattern that is more similar to normal platelets. For the remainder of our studies described here, K562 cells served as controls for Mpl behaviour in human cells with endogenous, wildtype JAK2.
These differential patterns suggested that wildtype Jak2 is more stably associated with Mpl than is mutant Jak2. Results in Figure 1F confirm that this assumption is true. Approximately 8 fold more Jak2WT co-immunoprecipitated with Mpl expressed as an mK02 fusion protein in K562 cells, by comparison to co-immunoprecipitation of Jak2V617F with endogenous Mpl in HEL cells.
Mpl accumulates in distinct intracellular compartments in the HEL and K562 hematopoietic cell lines
Cellular localization of Mpl is a critical factor in the biogenesis and responsiveness of platelets to Tpo (31). Here we used confocal immunofluorescence imaging to study the distribution of endogenous Mpl in HEL cells. For comparison, both HEL and K562 cells were also transiently transfected with vectors for recombinant Mpl fused to a fluorescent reporter (MplmOrange2). In JAK2V617F+/+ HEL cells, both endogenous Mpl and MplmOrange2 were largely restricted to intracellular foci throughout the cell. Mpl was barely detectable at the HEL cell surface (Figure 2A-B and Movie 1). In JAK2WT+/+ K562 cells, appreciable amounts of MplmOrange2 were observed at the plasma membrane (arrow, Figure 2C). However, MplmOrange2 was also found in a large perinuclear, intracellular pool. Thus, despite similar western blotting profiles of mature:immature Mpl in these two cell lines, MplmOrange2 surface expression is a marked feature only of K562 cells that express endogenous JAK2WT.
Figure 2.
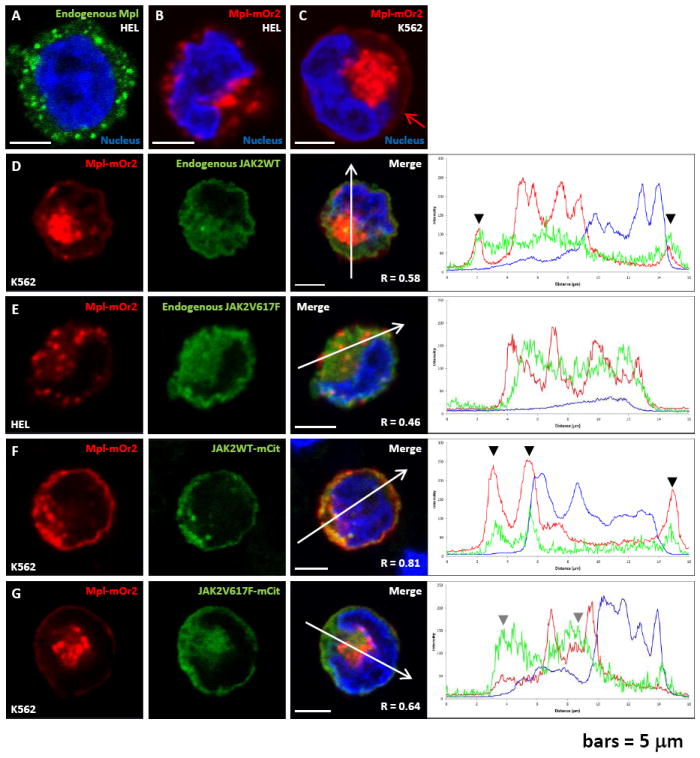
In JAK2WT cells, Mpl is detected at the cell surface and co-localizes with JAK2. A-B) Confocal fluorescent images of HEL cells (JAK2V617F+/+) after immuno-fluorescence labeling of endogenous Mpl (A), or after transient transfection with cDNA coding for a chimeric Mpl protein fused to mOrange2 (B). C) Confocal fluorescence image of K562 cells (JAK2WT+/+) transiently transfected with cDNA coding for MplmOrange2. Red arrow in this image points to plasma membrane localization. D-E) Confocal fluorescent images obtained after transient expression of MplmOrange2 and immuno-fluorescent labeling of endogenous JAK2 in K562 (D) or HEL (E) cells. Profiles of fluorescence (across individual cells and indicated by a white arrow) are showing a good co-localization of Mpl and JAK2WT in K562 cells at the plasma membrane (black arrow heads) but no co-localization have been observed between MplmOrange2 and JAK2V617F in HEL cells. F-G) Confocal fluorescent images obtained after transient co-expression of MplmOrange2 and either JAK2WTmCitrine (F) or JAK2V617FmCitrine (G) in K562 cells. Profiles of fluorescence (white arrows) are showing a very good co-localization of MplmOrange2 with JAK2WTmCitrine, especially at the plasma membrane (black arrow heads), but a very poor co-localization with JAK2V617FmCitrine (grey arrow heads). Nuclei are labeled with Hoechst and scale bars = 5 μm. R: Pearson’s correlation coefficient.
Jak2WT more consistently co-localizes with Mpl than Jak2V617F
We next evaluated the extent to which Mpl and Jak2 co-localize in both cell types. Results in Figures 2D-G show that transfected MplmOrange2 co-localizes at the plasma membrane with endogenous Jak2WT in K562 cells (Figure 2D), while co-localization is minimal with endogenous Jak2V617F in HEL cells (Figure 2E). Arrowheads in plots, at right, mark locations where strong co-localization was noted at the cell margin or intracellular vesicles. When co-transfected into K562 cells, MplmOrange2 and wildtype Jak2mCitrine show significant co-localization at the plasma membrane as well as the intracellular pool (Figure 2F). Weak co-localization was observed when MplmOrange2 was co-transfected with citrine-tagged mutant Jak2 in K562 cells (Figure 2G).
As another evaluation method for Mpl-Jak2 association, we used a proximity ligation assay that reports interactions within the <40 nm distance permissive for hybridization of the Mpl and Jak2 probes and amplification of the fluorescence reporter. As shown in Figure 3, control samples lacking Mpl (Figure 3A) or omitting the primary probes (Figure 3B) are devoid of green fluorescence for the hybridization signal. For HEL cells that express only Jak2V617F, and untransfected K562 cells, the plasma membrane was marked with anti-glycophorin A (CD235a) antibodies. In HEL cells, Mpl-Jak2 association was low, with a few intracellular spots (green arrowheads) and plasma membrane spots (white arrowheads) per cell (Figure 3C-D). Labeling was significantly more intense and abundant for K562 cells expressing MPLmKO2 and endogenous WTJak2, with numerous spots in the cell interior (green arrowheads) and on the plasma membrane (white arrowheads) (Figure 3E-F). Taken together, these results suggest that wildtype Jak2 may bind Mpl soon after synthesis, serving as a chaperone during trafficking to the cell surface and acting as a signaling partner at the plasma membrane. A role for Jak2 in receptor stabilization at the plasma membrane has been proposed by others (4).
Figure 3.
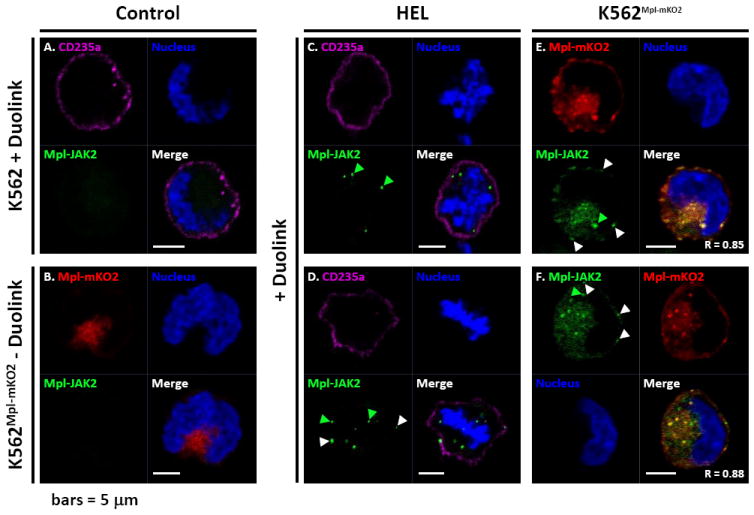
Mpl and Jak2 association is not limited to the plasma membrane. Proximity ligation assay (Duolink) in HEL, K562 and K562Mpl-mKO2 cells. A) K562 cells labelled live for 30 minutes with an anti-CD235a-Atto655 (glycophorin A) antibody before treatment with the Duolink reagents. B) K562Mpl-mKO2 cells non-treated with the Duolink reagents. C-D) HEL cells primed live for 30 minutes with an anti-CD235a-Atto655 antibody before treatment with the Duolink reagents. E-F) K562Mpl-mKO2 cells submitted to the Duolink reaction. White arrow heads are pointing at example of Duolink positive reactions at the plasma membrane, while green arrow heads are pointing at positive Duolink reactions within the cytoplasm.Nuclei are labeled with Hoechst and scale bars = 5 μm. R: Pearson’s correlation coefficient.
JAK2 is critical for Mpl sub-cellular localization
The effects of Jak2 knockdown on Mpl trafficking were addressed by treating the two cell lines with control (NT) or anti-JAK2 siRNA. Results are presented in Figure 4. We obtained about 50% Jak2 knock-down (KD) after 72 hr treatment of HEL cells, where the gene is amplified (Figure 4A). In the K562 cells, we achieved about 83% KD of endogenous Jak2 (Figure 4B). Confocal images in Figure 4C show that Mpl is predominantly found in reticular structures after Jak2 knockdown in K562 cells. Colocalization with calnexin confirms that this accumulation of Mpl is due to accumulation in the ER.It is noteworthy that calnexin was seen in clusters around the nuclear envelope in K562 cells (a subdomain of the ER) and redistributed to a more diffuse distribution in Jak2-depleted cells, including Jak2’s role in the ER may not be restricted to its chaperone role for Mpl receptors in transit. The clustering of ER proteins has previously been reported (32).
Figure 4.
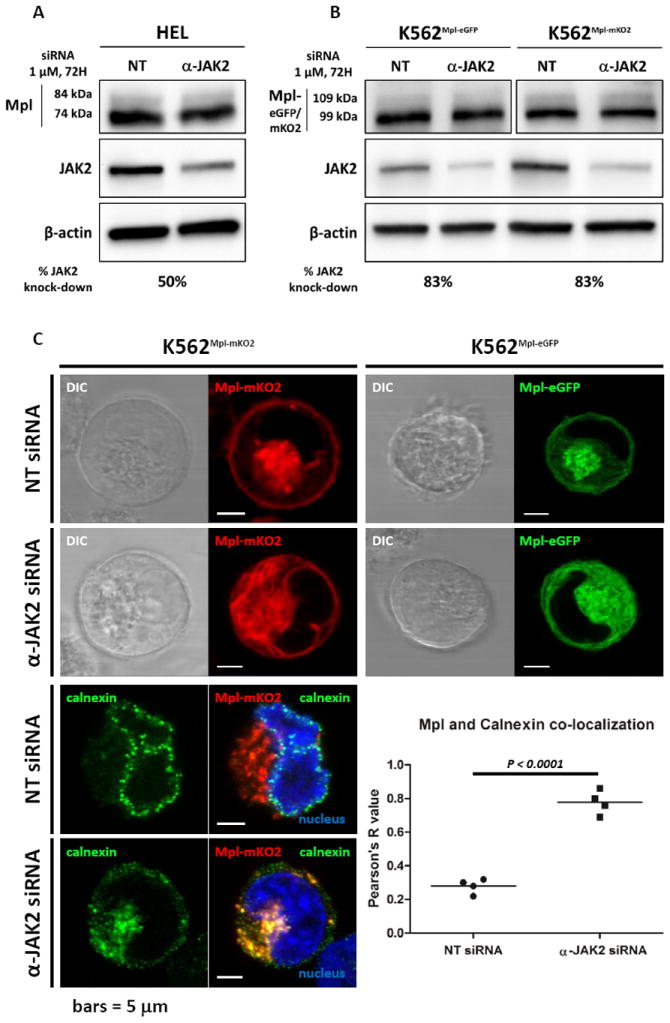
JAK2 is critical for Mpl sub-cellular localization. A-B) HEL (A) and K562 (B) cells treated with non-targeting (NT) or anti-JAK2 siRNA analysed by western blot. C) Confocal live cell (upper panel), or fixed cells (lower left panel), imaging of K562 cells expressing MpleGFP or MplmKO2 treated with NT or anti-JAK2 siRNA showing a difference of distribution of intra-cellular Mpl in the siRNA treated cells compare to the control cells. After treatment with anti-JAK2 siRNA, Mpl strongly co-localizes with the ER marker, calnexin. This co-localization in treated vs non-treated cells is quantified and shown Figure 4C lower right panel. Nuclei are labeled with Hoechst and scale bars = 5 μm.
Sub-cellular localization of Mpl
Our next task was to use immuno-labeling and confocal microscopy for initial characterization of the punctate structures that harbor both endogenous Mpl and transfected MplmOrange2 or MplmK02 in our model systems. Results are reported in Figure 5. These images examine colocalization of intracellular Mpl with markers for the ER, the recycling compartment or the autophagic pathway; each image is accompanied by associated line plots for fluorescence intensity and the Pearson’s correlation coefficient R value. Although colocalization was not significant between Mpl and antibodies to ERGIC-53 (ER-Golgi intermediate compartment), Golgin-97 (Golgi) or EEA1 (early endosome), we found partial colocalization with the intra-vital dye, ER Tracker, as well as Sec 31a (marker of ER exit sites) and Rab11 (MVBs; ref (33)). Strong co-localization was found for the autophagic marker, LC3, and for the lysosomal marker, LAMP1 (multiple arrowheads in plots for Figure 5E, F and 5L,M).
Figure 5.
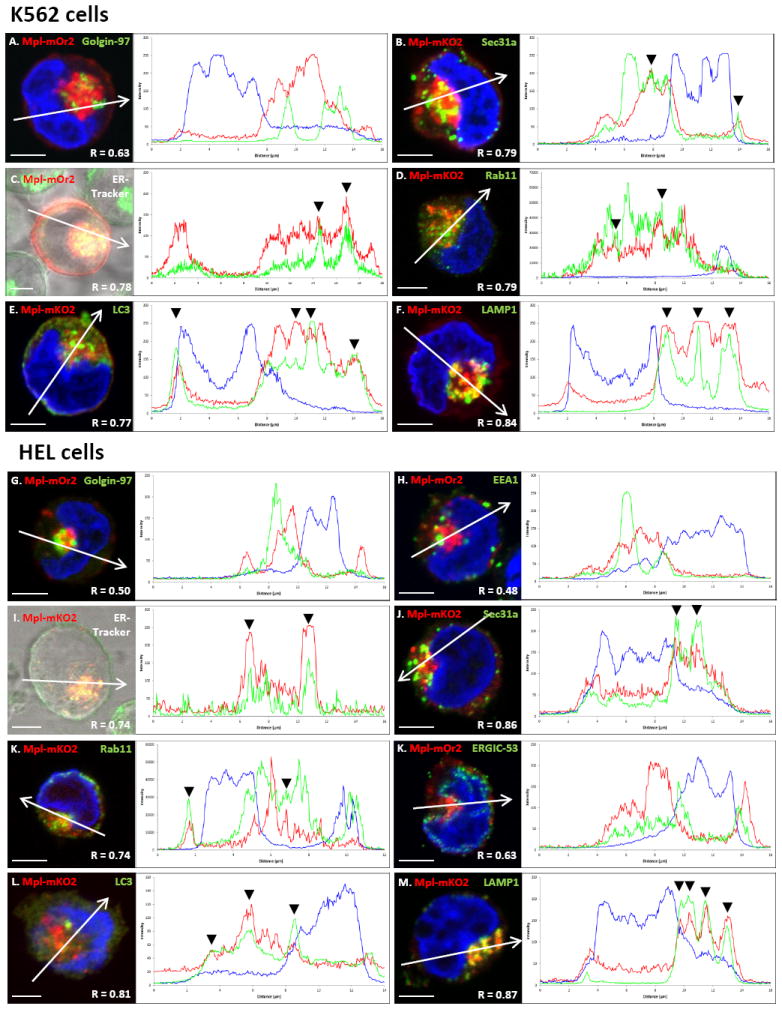
Intracellular Mpl pools partially co-localize with ER markers and co-localize with autophagic and lysosomal markers in K562 and HEL cells. A-M) Confocal fluorescent images obtained after transient expression of MplmOrange2 or MplmKO2 and indicated labeling in K562 (A-F) or HEL (G-M) cells. Plots at right of each image show fluorescence intensities for each emission spectrum at points along the line drawn across each image. Black arrows in the plots point to overlap between MplmKO2 and either ER-tracker (C,I), or Sec31a (B,J) that marks ER exit sites. MplmOrange2 did not co-localize with markers for the Golgi (Golgin-97) or early endosomes (EEA1) in either cell line. However MplmKO2 co-localized greatly with autophagic (LC3) (E,L) and lysosomal (LAMP1) (F,M) markers. Nuclei are labeled with Hoechst and scale bars = 5 μm. R: Pearson’s correlation coefficient.
Correlated light and electron microscopy localization studies show immature Mpl enters autophagosomes from transitional ER
The marked colocalization of Mpl with LC3 and LAMP1, as seen in Figure 5, suggested that Mpl is delivered to lysosomes through the autophagy pathway.Our next goal was to develop a reporter to confirm the intracellular location of Mpl with high spatial resolution. We focused on the innovative miniSOG technology, which is based upon mutagenesis of Arabidopsis phototropin 2. Only half the size of fluorescent proteins derived from Aequorea, miniSOG emits singlet oxygen upon photo-activation and can be used to catalyze the local precipitation of diaminobenzidine (DAB) for electron microscopy studies (34). Both HEL and K562 cell lines were transiently transfected with MplmK02-miniSOG. After 24hr, cells were allowed to settle on poly-L lysine coated coverslips and fixed with glutaraldehyde. Transfected cells were identified based upon miniSOG and mKO2 fluorescence (Figures 6A-B), followed by photo-oxidation in the presence of DAB and oxygen infusion. Samples were then embedded in epon and processed for ultrathin sectioning, as described in Shu et al (2011).
Figure 6.
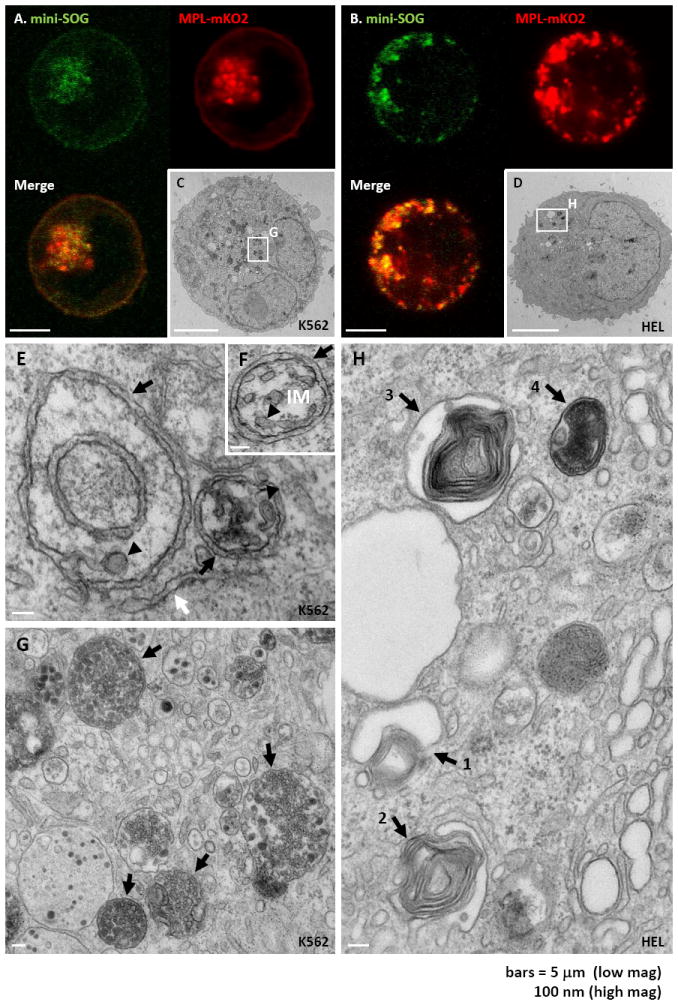
MiniSOG-tagged Mpl reveals that Mpl enters the autophagic pathway from specialized ER exit sites. A-B) Confocal fluorescent images obtained after transient expression of Mpl tagged with mKO2 and miniSOG in K562 (A) and HEL (B) cells. C-D): Electron micrographs of K562 (C) and HEL (D) cells transiently expressing MplmKO2-miniSOG. Dark precipitate indicates the presence of the miniSOG-tagged Mpl protein after photo-precipitation of DAB. White boxes indicate original location of higher magnification images with corresponding letters. E-G) High magnification electron micrograph of K562 cells treated in the same conditions as Fig. 4C, showing MplmKO2-miniSOG. Dark precipitate into autophagic carrier vesicles (black arrows, Figure 5E). Osmiophilic reaction products also heavily stain mature autophagosome in these preparations (black arrows, Figure 5G). H) High magnification electron micrograph of HEL cells treated in the same conditions as Fig. 5D, showing different stage of formation of autophagosomes (black arrows numbered from stages 1-4). Scale bars = 5 μm (A-D) or 100 nm (E-H).
Images in Figures 6C-H and Supplemental Figure 1 show that dark precipitates marking the location of MplmK02-miniSOG are predominantly in vesicular structures consistent with the punctate dots seen in fluorescent images of K562 and HEL cells. In high resolution micrographs of these structures in K562 cells (Figure 6E-G), it is apparent that these structures contain multivesicular bodies. Negative controls, showing these electron-dense reaction products as attributed to specific photo-oxidation of the MplminiSOG recombinant protein, are shown in Supplemental Figure 2.
Evidence that these Mpl-rich structures bud directly from the ER is found in micrographs in Figure 6E-F. The morphology is consistent with specialized transition regions, sometimes called isolated membrane, (26) that are initiation sites for autophagosome formation. These results provide the first evidence that, when retained in the ER, Mpl is concentrated and loaded into autophagic carrier vesicles (black arrows, Figure 6E). Osmiophilic reaction products also heavily stain mature autophagosome in these preparations (black arrows, Figure 6G). Note the presence of Mpl reaction product on small vesicles within these autolysosomes, consistent with results reported later in Figure 8C that Mpl is released from these cells in exosomes.
Figure 8.
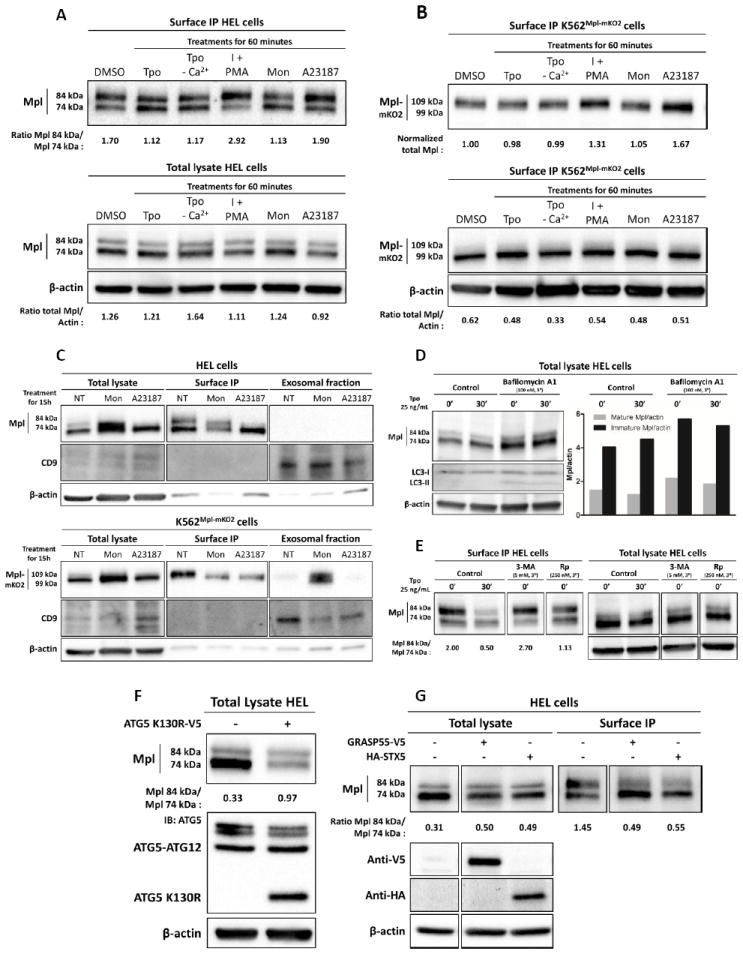
Complex Mpl trafficking includes conventional and autophagic secretory pathways, as well as multiple recycling pools. A-B) Surface biotinylation and total lysates western blot analysis of HEL or K562Mpl-mKO2 cells after treatments for 60 minutes with Tpo, ionomycin (I), PMA, Monensin or A23187. C) Total lysate, surface biotinylation and exosomal fraction western blot analysis of HEL and K562Mpl-mKO2 cells treated with Monensin or A23187 for 15 hours. D) Western blot analysis of HEL cells treated, or not, with Bafilomycin A1 at 100 nM for 3 hours and then stimulated or not with Tpo at 25 ng/mL for 30 minutes. The quantification of the blot is shown. E) Surface biotinylation and total lysates western blot analysis of HEL cells after treatments for 30 minutes with Tpo or for 3 hours with 3-MA (3-Methyladenine) or Rapamycin (Rp). F) Total lysate and surface biotinylation western blot analysis of HEL cells over-expressing ATG5 K130R-V5 6 hours post-transfection. G) Total lysate and surface biotinylation western blot analysis of HEL cells over-expressing either GRASP55-V5 or HA-STX5.
EM images of Mpl-containing structures in HEL cells (Figure 6H) also support routing into the autophagic pathway, where the numbered black arrows appear to represent stages of autophagosome maturation (35). The morphology of autophagosomes in HEL cells is slightly different from those in K562 cells, with electron dense precipitate from photo-oxidized MplmK02-miniSOG labeling highly convoluted structures or “whirls”. The micrograph in Supplemental Figures 3A indicates that these Mpl-containing membranes originate from the ER.
As confirmation, K562 and HEL cells were co-transfected with MpleCFP and a pH-sensitive fluorescent reporter for LC3 localization (Supplemental Figures 4A-B). We employed the mRFP-GFP version of LC3 (tfLC3), that distinguishes between LC3 that is associated with classical autophagic vesicles (where both mRFP and GFP fluorescence is observed) and LC3 that is associated with lysosomes (where the acidic lumen quenches only the GFP fluorescence). (36) We found cases of vesicles positive for MpleCFP and LC3mRFP-GFP in both cell lines (black arrow heads), consistent with Mpl trafficking within neutral pH autophagosomes. Fluorescence imaging also captured vesicles positives for MpleCFP and red-fluorescent LC3, indicating delivery of Mpl to the low pH compartment of the lysosome, where green fluorescence from the tfLC3 chimera was quenched.
Both mature and ER-core glycosylated Mpl reach the cell surface
To confirm prior reports that the 74 kDa immature form of Mpl reaches the cell surface, HEL and K562 surface proteins were biotinylated by incubating live, chilled cells with NHS-biotin, followed by cell lysis and immunoprecipitation using streptavidin-conjugated beads. After SDS-PAGE and electrophoretic transfer to nitrocellulose, blots were probed with an anti-Mpl antibody to evaluate the relative proportions of immature and mature Mpl glycoprotein at the cell surface. The biotinylation protocol was performed on cells with or without specified intervals of ligand addition (Tpo, 25 ng/ml) to evaluate the extent of receptor recycling.
Results of these experiments are shown in Figure 7A. In HEL cells, that express only mutant Jak2, both forms of the Mpl glycoprotein are present at the cell surface prior to ligand addition. Consistent with a recent report by Pecquet and colleagues (37), the Mpl receptor is rapidly internalized after Tpo addition. Remarkably, after the initial loss of total surface Mpl, the levels of Mpl begin to recover within 30 min. The newly-arrived Mpl is primarily the ER-core glycosylated form, increasing the ratio of biotinylated immature/mature Mpl as shown in the plot in Figure 7A.
Figure 7.
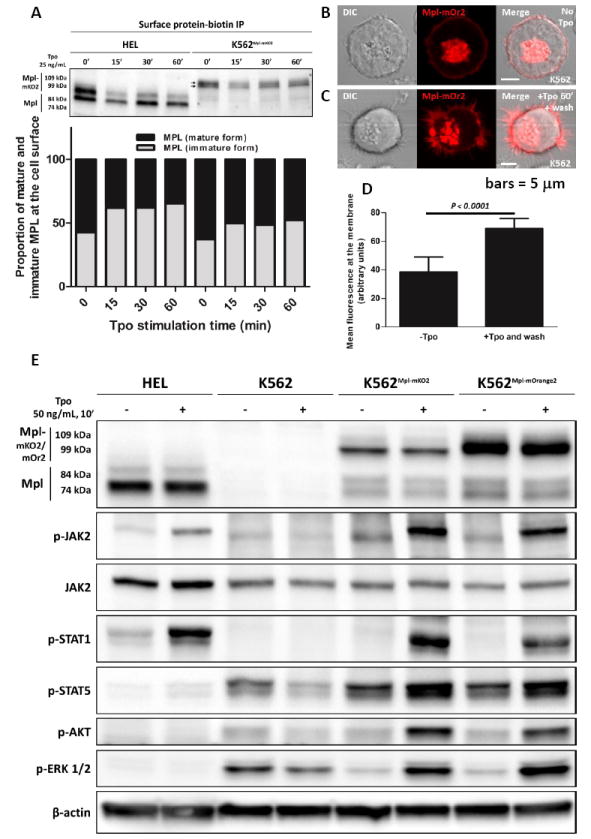
Surface Mpl is rapidly internalized after Tpo addition, followed by recovery from recycling and autolysosome compartments. A) Western blot analysis and quantification of streptavidin-immunoprecipitated surface-biotinylated proteins, probed with an anti-Mpl antibody. B-C) Confocal images of K562 cells transiently expressing MplmOrange2. Cells in (B) are untreated. Cells in (C) were stimulated 60’ with Tpo, washed and chased for 15 min before imaging. D) Quantification of plasma membrane fluorescence in resting and Tpo-stimulated cells. Mean fluorescence at the membrane was estimated by averaging the total fluorescence after values equal to or below background. A two-tailed Student t-test shows a significant difference in the membrane fluorescence after stimulation with Tpo followed by Tpo removal. Scale bars = 5 μm. E) Western blot analysis of protein phosphorylation in HEL, K562Mpl-mKO2 and K562Mpl-mOrange2 cells after Tpo stimulation (50 ng/mL Tpo, 10 min).
Although less dramatic for MplmK02 fusion proteins expressed in the background of wildtype Jak2, we were also able to document the presence of immature Mpl at the surface of K562 cells. These results are shown in Figure 7A, where gradient gel electrophoresis can distinguish 2 distinct bands for the larger MplmK02 fusion protein. After Tpo stimulation, there is a net loss of Mpl from the surface due to internalization. As new Mpl arrives at the cell surface, the biotinylation assays shows there is an increase in the ratio of immature (core-glycosylated) to mature Mpl fusion protein.
Pecquet et al (2012) showed that Mpl constitutively cycles between the plasma membrane and an early endocytic compartment. To evaluate the total levels of Mpl at the surface of K562 cells, with and without Tpo stimulation, quantitative confocal imaging was performed for cells after transient MplmOrange2 expression. Results are shown in Figures 7B-D, where receptor fluorescence at the membrane was measurable in resting cells, followed by a significant increase in fluorescence after 1 hr exposure to Tpo stimulation and 15 minutes incubation without Tpo. These data confirm that, after a pulse of Tpo stimulation, surface receptor levels recover and can even exceed resting levels (plot in Figure 7D).
Results in Figure 7E are important controls to show that Tpo activates Jak-Stat pathways under the conditions used in this study. After transfection with expression vectors for Mpl-fluorescent fusion proteins (MplmKO2 and MplmOrange2), stimulation of K562 cells with Tpo leads to robust phosphorylation of wildtype Jak2, Stat1, Stat5, Akt and Erk. Moreover, K562 cells transfected with Mpl fusion proteins are capable of binding AF488-Tpo and respond by membrane ruffling and internalization of the fluorescent ligand (Supplemental Figure 5A-B).
In HEL cells, it is noteworthy that all copies of Jak2 are in the presumed “constitutively-active” form (V617F). However, basal levels of Jak2 phosphorylation are low and there is a detectable increase after Tpo addition (Fig.7E). Despite low fluorescence intensity for surface Mpl in these cells (see Figure 2A-B,E), there are sufficient receptors available for initiating signals. Tpo stimulation of HEL cells activates poorly Stat5 and does not lead to activation of Akt or Erk, but does markedly elevate phospho-Stat1 levels.
Lanes at top of Figure 7E report the relative levels of endogenous Mpl (HEL cells) or transfected Mpl fusion proteins (K562) in these experiments. Note that the lower bands in K562 cells appear to be degradation products of the Mpl fusion protein, since PCR analysis ruled out endogenous expression (Supplemental Figure 6C).
Complex Mpl trafficking includes conventional and autophagic secretory pathways, as well as multiple recycling pools
Our final goal was to gain insight into the routes by which the ER core-glycosylated form of Mpl traffics to the cell surface, using both pharmacologic and transfection approaches. Results presented in Figure 8 focus on the source of Mpl for recovery after ligand-mediated endocytosis, as well as support the conclusion that immature Mpl utilizes the unconventional autophagic secretory pathway to reach the cell surface (27).
Data in Figure 7 showed that Mpl surface levels drop after exposure to ligand and the recovery seen within 60 min is mostly due to the immature, 74 kDa form. Based upon evidence that fusion of secretory lysosomes in other hematopoietic lineages is calcium dependent (38), we compared 1 hr recovery in cells treated with Tpo in the absence and presence of extracellular calcium. We observed no change, indicating that fusion of the stimulus-recovery pool is not linked to receptor-induced calcium influx (lanes 2-3 in Fig. 8A-B). However, when cells were acutely treated with calcium ionophores (ionomycin plus PMA or A23187; lanes 4,6 in each blot series), we noted an increase in the surface-biotinylated 84 kDa mature form of Mpl. This suggests that a fraction of mature Mpl is also stored in a pool that can translocate to the plasma membrane in response to elevated calcium. These treatments do not change overall levels of Mpl, based upon western blots of total lysates (lower panels, Figure 8A-B).
A prior report by Colombo and colleagues implicated calcium in the fusion of exosome-containing MVBs (multivesicular bodies) with the plasma membrane of K562 cells (39). Monensin, an inhibitor of the Na+/H+ changer, led to accumulation of MVB and enhanced exosome release. In Fig. 8C, we show that 15 hr incubation of K562 cells with monensin leads to an increase in intracellular Mpl, reduction of surface Mpl and release of both Mpl forms as exosomes. In contrast, HEL cells fail to release Mpl in exosomes, accumulating a large pool of both forms intracellularly. In both cell types, calcium ionophore treatment for the same duration depletes cells of the mature 84 kDa Mpl form. Immunoblotting for CD9 serves as a marker of exosomes in this assay (middle panels, Fig 8C).
Data in Figure 8D reports results from HEL cells treated for 3 hrs with Bafilomycin A1, that blocks maturation of autophagosomes (40). This treatment led to accumulation of both forms of Mpl notably the immature 74 kDa form, consistent with a block in autophagic processing. Pharmacological perturbation with 3-Methylamine (3-MA, a PI3K inhibitor preventing autophagosome formation) (41), or rapamycin (an activator of autophagy); (42) confirmed the role of an autophagy-based pathway for delivery of core-glycosylated Mpl to the cell surface. HEL cells treated with 3-MA for 3 hrs (Figure 8E) show a clear decrease in the surface level of core-glycosylated Mpl which is confirmed by the shift in the mature/immature Mpl ratio at the cell surface from 2 to 2.7. On the other hand, rapamycin-mediated induction of autophagy in HEL cells led to a decrease in the mature Mpl form at the cell surface as well as to a slight increase of the immature Mpl levels; the ratio of mature to immature accordingly drops from 2 to 1.13.
As additional confirmation for the involvement of autophagy in the routing of immature Mpl to the cell surface, we over-expressed a mutant form of ATG5 (ATG5-K130R) that cannot associate with ATG12 (43). Results in HEL cells transfected with ATG5-K130R and analysed 6 hrs post-transfection show a strong decrease in the accumulated total Mpl. The most dramatic decrease was in the ER core-glycosylated form, suggesting that a large fraction of the ER form of Mpl was rapidly degraded in the absence of an autophagic route. Although the mechanism for this rapid degradation is not known, it could possibly occur by retrograde transport of misfolded protein from the ER to the cytosol followed by proteasome-mediated proteoloysis (44-46).
Further support for autophagic trafficking of immature form of Mpl from the ER is provided in Figure 8G. HEL cells were transiently transfected with a V5-tagged version of GRASP55 (GRASP55-V5) or with an HA-tagged version of Syntaxin 5 (HA-STX5). Overexpression of these proteins both resulted in markedly lowered levels of mature Mpl at the cell surface and an increase in the core-glycosylated form, especially when overexpressing GRASP55 (compared to STX5) (Lanes 5-6, upper panel, Figure 7G), albeit through different mechanisms.Syntaxin 5 overexpression inhibits the transit of cargo from the ER to the Golgi along the conventional secretory route, while GRASP55 overexpression is expected to actively enhance the autophagic route (23).
DISCUSSION
Mpl, a new addition to list of proteins delivered to surface via unconventional secretion
The present study provides the following evidence that the thrombopoietin receptor, Mpl, can traffic to the plasma membrane via an unconventional, autophagy-dependent route: 1) Mpl colocalizes in low-pH compartments with autophagic and MVG markers LC3, LAMP1 and Rab11 (33) (47); 2) correlated light-EM imaging of cells expressing MplminiSOG fusion proteins show accumulation in organelles with classical features of autophagy; 3) blockade of autophagosome formation with 3-MA, or maturation with bafilomycin A1, lead to accumulation of immature Mpl and a decrease level of surface immature Mpl; and 4) surface levels of ER-core glycosylated Mpl are elevated in cells treated with rapamycin or after overexpression of Syntaxin 5 or Grasp55.These novel observations appear to resolve the unexplained presence of ER-core glycosylated Mpl at the surface of platelets and cells of hematopoietic lineage, where it can be labeled through biotinylation reactions (ref (37) and Figures 7-8, this paper).
The imperative to understand receptor trafficking in the disease state has been underscored by the recent discoveries that calreticulin mutations are also linked to myeloproliferative neoplasms (12, 13). The discovery of Mpl’s alternative routing to the surface adds a new chapter to the remarkable and very recent progress in the field of autophagy-based unconventional protein secretion (24, 48, 49). Newly coined by the term “autosecretion”, the regulated exocytosis of autophagic cargo has been shown in Paneth cells in the context of Crohn’s disease (50), in cathepsin K secretion by osteoclasts (51) and in degranulating mast cells (52). Our observation of partial colocalization between intracellular Mpl and Sec31 (Figure 5), as well as the mammalian paralog of Atg8 (LC3; Figure 5), is consistent with prior evidence that autophagosome formation is dependent on functional ER exit sites (53) and Atg machinery. We note the similarity between the trafficking of ER-trapped Mpl to that of mutant CFTR (54). Gee and colleagues reported improved delivery of immature core-glycosylated CFTR to the plasma membrane in cells overexpressing a member of the GRASP family of proteins (23). GRASPs are strongly implicated in unconventional secretion linked to ER stress responses, reviewed in (27, 55). This raises the potential for trapped Mpl to initiate stress response signals that contribute to MPN pathogenesis, as has recently been implicated in the context of breast cancer (56). Notably, Albu and Constantinescu (2011) recently showed that N-glycosylation mutants of Mpl bind and respond to Tpo, suggesting that the immature Mpl glycoproteins are likely to be functional once delivered from autophagosomal compartments (57).
Aberrant Mpl trafficking is linked to mutant Jak2 associated with MPN
Mpl transmits survival signals in hematopoietic progenitors (1), underscoring the need for tight regulation to maintain surface expression at low to modest levels. There is keen interest in the relationship between JAK2V617F and other JAK2 mutations in the pathogenesis of MPN (58). Roles for Jak2 in the regulation of Mpl trafficking are well established, beginning with observations that interactions between juxta-membrane (“Box 1 and 2”) features in the receptor and the Jak2 FERM domain were required to promote cell surface localization and stabilization of mature Mpl (4, 17). At least for the closely related Epo receptor, residues in the N-terminal pseudokinase domain linker also mediate interactions between Jak2 and its cognate receptor (59). Interactions between Jak2 and Mpl are influenced by the receptor’s N-glycosylation status, particularly at Asn117 (57). This may be related to conformational restrictions on the dimerization competency of the newly synthetized receptor in the ER, as suggested by recent structural studies (60).
We show here that the presence of endogenous wildtype Jak2, or its introduction via transient transfection, raises the ratio of mature:immature Mpl in the two human hematopoietic cells lines studied here. Mpl-Jak2 association likely occurs early after Mpl protein synthesis and co-translational translocation, based upon the Jak2 knockdown studies showing Mpl accumulation in the ER of K562 cells (Figure 4). Based upon the punctate signature using the proximity ligation assay (Figure 3), Mpl and Jak2 may also associated in post-ER structures. Jak2 kinase activity may also influence the function and distribution of other ER resident proteins, based upon our observation that calnexin patterns were altered in Jak2-depleted cells (Figure 4C).
By comparison to the wild type form of Jak2, our co-precipitation studies suggest that mutant JAK2V617F has at least an 8-fold weaker association with Mpl (Fig. 1F). Although the explanation for this weak association is unknown, it may be linked to the fact that the V617F mutation is in the pseudokinase domain thought to provide at least one component of coupling (59). We introduced wildtype Jak2 in HEL cells, where the endogenous pool is comprised solely of mutant Jak2. This manipulation shifted the ratio of mature:immature Mpl, presumably by routing more Mpl from the ER through the conventional pathway. Overall levels of Mpl were not elevated in this scenario, possibly because Mpl at the cell surface is subject to constitutive recycling and associated degradative loss occurs during the recycling process (37). An alternative explanation may be that Jak2 actively promotes proteolytic processing of core-glycosylated Mpl within the degradative arm of the autophagic pathway (Figure 9).
Figure 9.
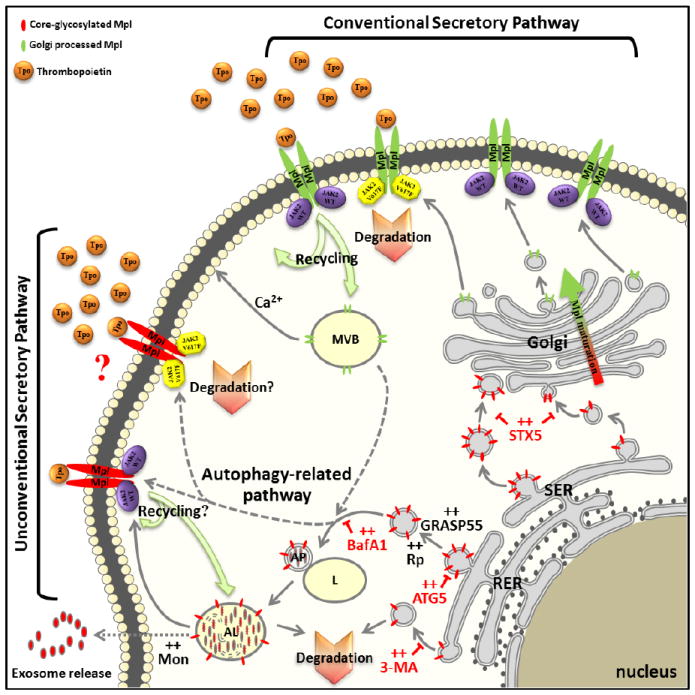
The complex intracellular trafficking of Mpl. Mature Mpl reaches the surface through the conventional ER-Golgi pathway and bears fully processed N-linked carbohydrate. Immature Mpl appears to be routed through an autophagic pathway for unconventional secretion, providing an alternate pathway for membrane expression of core-glycosylated Mpl. Once delivered to the cell surface, both forms of Mpl can be recycled or stored in calcium-dependent and independent compartments capable of fusion with the plasma membrane. In some cells, such as K562 cells, Mpl is packaged into exosomes and secreted.
Tpo: Thrombopoietin; SER: Smooth Endoplasmic Reticulum; RER: Rough Endoplasmic Reticulum; L: Lysosome; AP: Autophagosome; AL: Auto-Lysosome; STX5: Syntaxin 5; GRASP55: Golgi Re-Assembly Stacking Protein 2, 55kDa; ATG5: Autophagy protein 5; MVB: Multi-Vesicular Body; Mon: Monensin; BafA1: Bafilomycin A1; 3-MA: 3-Methyladenine; Rp: Rapamycin.
Relevance to Platelet Function
We and others have taken advantage of nucleated hematopoietic cells to study Mpl expression levels and its unique relationship with Jak2. Previously, the V617F mutation has been only connected to Mpl levels through miR-28-mediated feedback control of Mpl message stability (61). Moliterno et al. reported a weak inverse correlation between the JAK2V617F allelic ratio and the amount of Mpl in platelets of JAK2V617F-positive MPN patients (62). These authors also noted decreased Mpl expression in MPN patients lacking JAK2 mutations.We show here that Jak2 knockdown traps Mpl in the ER of nucleated hematopoietic cells. Patient studies have thus far been limited to analysis of circulating platelets, which are devoid of nuclei and maintain only a subset of intracellular organelles. Since platelets lack ER, it is clear that a missing piece of the puzzle is the role for Jak2 in platelet biogenesis. Although the transcriptional control of Mpl synthesis begins in megakaryocyte precursors, it is intriguing to note that platelets contain at least three storage compartments (α and dense core granules, as well as secretory lysosomes) that are differentially dependent upon calcium for degranulation (63). Our data in Figure 8 suggest that both calcium-dependent and calcium-independent reservoirs for Mpl could co-exist, arising from complex trafficking patterns from conventional and unconventional secretory pathways as well as from recycling endosomes. The potential for platelets or megakaryocytes to release Mpl in exosomes has not yet been evaluated; if it occurs, exosomal Mpl could serve as a sink for Tpo in vivo.
Therapeutic benefit of Jak inhibitors may be limited by effects on Mpl trafficking
The recent FDA approval of the Jak1-2 inhibitor, ruxolitinib, has led to significant clinical benefit for MPN patients (64). This and other Jak-targeted therapies are based on the high incidence of activating Jak2 mutations in BCR-ABL1-negative MPN (65). However, this class of kinase inhibitors do not spare wildtype Jak2 and its normal role in hematopoiesis. Here, we contribute to growing evidence that Jak2 serves an important chaperone role for the thrombopoietin receptor, which may be connected to thrombocytopenia and other significant side effects in patients treated with this class of drugs (66). This relationship in a therapeutic setting will not be easy to evaluate, due to the complex feedback loop for Mpl in the control of circulating Tpo levels (5). In addition, JAK2V617F allele status does not correlate as strongly with clinical phenotype as does the degree of impaired platelet Mpl expression (62). Although a large fraction of MPN patients present with low Mpl levels, it is not solely restricted to cases with mutations in Jak2. In murine models, expression of JAK2V617F alone can induce thrombocytosis (6, 7). This is not generalizable to humans, since only a fraction of JAK2V617F+ primary myelofibrosis patients have low platelet counts. Our study highlights the fact that mechanisms of action for Jak inhibitors are not straightforward and calls for further investigation into the links between Jak2 chaperone function, unconventional Mpl secretion and the pathogenesis of MPNs. Additional indirect mechanisms, such as the overproduction of anti-inflammatory cytokines, drive abnormal myelopoiesis in these diseases (58). These complex considerations may be expected to alter strategies for Jak-targeted drug delivery and combination therapy.
Materials and Methods
Platelet isolation
Buffy coats (45 ml) pooled from healthy donors (HD) was supplemented with 5 mL of sodium citrate buffer (25 g of sodium citrate, 8 g of citric acid, 500 mL of ddH2O), then diluted three times in RPMI. 35 ml was carefully layered on the top of 15 ml of Ficoll-Paque Plus (GE Healthcare, Piscataway, NJ, USA). After centrifugation at 400xg for 30 min, the intermediate layer containing PBMCs (leucocytes, monocytes) and platelets was collected and resuspended in 50 mL of RPMI supplemented with 1/10 volume of ACD buffer (6.25 g of sodium citrate, 3.1 g of citric acid, 3.4 g of D-glucose in 250 mL of ddH2O). PBMCs were pelleted by centrifugation at 200xg for 15 minutes and the platelet rich supernatant was collected. Platelets were pelleted by centrifugation at 900xg for 20 minutes and processed for western blotting as described below.
Exosome isolation
Exosomes were isolated as described (39).
cDNA constructs
Construction of plasmids coding for JAK2WT and JAK2V617F, as well as JAK2WTmCitrine and JAK2V617FmCitrine, have been described (67, 68). MPL cDNA fused to eCFP,eGFP, mOrange2, mKO2 or miniSOG was generated by gene fusion PCR (69) using pfu ultra DNA polymerase (Stratagene, Amsterdam, The Netherlands). Total cDNA was amplified by PCR before sub-cloning into pcDNA3.1 directional topo vector (Life Technologies, Carlsbad, CA, USA). All constructs were checked by sequencing. cDNAs coding for GRASP55, STX5 and ATG5 were purchased from Thermo Scientific. HASTX5 was obtained by gene fusion PCR before sub-cloning into pcDNA 3.1. GRASP55-V5 was obtained by removing GRASP55 stop codon and then sub-cloning of the cDNA into pcDNA 3.1 containing the V5 tag. ATG5-K130R-V5 was obtained by standard mutagenesis of codon 130 and the V5 tag was inserted as described for GRASP55. The mRFP-GFPLC3 cDNA and mKO2-miniSOG cDNA containing plasmids were gifts from Drs T. Yoshimori (36, 70) and Roger Tsien (34), respectively.
Transient transfection
Transient transfections of HEL and K562 cells were performed using the Amaxa Nucleofector device (Amaxa, Cologne, Germany). Cells (10 × 106) were transfected with 25 μg DNA plasmid following manufacturer’s protocol. Cells were harvested for experiments 24hr later.
Stably transfected cell lines
K562 cells were transfected with cDNA coding for MPL-mKO2, MPL-mOr2 or MPL-eGFP, as described above, selected using geneticin, sorted by FACS and maintained in media supplemented with geneticin at 1000 μg/mL.
Immunoblots
Cells (10 × 106), or platelets, were lysed in RIPA buffer and 25 μg of total proteins were loaded on 4-15% polyacrylamide precast gradient gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose membrane (IBlot device, Life Technologies, Carlsbad, CA, USA). After blocking, membranes were probed with 1° antibodies for Mpl (H-300) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-Jak2, Jak2, p-Stat1, Stat1, pStat5, Stat5, p-Akt, Akt, p-Erk1/2 or Erk (Cell Signaling Technology, Danvers, MA, USA). Blots probed for total Jak2 and β-actin were used for normalization (Millipore, Billerica, MA, USA). In some cases, membranes were stripped 1x (Thermo Fischer Scientific) and then re-probed. After incubation with HRP-conjugated 2° antibodies, reactive bands were revealed with a chemiluminescent substrate (Thermo Fischer Scientific) and imaged with a Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA, USA), equipped with ImageLab 4.0.1 software.
siRNA experiment
Anti-JAK2 (#E-003146-00-0005) and non-targeting (NT) (#D-001910-10-05) siRNA were purchased from Thermo Scientific. 0.5 × 106 cells (HEL, K562Mpl-mKO2, K562Mpl-eGFP) were incubated with either anti-JAK2 or NT siRNA at 1 μM in Accel delivery media (Thermo Scientific) following manufacturer recommendations. After 72 hours, an aliquot of live cells was imaged by confocal microscopy and the remaining cells were lysed in RIPA buffer and analysed by western blot.
Co-Immuno Precipitation of JAK2 and Mpl
25 × 106 cells (HEL and K562Mpl-mKO2) were lysed in 1 mL of RIPA buffer (without vortexing). Lysates were then incubated for 3 hours 4°C with 60 μL of protein A/G slurry (Thermo Scientific) bound to 5 μg of anti-Mpl antibody (Millipore, #06-944). Beads were washed 3 times in RIPA buffer and re-suspended in 60 μL of Laemmli buffer before boiling and western blot analysis.
Intracellular labeling
Intracellular labeling steps were done at RT. Cells (up to 107) were harvested, fixed with 2% PBS-PFA for 20 min, rinsed 3x in PBS, permeabilized in 0.05% Triton X-100, 3% PBS-BSA for 10 min, nuclei have been stained with Hoechst dye during the permeabilization step; rinsed 3 times in PBS for 5 minutes; incubated with the primary antibody (all primary and secondary antibodies used are listed below) at 1/100 diluted in 3% PBS-BSA for 1H30; rinsed 3 times in PBS for 5 minutes; incubated with the fluorescent secondary antibody at 1/200 diluted in 3% PBS-BSA for 1 hour; rinsed 3 times in PBS for 5 minutes; mounted in between a glass slide and a glass coverslip using Prolong Gold (Life Technologies, Carlsbad, CA, USA). Mounted slides were kept at 4°C before imaging.Primary antibodies directed against: Jak2 (D2E12), calnexin (C5C9), EEA1 (C45B10), Rab11 (D4F5) and PDI (C81H6) were purchased from Cell Signaling Technology (CST, Danvers, MA, USA); ERGIC-53; anti-Sec31A and c-Mpl were from Santa Cruz Biotechnololy (SCB, Santa Cruz, CA, USA); anti-golgin-97 was from Life Technologies (Carlsbad, CA, USA); anti-LC3 was from MBL (MBL International, Woburn, MA, USA); anti-CD107a (LAMP-1) was from BioLegend (San Diego, CA, USA). Secondary antibodies used: DyLight-488 goat anti-rabbit and Capeli-488 goat anti-mouse were from Jackson ImmunoResearch (West Grove, PA, USA). Live cell labelling with ER-Tracker green have been done following manufacturer recommendation (Life Technologies, Carlsbad, CA, USA).
Tpo labeling with AlexaFluor 488
20 μg of lyophilized human recombinant Tpo (Life Technologies, Carlsbad, CA, USA) were reconstituted in 86 μL of PBS, mixed with 10 μL of NaHCO3 at 1M and 4 μL of Alexa Fluor 488 dye at 2.94 mM (Life Technologies, Carlsbad, CA, USA) and incubated at room temperature for 1 hour with very low magnetic agitation. Labelled Tpo was then separated from free dye using a G-25 spin column (GE, Waukesha,WI, USA) and labeled Tpo (Tpo-AF488) was eluted in PBS plus ultra-pure BSA. The Degree Of Labeling (DOL) = 4.22 moles of dye per mole of Tpo. Cm Tpo-AF488 = 150 ng/μL.
Statistical analysis
Statistical analysis and diagrams were made using GraphPad Prism 5. Statistical tests are described in legends. The Pearson’s pixel intensity correlation over space coefficient, R, of 2D bi-color confocal images were calculated using the Coloc_2 plugin of FIJI.
Confocal imaging, processing
Confocal images were acquired on a LSM 510 META Zeiss microscope equipped with a 405 nm laser diode, argon and HeNe1-543 nm lasers, 40xDIC or a 63xDIC oil objectives and ZEN software. Membrane fluorescence was based upon use of a membrane mask on unprocessed confocal images in Slide Book 5.
Photo-oxidation and electron microscopy
HEL and K562 cells were transiently transfected with MPL-mKO2-miniSOG and fixed with 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) in 0.1 M sodium cacodylate buffer (Electron Microscopy Sciences, Hatfield, PA), pH 7.4 at 4°C overnight. Cells were treated with blocking buffer, photo-oxidized, fixed, stained and embedded in resin for electron microscopy studies as described by (34). Ultrathin sections were prepared using a microtome (Leica UCT) and placed on formvar-carbon-coated nickel grids. Sections were post-stained with 2% aqueous uranyl acetate (Electron Microscopy Sciences, Hatfield, PA) and Ranolds lead citrate for 20 and 6 min respectively. Electron micrographs were captured in a Hitachi H7500 TEM equipped with an Advanced Microscopy Sciences XR60 camera.
Biotinylation assay
For each condition, 25×106 cells were treated +/- Tpo (25 ng/mL, 37°C) for specified times. Cells were harvested, washed 3x in ice cold PBS and re-suspended in 1 mL of PBS containing 1.5 mg of Sulfo-NHS-Biotin (Thermo Fischer Scientific). Biotinylation reactions went 30 min on ice, followed by 3 ice cold PBS washes supplemented with 100 mM of glycine in order to quench free biotin. Cells were lysed in RIPA buffer and biotinylated proteins immunoprecipitated using streptavidin-agarose beads (Life Technologies, Carlsbad, CA, USA). Samples were evaluated by SDS-PAGE and western blotting.
Proximity ligation assay (Duolink)
Duolink reagents and green detection kit were purchased from Sigma-Aldrich. HEL, K562 and K562-Mpl-mKO2 cells were treated following the manufacturer’s recommendation. Primary antibodies used for the Duolink experiment were anti-Mpl and anti-Jak2 antibodies purchased from Millipore. Prior to the duolink treatment, HEL and untransfected K562 cells were primed live with an anti-CD235a antibody (Pierce Antibodies) labelled in house with an Atto655 dye (Sigma-Aldrich) (2.5 dyes/antibody) in order to visualize the plasma membrane.
Drug treatments
Cells were treated as indicated in the corresponding figure legends. Ionomycin, PMA, monensin and A23187 were purchased from Sigma-Aldrich. 3-MA Bafilomycin A1 and rapamycin was from Invivogen.
Supplementary Material
Movie 1: Maximal intensity projection of a confocal z-stack analysis of HEL cells. Nuclei are stained with Hoechst (blue fluorescence) and endogenous Mpl receptor is immuno-stained with an antibody anti-Mpl revealed by a secondary antibody coupled to a Capelli-488 dye (green fluorescence). In this movie, the endogenous Mpl receptor appears to be sequestered in vesicles throughout the cell. No obvious evidence of its presence at the membrane can be observed.
Supplemental Figure 1: Correlated fluorescent and electron microscopy of mKO2-miniSOG tagged Mpl proteins. A) Schematic representation of the DNA construct coding for the MplmKO2-miniSOG fusion protein. B-C) Examples of K562 and HEL cells transiently expressing MplmKO2-miniSOG and imaged using both mKO2 and miniSOG fluorescent properties. D-E) Correlated light, before and after photo-oxidation, and electron micrograph images from a single K562 (B) or HEL (C) cell. Scale bars = 5 μm.
Supplemental Figure 2: TEM images of control experiments on K562 and HEL cells showing the absence of dark precipitate in the absence of photo-oxidation or in the absence of MplmKO2-miniSOG. A-B) TEM images of un-transfected K562 (A) or HEL (B) cells. No dark precipitate can be observed. C-D) TEM images of K562 (C) or HEL (D) cells transfected with MplmKO2-miniSOG in absence of photo-oxidation showing the absence of obvious dark precipitate. E-F) High magnification TEM images of un-transfected K562 (E) or HEL (F) cells submitted to the photo-oxidation process. Small dark spots can be observed in vesicles identified as lysosomes and are probably due to the presence of oxidative enzymes. The same observation has been made concerning mitochondria’s membranes. Scale bars = 1 μm (A-D) or 100 nm (E-F).
Supplemental Figure 3: TEM micrographs showing Mpl-positive structures budding from ER in HEL A-C), and K562 (D-F) cells. A,D) Involuted ER compartments in non-photo-oxidized HEL (A) and K562 (D) cells. B,E) double membrane autophagosomes (arrows) in HEL (B) and K562 (E) cells containing dark precipitates produced as a result of MplmKO2-miniSOG photo-oxidation. Multi lamellar body (black arrowhead) in HEL (C) and multivesicular bodies (white arrowheads) in K562 cells (F). Scale bar = 100 nm.
Supplemental Figure 4: Intracellular MpleCFP co-localizes with autophagosomes and autolysosomes. A-B) K562 (A) and HEL (B) cells stably expression LC3mRFP-GFP were co-transfected with MpleCFP. Cells were then stimulated, or not, with Tpo at 25ng/mL for the indicated time. In both cell lines, traces of fluorescence profile (white arrows) are showing the presence of MpleCFP in autophagosomes (black arrow heads) and in autolysosomes (violet arrow heads). Scale bars = 5 μm.
Supplemental Figure 5: Alexa Fluor-488 labeling of human recombinant Tpo. A) Non-reducing PAGE experiment showing efficiently labelled Tpo with alexa fluor-488 dye. The labeling reaction was done as described in the Materiel & Methods section. We can also note the presence of trace contamination. B) Confocal image of K562 cells transiently expressing MplmOrange2 and stimulated with Tpo chemically labeled with Alexa Fluor-488 dye (AF488). Tpo-488 binds specifically to MplmOrange2 and upon activation the receptor is internalized. As plotted on the attached fluorescence graph, there is a very good co-localization of MplmOrange2 and Tpo-488 at the plasma membrane as well as in endocytic vesicles (black arrow heads). Intracellular vesicles containing MplmOrange2 only (red arrow heads) were also easily identified. As a control, white stars indicate non-transfected cells that do not bind Tpo-AF488, showing the specificity of the binding. Scale bars = 5 μm.
Supplemental Figure 6: A) Western blot analysis and quantification of HEL and K562Mpl-mKO2 total lysate (prior to surface IP as presented in Figure 7A) probed with an anti-Mpl and an anti-actin antibodies. The quantification of this western blot is shown below and show that at the total lysate level there is no change in the distribution of Mpl forms. B) The same total lysates as in A) were run on the same gel as the surface IP lysate shown in Figure 7A and the membranes were probed with an anti-Grb2 and an anti-actin antibodies. This western blot shows the minor contamination of the surface IP proteins by total lysate proteins. C) Semi-quantitative RT-PCR assay showing the absence of endogenous MPL transcript in K562 cells, transfected or not with MplmKO2/mOrange2. RPLP0 is used as a housekeeping gene. Methods for (C): 5×106 cells of each cell line tested (HEL, K562, K562Mpl-mOrange2 and K562Mpl-mKO2) were lysed, and total RNA were extracted using a Nucleospin RNA extraction kit (Macherey-Nagel, Düren, Germany). Qualities of total RNA extracts were then assessed by spectrophotometry and agarose gel electrophoresis. Normalized quantities of RNA were then submitted to an oligodT RT-PCR using a first strand cDNA synthesis kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer recommendations. 5 μL of a 1/10 dilution (1/10 for RPLP0) of the newly synthesized cDNA were used to perform a PCR amplification specific for endogenous MPL, recombinant MPL and RPLP0 (housekeeping gene) using the primers listed below. The PCR reactions were done using a platinum Taq DNA polymerase (Life Technologies, Carlsbad, CA, USA) as follows: Activation at 95°C for 5 minutes; 35 cycles of 1) Denaturation at 95°C for 5 minutes, 2) Hybridization at 62°C for 30 seconds, 3) Elongation at 72°C for 45 seconds; elongation at 72°C for 5 minutes. For PCR reactions that were submitted to only 20, 25 or 30 cycles, the PCR tubes were removed from the thermocycler during the elongation step of the corresponding cycle number and then incubated 5 minutes in a water bath at 72°C to ensure the full extension of the DNA strands. Finally, PCR reactions were cooled down to 4°C, mixed with 10x loading buffer and 15 μL of each PCR were loaded on a 1.5% agarose gel. After migration DNA was stained using the GelRed dye (Biotium, Hayward, CA, USA) and imaged with a Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA, USA). Primers: 1) Amplification of the endogenous MPL gene : Fwdcommon hMPL 5’-CACTTCCAGACCTGCACCGG-3’, Rvse3’UTR-hMPL 5’-GGGGAACTAATTGAAGTAGTCTCAAGAGTAAATGGG -3’, Amplicon size = 658 bp. 2) Amplification of recombinant MPL-mOrange2 :Fwdcommon hMPL 5’-CACTTCCAGACCTGCACCGG-3’, RvseMPL-mOrange2 5’-GGAGTCCTGGGTCACGGTC-3’, Amplicon size = 662 bp. 3) Amplification of recombinant MPL-mKO2 :FwdcommonhMPL 5’-CACTTCCAGACCTGCACCGG-3’, RvseMPL-mKO2 5’-CCACATCAGCCTGAGGGGC-3’, Amplicon size = 656 bp. 4) Amplification of RPLP0 :FwdhRPLP0 5’-CTCTGGAGAAACTGCTGCCTCATATCC-3’, RvsehRPLP0 5’-AGCAGCAGC AGGAGCAGCTGTG-3’, Amplicon size = 656 bp.
Acknowledgments
This work was supported by grants from NIH P50GM065794 to BSW and the Ligue Nationale contre le Cancer (comités des Départements du Morbihan, d’Ille-et-Vilaine, de Vendée, des Charentes) to SH. MV is a recipient of a scholarship from the French Ministry of Research (2009-2012). Images in this paper were generated in the UNM Cancer Center Fluorescence Microscopy Shared Resource, funded as detailed on: http://hsc.unm.edu/crtc/microscopy/Facility.html. This project involved the National Center for Microscopy and Imaging Research, which was supported by grants from the NIH National Center for Research Resources (5P41RR004050-24) and now by the National Institute of General Medical Sciences (8 P41 GM103412-24) to MHE.
Footnotes
The authors have no conflict of interest to disclose.
Author Contributions: CC, AD, MPS, DB and MV performed research, analysed data and helped write the manuscript. CC, SH, MHE and BSW designed research, analysed data and wrote the manuscript.
References
- 1.Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115(12):3339–3347. doi: 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, Moore MW. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183(2):651–656. doi: 10.1084/jem.183.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solar GP, Kerr WG, Zeigler FC, Hess D, Donahue C, de Sauvage FJ, Eaton DL. Role of c-mpl in early hematopoiesis. Blood. 1998;92(1):4–10. [PubMed] [Google Scholar]
- 4.Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem. 2005;280(29):27251–27261. doi: 10.1074/jbc.M501376200. [DOI] [PubMed] [Google Scholar]
- 5.Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver-Moore K, de Sauvage FJ. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood. 1996;87(6):2154–2161. [PubMed] [Google Scholar]
- 6.Stoffel R, Wiestner A, Skoda RC. Thrombopoietin in thrombocytopenic mice: evidence against regulation at the mRNA level and for a direct regulatory role of platelets. Blood. 1996;87(2):567–573. [PubMed] [Google Scholar]
- 7.Tiedt R, Coers J, Ziegler S, Wiestner A, Hao-Shen H, Bornmann C, Schenkel J, Karakhanova S, de Sauvage FJ, Jackson CW, Skoda RC. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood. 2009;113(8):1768–1777. doi: 10.1182/blood-2008-03-146084. [DOI] [PubMed] [Google Scholar]
- 8.Lannutti BJ, Epp A, Roy J, Chen J, Josephson NC. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood. 2009;113(8):1778–1785. doi: 10.1182/blood-2007-11-124859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moliterno AR, Spivak JL. Posttranslational processing of the thrombopoietin receptor is impaired in polycythemia vera. Blood. 1999;94(8):2555–2561. [PubMed] [Google Scholar]
- 10.Moliterno AR, Hankins WD, Spivak JL. Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N Engl J Med. 1998;338(9):572–580. doi: 10.1056/NEJM199802263380903. [DOI] [PubMed] [Google Scholar]
- 11.Kralovics R, Buser AS, Teo SS, Coers J, Tichelli A, van der Maas AP, Skoda RC. Comparison of molecular markers in a cohort of patients with chronic myeloproliferative disorders. Blood. 2003;102(5):1869–1871. doi: 10.1182/blood-2003-03-0744. [DOI] [PubMed] [Google Scholar]
- 12.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 13.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 15.Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014 doi: 10.1182/blood-2014-03-530865. [DOI] [PubMed] [Google Scholar]
- 16.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8(6):1327–1338. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 17.Tong W, Sulahian R, Gross AW, Hendon N, Lodish HF, Huang LJ. The membrane-proximal region of the thrombopoietin receptor confers its high surface expression by JAK2-dependent and -independent mechanisms. J Biol Chem. 2006;281(50):38930–38940. doi: 10.1074/jbc.M607524200. [DOI] [PubMed] [Google Scholar]
- 18.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 19.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7(8):766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 21.Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9(6):861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16(6):618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 23.Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146(5):746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 24.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30(23):4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruns C, McCaffery JM, Curwin AJ, Duran JM, Malhotra V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol. 2011;195(6):979–992. doi: 10.1083/jcb.201106098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 27.Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012 doi: 10.1016/j.tcb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang M, Schekman R. Cell biology. Unconventional secretion, unconventional solutions. Science. 2013;340(6132):559–561. doi: 10.1126/science.1234740. [DOI] [PubMed] [Google Scholar]
- 29.Cleyrat C, Vilaine M, Boissinot M, Girodon F, Steinkamp MP, Dobo I, Lippert É, Corre I, Wilson BS, Hermouet S. Surface Expression of Mpl: Aberrant Mpl Trafficking In Myeloproliferative Neoplasms Is Linked to Insufficient Expression of Wild Type Jak2. Blood (ASH Annual Meeting Abstracts); 2011. p. 2820. [Google Scholar]
- 30.Xie S, Wang Y, Liu J, Sun T, Wilson MB, Smithgall TE, Arlinghaus RB. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20(43):6188–6195. doi: 10.1038/sj.onc.1204834. [DOI] [PubMed] [Google Scholar]
- 31.Porteu F, Rouyez MC, Cocault L, Benit L, Charon M, Picard F, Gisselbrecht S, Souyri M, Dusanter-Fourt I. Functional regions of the mouse thrombopoietin receptor cytoplasmic domain: evidence for a critical region which is involved in differentiation and can be complemented by erythropoietin. Mol Cell Biol. 1996;16(5):2473–2482. doi: 10.1128/mcb.16.5.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson BS, Pfeiffer JR, Smith AJ, Oliver JM, Oberdorf JA, Wojcikiewicz RJ. Calcium-dependent clustering of inositol 1,4,5-trisphosphate receptors. Mol Biol Cell. 1998;9(6):1465–1478. doi: 10.1091/mbc.9.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9(2):230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 34.Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9(4):e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol. 2012;24(4):496–501. doi: 10.1016/j.ceb.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 37.Pecquet C, Diaconu CC, Staerk J, Girardot M, Marty C, Royer Y, Defour JP, Dusa A, Besancenot R, Giraudier S, Villeval JL, Knoops L, Courtoy PJ, Vainchenker W, Constantinescu SN. Thrombopoietin receptor down-modulation by JAK2 V617F: restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood. 2012;119(20):4625–4635. doi: 10.1182/blood-2011-08-372524. [DOI] [PubMed] [Google Scholar]
- 38.Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol. 2002;3(2):122–131. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 39.Savina A, Furlan M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278(22):20083–20090. doi: 10.1074/jbc.M301642200. [DOI] [PubMed] [Google Scholar]
- 40.Musiwaro P, Smith M, Manifava M, Walker SA, Ktistakis NT. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy. 2013;9(9) doi: 10.4161/auto.25455. [DOI] [PubMed] [Google Scholar]
- 41.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 42.Tanemura M, Ohmura Y, Deguchi T, Machida T, Tsukamoto R, Wada H, Kobayashi S, Marubashi S, Eguchi H, Ito T, Nagano H, Mori M, Doki Y. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102–114. doi: 10.1111/j.1600-6143.2011.03771.x. [DOI] [PubMed] [Google Scholar]
- 43.Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun. 2013;4:2130. doi: 10.1038/ncomms3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 45.Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22(10):2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCracken AA, Brodsky JL. Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD) Bioessays. 2003;25(9):868–877. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- 47.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18(8):1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 48.Pfeffer SR. Unconventional secretion by autophagosome exocytosis. J Cell Biol. 2010;188(4):451–452. doi: 10.1083/jcb.201001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10(2):148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 50.Cadwell K, Patel KK, Komatsu M, Virgin HWt, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009;5(2):250–252. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21(5):966–974. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, Ichimura Y, Ezaki J, Nishida K, Komazawa-Sakon S, Niyonsaba F, Ishii T, Yanagawa T, Kominami E, Ogawa H, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127(5):1267–1276 e1266. doi: 10.1016/j.jaci.2010.12.1078. [DOI] [PubMed] [Google Scholar]
- 53.Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11(9):1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 54.Luo Y, McDonald K, Hanrahan JW. Trafficking of immature DeltaF508-CFTR to the plasma membrane and its detection by biotinylation. Biochem J. 2009;419(1):211–219. doi: 10.1042/BJ20081869. 212 p following 219. [DOI] [PubMed] [Google Scholar]
- 55.Giuliani F, Grieve A, Rabouille C. Unconventional secretion: a stress on GRASP. Curr Opin Cell Biol. 2011;23(4):498–504. doi: 10.1016/j.ceb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, Warri A, Shajahan AN. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72(6):1321–1331. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Albu RI, Constantinescu SN. Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels. Front Endocrinol (Lausanne) 2011;2:71. doi: 10.3389/fendo.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boissinot M, Cleyrat C, Vilaine M, Jacques Y, Corre I, Hermouet S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene. 2011;30(8):990–1001. doi: 10.1038/onc.2010.479. [DOI] [PubMed] [Google Scholar]
- 59.Zhao L, Dong H, Zhang CC, Kinch L, Osawa M, Iacovino M, Grishin NV, Kyba M, Huang LJ. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J Biol Chem. 2009;284(39):26988–26998. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Staerk J, Defour JP, Pecquet C, Leroy E, Antoine-Poirel H, Brett I, Itaya M, Smith SO, Vainchenker W, Constantinescu SN. Orientation-specific signalling by thrombopoietin receptor dimers. EMBO J. 2011;30(21):4398–4413. doi: 10.1038/emboj.2011.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Girardot M, Pecquet C, Boukour S, Knoops L, Ferrant A, Vainchenker W, Giraudier S, Constantinescu SN. miR-28 is a thrombopoietin receptor targeting microRNA detected in a fraction of myeloproliferative neoplasm patient platelets. Blood. 2010;116(3):437–445. doi: 10.1182/blood-2008-06-165985. [DOI] [PubMed] [Google Scholar]
- 62.Moliterno AR, Williams DM, Rogers O, Spivak JL. Molecular mimicry in the chronic myeloproliferative disorders: reciprocity between quantitative JAK2 V617F and Mpl expression. Blood. 2006;108(12):3913–3915. doi: 10.1182/blood-2006-03-008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen D, Bernstein AM, Lemons PP, Whiteheart SW. Molecular mechanisms of platelet exocytosis: role of SNAP-23 and syntaxin 2 in dense core granule release. Blood. 2000;95(3):921–929. [PubMed] [Google Scholar]
- 64.Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012;119(12):2721–2730. doi: 10.1182/blood-2011-11-395228. [DOI] [PubMed] [Google Scholar]
- 65.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. doi: 10.1038/leu.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Santos FP, Verstovsek S. JAK2 inhibitors: what’s the true therapeutic potential? Blood Rev. 2011;25(2):53–63. doi: 10.1016/j.blre.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cleyrat C, Jelinek J, Girodon F, Boissinot M, Ponge T, Harousseau JL, Issa JP, Hermouet S. JAK2 mutation and disease phenotype: a double L611V/V617F in cis mutation of JAK2 is associated with isolated erythrocytosis and increased activation of AKT and ERK1/2 rather than STAT5. Leukemia. 2010;24(5):1069–1073. doi: 10.1038/leu.2010.23. [DOI] [PubMed] [Google Scholar]
- 68.Girodon F, Steinkamp MP, Cleyrat C, Hermouet S, Wilson BS. Confocal imaging studies cast doubt on nuclear localization of JAK2V617F. Blood. 2011;118(9):2633–2634. doi: 10.1182/blood-2011-02-336479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 70.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1: Maximal intensity projection of a confocal z-stack analysis of HEL cells. Nuclei are stained with Hoechst (blue fluorescence) and endogenous Mpl receptor is immuno-stained with an antibody anti-Mpl revealed by a secondary antibody coupled to a Capelli-488 dye (green fluorescence). In this movie, the endogenous Mpl receptor appears to be sequestered in vesicles throughout the cell. No obvious evidence of its presence at the membrane can be observed.
Supplemental Figure 1: Correlated fluorescent and electron microscopy of mKO2-miniSOG tagged Mpl proteins. A) Schematic representation of the DNA construct coding for the MplmKO2-miniSOG fusion protein. B-C) Examples of K562 and HEL cells transiently expressing MplmKO2-miniSOG and imaged using both mKO2 and miniSOG fluorescent properties. D-E) Correlated light, before and after photo-oxidation, and electron micrograph images from a single K562 (B) or HEL (C) cell. Scale bars = 5 μm.
Supplemental Figure 2: TEM images of control experiments on K562 and HEL cells showing the absence of dark precipitate in the absence of photo-oxidation or in the absence of MplmKO2-miniSOG. A-B) TEM images of un-transfected K562 (A) or HEL (B) cells. No dark precipitate can be observed. C-D) TEM images of K562 (C) or HEL (D) cells transfected with MplmKO2-miniSOG in absence of photo-oxidation showing the absence of obvious dark precipitate. E-F) High magnification TEM images of un-transfected K562 (E) or HEL (F) cells submitted to the photo-oxidation process. Small dark spots can be observed in vesicles identified as lysosomes and are probably due to the presence of oxidative enzymes. The same observation has been made concerning mitochondria’s membranes. Scale bars = 1 μm (A-D) or 100 nm (E-F).
Supplemental Figure 3: TEM micrographs showing Mpl-positive structures budding from ER in HEL A-C), and K562 (D-F) cells. A,D) Involuted ER compartments in non-photo-oxidized HEL (A) and K562 (D) cells. B,E) double membrane autophagosomes (arrows) in HEL (B) and K562 (E) cells containing dark precipitates produced as a result of MplmKO2-miniSOG photo-oxidation. Multi lamellar body (black arrowhead) in HEL (C) and multivesicular bodies (white arrowheads) in K562 cells (F). Scale bar = 100 nm.
Supplemental Figure 4: Intracellular MpleCFP co-localizes with autophagosomes and autolysosomes. A-B) K562 (A) and HEL (B) cells stably expression LC3mRFP-GFP were co-transfected with MpleCFP. Cells were then stimulated, or not, with Tpo at 25ng/mL for the indicated time. In both cell lines, traces of fluorescence profile (white arrows) are showing the presence of MpleCFP in autophagosomes (black arrow heads) and in autolysosomes (violet arrow heads). Scale bars = 5 μm.
Supplemental Figure 5: Alexa Fluor-488 labeling of human recombinant Tpo. A) Non-reducing PAGE experiment showing efficiently labelled Tpo with alexa fluor-488 dye. The labeling reaction was done as described in the Materiel & Methods section. We can also note the presence of trace contamination. B) Confocal image of K562 cells transiently expressing MplmOrange2 and stimulated with Tpo chemically labeled with Alexa Fluor-488 dye (AF488). Tpo-488 binds specifically to MplmOrange2 and upon activation the receptor is internalized. As plotted on the attached fluorescence graph, there is a very good co-localization of MplmOrange2 and Tpo-488 at the plasma membrane as well as in endocytic vesicles (black arrow heads). Intracellular vesicles containing MplmOrange2 only (red arrow heads) were also easily identified. As a control, white stars indicate non-transfected cells that do not bind Tpo-AF488, showing the specificity of the binding. Scale bars = 5 μm.
Supplemental Figure 6: A) Western blot analysis and quantification of HEL and K562Mpl-mKO2 total lysate (prior to surface IP as presented in Figure 7A) probed with an anti-Mpl and an anti-actin antibodies. The quantification of this western blot is shown below and show that at the total lysate level there is no change in the distribution of Mpl forms. B) The same total lysates as in A) were run on the same gel as the surface IP lysate shown in Figure 7A and the membranes were probed with an anti-Grb2 and an anti-actin antibodies. This western blot shows the minor contamination of the surface IP proteins by total lysate proteins. C) Semi-quantitative RT-PCR assay showing the absence of endogenous MPL transcript in K562 cells, transfected or not with MplmKO2/mOrange2. RPLP0 is used as a housekeeping gene. Methods for (C): 5×106 cells of each cell line tested (HEL, K562, K562Mpl-mOrange2 and K562Mpl-mKO2) were lysed, and total RNA were extracted using a Nucleospin RNA extraction kit (Macherey-Nagel, Düren, Germany). Qualities of total RNA extracts were then assessed by spectrophotometry and agarose gel electrophoresis. Normalized quantities of RNA were then submitted to an oligodT RT-PCR using a first strand cDNA synthesis kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer recommendations. 5 μL of a 1/10 dilution (1/10 for RPLP0) of the newly synthesized cDNA were used to perform a PCR amplification specific for endogenous MPL, recombinant MPL and RPLP0 (housekeeping gene) using the primers listed below. The PCR reactions were done using a platinum Taq DNA polymerase (Life Technologies, Carlsbad, CA, USA) as follows: Activation at 95°C for 5 minutes; 35 cycles of 1) Denaturation at 95°C for 5 minutes, 2) Hybridization at 62°C for 30 seconds, 3) Elongation at 72°C for 45 seconds; elongation at 72°C for 5 minutes. For PCR reactions that were submitted to only 20, 25 or 30 cycles, the PCR tubes were removed from the thermocycler during the elongation step of the corresponding cycle number and then incubated 5 minutes in a water bath at 72°C to ensure the full extension of the DNA strands. Finally, PCR reactions were cooled down to 4°C, mixed with 10x loading buffer and 15 μL of each PCR were loaded on a 1.5% agarose gel. After migration DNA was stained using the GelRed dye (Biotium, Hayward, CA, USA) and imaged with a Bio-Rad ChemiDoc XRS+ (Bio-Rad, Hercules, CA, USA). Primers: 1) Amplification of the endogenous MPL gene : Fwdcommon hMPL 5’-CACTTCCAGACCTGCACCGG-3’, Rvse3’UTR-hMPL 5’-GGGGAACTAATTGAAGTAGTCTCAAGAGTAAATGGG -3’, Amplicon size = 658 bp. 2) Amplification of recombinant MPL-mOrange2 :Fwdcommon hMPL 5’-CACTTCCAGACCTGCACCGG-3’, RvseMPL-mOrange2 5’-GGAGTCCTGGGTCACGGTC-3’, Amplicon size = 662 bp. 3) Amplification of recombinant MPL-mKO2 :FwdcommonhMPL 5’-CACTTCCAGACCTGCACCGG-3’, RvseMPL-mKO2 5’-CCACATCAGCCTGAGGGGC-3’, Amplicon size = 656 bp. 4) Amplification of RPLP0 :FwdhRPLP0 5’-CTCTGGAGAAACTGCTGCCTCATATCC-3’, RvsehRPLP0 5’-AGCAGCAGC AGGAGCAGCTGTG-3’, Amplicon size = 656 bp.