

Abstract
Background
Epigenetics is the study of gene expression changes that are not caused by changes in the deoxyribonucleic acid (DNA) sequence. DNA methylation is an epigenetic mark occurring in C–phosphate–G sites (CpGs) that leads to local or regional gene expression changes. Reduced-representation bisulfite sequencing (RRBS) is a technique that is used to ascertain the DNA methylation of millions of CpGs at single-nucleotide resolution. The genomic coverage of RRBS is given by the restriction enzyme combination used during the library preparation and the throughput capacity of the next-generation sequencer, which is used to read the generated libraries. The four-nucleotide cutters, MspI and TaqαI, are restriction enzymes commonly used in RRBS that, when combined, achieve ~12% genomic coverage. The increase in throughput of next-generation sequencers allows for novel combinations of restriction enzymes that provide higher CpG coverage.
Results
We performed a near-neighbor analysis of the four nucleotide sequences most frequently found within 50 nt of all genomic CpGs. This resulted in the identification of seven methylation-insensitive restriction enzymes (AluI, BfaI, HaeIII, HpyCH4V, MluCI, MseI, and MspI) that shared similar restriction conditions suitable for RRBS library preparation. We report that the use of two or three enzyme combinations increases the theoretical epigenome coverage to almost half of the human genome.
Conclusions
We provide the enzyme combinations that are more likely to increase the CpG coverage in human, rat, and mouse genomes.
Electronic supplementary material
The online version of this article (doi:10.1186/1756-0500-7-534) contains supplementary material, which is available to authorized users.
Keywords: RRBS, DNA methylation, Epigenetics, Restriction enzyme, Next-generation sequencing
Background
Epigenetics is the study of gene expression changes that are not caused by changes in the deoxyribonucleic acid (DNA) sequence. Methylation of cytosine at CG dinucleotides is an epigenetic mark that is shown to modulate local and regional gene expression [1]. Multiple techniques have been developed to quantify DNA methylation, which center around the treatment of DNA with bisulfite, the use of restriction enzymes sensitive to DNA methylation, or the use of methylation-binding proteins [2]. The reduced-representation bisulfite sequence (RRBS) is a robust technique that provides DNA methylation levels at a nucleotide resolution of millions of CpGs with little DNA input [3]. RRBS is becoming increasingly popular because it provides a higher resolution and greater genomic coverage than array-based technologies, and it is cheaper than whole-genome bisulfite sequencing. The CpG coverage of RRBS has been improved by the increase in sequencing throughput and the depth of sequencing of next-generation sequencers (NGS).
RRBS was originally described as using a DNA methylation-insensitive restriction enzyme with a consensus sequence that is often found in C–phosphate–G (CpG)-rich regions to digest genomic DNA. The fragments that are generated are selected for size and contain a “reduced representation” of the starting genomic DNA. The size-selected fragments are ligated to chemically modified sequencing adapters, treated with bisulfite, and are amplified via polymerase chain reaction (PCR). The resulting RRBS library continues to a standard next-generation sequencing pipeline. The sequencing reads are aligned against a reference genome, while DNA methylation levels are ascertained by counting the frequency of CGs (methylated) and TGs (demethylated) at the various CpG sites. Please refer to Gu et al. for a detail description of the RRBS library preparation process [4].
The CpG coverage achieved by RRBS is dependent on the restriction enzymes used to digest the genomic DNA and the sequencing throughput. The use of MspI (C|CGG), which is frequently found in CpG islands (CGIs), generates few CG-rich DNA fragments that provide coverage to most of the CGI islands [5]. The coverage of high CG density regions was expanded by the use of a combination of MspI and TaqαI (T|CGA). This enzyme combination was reported to cover approximately 1.8 million CpGs (sequencing depth of 10 nt), representing approximately 6.6% of the total human CGs [6]. A significant improvement in low density CG region coverage, which includes shore regions and coding sequences (CDS), was achieved by the combined use of MspI and ApeKI (G|CWGC) [7]. This enzyme combination expanded CpG coverage by approximately 2-fold, while limiting the increase in sequencing cost.
NGS advances have resulted in higher reads and increased sequencing depth, thus allowing novel enzyme combinations to be used. Here, we set out to describe the various enzymes that result in higher genomic and read coverage.
Methods
MATLAB® 2014a (The MathWorks, Inc., Natick, MA, USA), equipped with a bioinformatics toolbox, was used to create scripts that identify the various parameters measured. Databases were downloaded from the National Center for Biotechnology Information (NCBI) file transfer protocol site, and they were used to ascertain the assembled genomes for Homo sapiens (HuRef; annotation release 106), Rattus novergicus (NCBI build 4.2), and Mus musculus (GRCm38.p2; annotation release 104).
CpGs within fragments were counted from the 5’ and 3’ ends. We established a 40–400 bp fragment cutoff and a sequencing depth of 50 nt to carry out our comparative analysis. For genomic CpG distribution studies, shore regions were up to 2 kb from a CGI, and shelf regions were 2 kb to 4 kb from a CGI. Open sea regions were the genomic CpGs not contained in genes, promoters (2 kb from transcription start site), CGIs, and shore and shelf regions.
Synthetic DNA bearing the seven restriction sites approximately every 50 nt was obtained from Integrated DNA Technologies, Inc. (Coralville, IA, USA). Restriction enzymes were obtained from New England Biolabs (Ipswich, MA, USA). The synthetic insert was amplified by PCR using the following cycle parameters: initial 95°C incubation, 30 cycles of 95°C/10 s denaturing, 66°C/10 s annealing, and 72°C/20 s extension, and final 72°C for 7 min incubation. Primer pairs used are shown in Figure 1. Unique band amplification was confirmed by gel electrophoresis and PCR products were purified using the MinElute PCR kit (Qiagen). Restriction digestion of 1 μg of custom-designed DNA was carried out at 37°C for 4 h using 30U AluI, 80U BfaI, 30U HaeIII, 15U HpyCH4V, 10U MluCI, 10U MseI, and 10U MspI. Restriction products were size separated in 2% agarose.
Figure 1.

CpGs covered using selected enzymes with respect to MspI or MspI ApeKI in (A) Homo sapiens , (B) Rattus novergicus , and (C) Mus musculus .
Results and discussion
We began the search for novel enzyme combinations by identifying the 4-nt sequences most frequently found within 50-nt of all genomic CGs. This flanking distance was chosen since it is a common sequencing depth used in NGS (i.e. Illumina HiSeq; Illumina, Inc., San Diego, CA, USA). The user can expect greater CG coverage as the sequencing depth increases. Additional file 1: Tables S1, S2, S3 show the results of the near-neighbor analysis for human, rat, and mouse genomes, respectively. Only the sites for which a restriction enzyme has been described are shown in the analysis. The near-neighbor analysis identified the order of the enzymes that cover the most CGs per organism. Table 1 shows the properties of eleven 4-nt and 5-nt cutting enzymes that are suitable for RRBS use. Seven restriction enzymes (AluI, BfaI, HaeIII, HpyCH4V, MluCI, MseI, and MspI) were chosen for our analysis since they share the same reaction conditions, and thus simplify the library preparation process. Moreover, we compared the results of selected enzyme combinations with the outcome of an MspI or MspI ApeKI combination.
Table 1.
Properties of the identified enzymes useful in RRBS
| Enzyme | Consensus sequence | Restriction temp. (°C) | Digestion buffer | Heat inactivation (°C) | Methylation sensitive | Catalog number |
|---|---|---|---|---|---|---|
| AluI | AG|CT | 37 | CutSmart | 80 | No | R0137S |
| BfaI | C|TAG | 37 | CutSmart | 80 | No | R0568S |
| HaeIII | GG|CC | 37 | CutSmart | 80 | No | R0108S |
| HpyCH4V | TG|CA | 37 | CutSmart | 65 | No | R0620S |
| MluCI | |AATT | 37 | CutSmart | No | No | R0538S |
| MseI | T|TAA | 37 | CutSmart | 65 | No | R0525S |
| MspI | C|CGG | 37 | CutSmart | No | No | R0106S |
| TaqI | T|CGA | 65 | CutSmart | 80 | dam | R0149S |
| CviQI | G|TAC | 25 | NEB 3.1 | No | No | R0639S |
| CviAII | C|ATG | 25 | CutSmart | 65 | No | R0640S |
| ApeKI | G|CWGC | 75 | NEB 3.1 | No | Yes | R0643S |
We performed in silico restriction digestions with a combination of the seven enzymes selected, and we registered the number of CpGs covered, the number of fragments generated, and identified the genomic coverage (assuming 27 M, 23.9 M, and 21.9 M CpGs for human, rat, and mouse genomes, respectively). In addition, we calculated the CpG/fragment ratio, which represents the density of CGs within a fragment, as generated by a given enzyme. The results show that in the human genome, MspI (C|CGG) is the single enzyme that has the highest CpG/fragment ratio, followed by HaeIII (GG|CC) and AluI (AG|CT). In contrast, MseI (T|TAA) and MluCI (|AATT) show the lowest CpG/fragment ratio. We also counted the number of fragments that contained no CpGs within the chosen sequencing depth and identified them as CpG-free fragments. We found that, aside from the use of MspI alone, the MspI ApeKI combination had the least CpG-free fragments (959 K) followed by HaeIII (1.5 M) and MspI HaeIII (1.7 M). It is important to note that only the use of restriction enzymes that contain CG in their consensus sequence will not generate CpG-free fragments. However, since CG-containing enzymes provide reduced coverage of low density CpG regions, the use of non CG-containing enzymes generates a large number of CpG-free fragments as a byproduct of the increased coverage of low density CpG regions. This has a dramatic impact on the sequencing cost because CpG-free fragments, which are of no value for RRBS, were 31 to 65% of the total fragments generated. Thus, it is up to the end-user to evaluate the benefit of expanding CpG coverage at the expense of increasing sequencing cost.
The individual CpG read coverage was calculated by dividing 150 M (typical reads for the Illumina HiSeq) by the number of fragments generated. The read coverage is critical in selecting an optimal enzyme combination because higher reads have a direct impact on the accuracy of the methylation call. Moreover, the more times a CG is read, it will ultimately improve the subsequent statistics that are applied to find differentially methylated CpGs. We set a read coverage cut-off of 10×, but it is important to note that this threshold is only a baseline used for comparisons and, in practice, is expected to change depending on the mapping rate. We abstained from including a mapping rate into our data because this number varies according to the alignment algorithm used, the size selection of the fragments, and repeat sequences generated for each enzyme combination [8]. However, the user should expect that approximately 70% of the sequences experimentally obtained will be mapped, and thus, result in a proportional decrease of CpG read coverage. Additional file 1: Tables S4, S5, S6 present the full combinatorial analysis, so as to facilitate the choice of the enzyme combination that best suits the investigator’s needs. In Table 2, we present selected enzyme combinations that offer high CpG coverage and at least 10× predicted read coverage. Of note, in the human genome, HaeIII HpyCH4V offers the best CpG coverage (53.2%; 10.3× read coverage), but MspI HypCH4V offers better read coverage (14.2×) while maintaining high CpG coverage (42.3%). Moreover, recent reports suggest that MspI restriction is affected by hydroxymethylation [9], in which case AluI HaeIII (CpG coverage, 51.6%; read coverage, 11×) is a suitable substitutions. Higher read coverage can be achieved using MspI HaeIII (CpG coverage, 32.2%, read coverage 26.7×) or HaeIII (genomic coverage, 25%, read coverage 33.2×). Similar options are also available for the rat and mouse genomes.
Table 2.
Summary of enzyme combinations that achieve the best genomic coverage in human, rat, and mouse genomes
| Enzyme combinations | CpGs covered | Fragments generated | Genomic CpG coverage (%) | CpG/fragment ratio | Read coverage |
|---|---|---|---|---|---|
| I. Homo sapiens | |||||
| HaeIII | 6,755,710 | 4,521,945 | 25.0 | 1.49 | 33.2× |
| MspI HaeIII | 8,703,801 | 5,613,529 | 32.2 | 1.55 | 26.7× |
| MspI AluI | 10,642,461 | 9,505,806 | 39.4 | 1.12 | 15.8× |
| MspI HpyCH4V | 11,443,197 | 10,567,403 | 42.3 | 1.08 | 14.2× |
| MspI BfaI HaeIII | 12,111,370 | 11,319,689 | 44.8 | 1.07 | 13.3× |
| MspI AluI HaeIII | 13,769,398 | 14,064,014 | 50.9 | 0.98 | 10.7× |
| AluI HaeIII | 13,957,636 | 13,633,940 | 51.6 | 1.02 | 11.0× |
| HaeIII HpyCH4V | 14,395,279 | 14,532,544 | 53.2 | 0.99 | 10.3× |
| II. Rattus novergicus | |||||
| HaeIII | 4,292,555 | 3,206,578 | 17.9 | 1.34 | 46.8× |
| MspI HaeIII | 5,859,114 | 4,154,939 | 24.5 | 1.41 | 36.1× |
| MspI AluI | 9,423,433 | 8,934,858 | 39.4 | 1.05 | 16.8× |
| MspI BfaI HaeIII | 9,643,490 | 9,579,581 | 40.3 | 1.01 | 15.7× |
| HaeIII HpyCH4V | 10,937,751 | 11,405,956 | 45.7 | 0.96 | 13.2× |
| MspI AluI BfaI | 11,837,958 | 13,122,827 | 49.5 | 0.90 | 11.4× |
| MspI BfaI HpyCH4V | 12,246,891 | 13,543,165 | 51.2 | 0.90 | 11.1× |
| AluI HpyCH4V | 12,674,196 | 14,657,325 | 53.0 | 0.86 | 10.2× |
| III. Mus musculus | |||||
| MspI HaeIII | 5,708,849 | 4,591,528 | 26.1 | 1.24 | 32.7× |
| MspI AluI | 8,524,374 | 9,393,465 | 39.0 | 0.91 | 16.0× |
| MspI BfaI HaeIII | 9,221,025 | 10,607,497 | 42.2 | 0.87 | 14.1× |
| HaeIII HpyCH4V | 10,164,473 | 12,447,516 | 46.5 | 0.82 | 12.1× |
| AluI HaeIII | 10,229,379 | 12,588,763 | 46.8 | 0.81 | 11.9× |
| MspI AluI HaeIII | 10,706,690 | 13,170,774 | 49.0 | 0.81 | 11.4× |
| MspI HaeIII HpyCH4V | 10,759,254 | 13,095,284 | 49.2 | 0.82 | 11.5× |
| MspI BfaI HpyCH4V | 11,347,760 | 14,601,495 | 51.9 | 0.78 | 10.3× |
Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput.
We compared the CpGs covered by the enzymes shown in Table 2 with respect to MspI or MspI ApeKI. Figure 1 shows that, in humans, the use of ApeKI doubles the CpGs covered by MspI. Similarly, the selected enzyme combinations contained a significant amount of CpGs covered by MspI alone or in combination with ApeKI, while dramatically increasing the coverage of new CpGs. Interestingly, HaeIII showed the least overlapping CpGs when compared to MspI or MspI ApeKI, but maintained a significant amount of new CpGs covered. Figure 2 shows the genomic distribution of the CpGs covered by the selected enzymes. The results show CGI coverage was similar in all enzymes selected. However, low density CG regions (shore, shelf, open sea, and intron) coverage was significantly improved by the selected enzyme combinations. HaeIII showed a similar genomic distribution profile as MspI ApeKI. The CpG cover and distribution data suggests that HaeIII or MspI HaeIII may be used as an alternative to MspI ApeKI since they covered a significant amount of new CpGs, while limiting the generation of CpG-free fragments.
Figure 2.
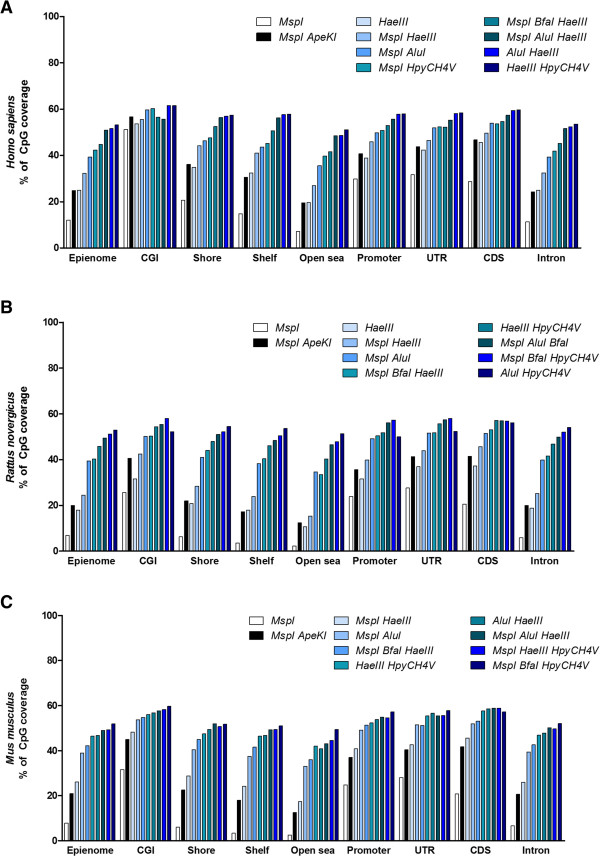
Genomic distribution of CpGs covered by selected enzymes in (A) Homo sapiens , (B) Rattus novergicus , and (C) Mus musculus .
We chose to analyze the restriction fragments between 40 – 400 bp to simplify the comparison between enzymes. However, in practice, fragments are size-selected based in their fragment distribution profile. Additional file 2: Figure S1, Additional file 3: Figure S2, Additional file 4: Figure S3 depict the fragment distribution profiles of the enzymes selected in Table 2. The figures show that the majority of the fragments are contained between 40 – 200 bp independently of the enzyme combination used.
We performed a restriction assay alone and in combination to assess restriction enzyme efficiency and compatibility. Our initial results using 10U/enzyme for 1 hr at 37°C showed that BfaI underperformed in restricting 200 ng of template. Full digestion of 1 μg template was achieved using 80U of BfaI for 4 hrs at 37°C in a 50 μl reaction (Figure 3). Here, our aim was to show enzyme compatibility, but the exact conditions for enzyme restriction will have to be determined by the end-user.
Figure 3.
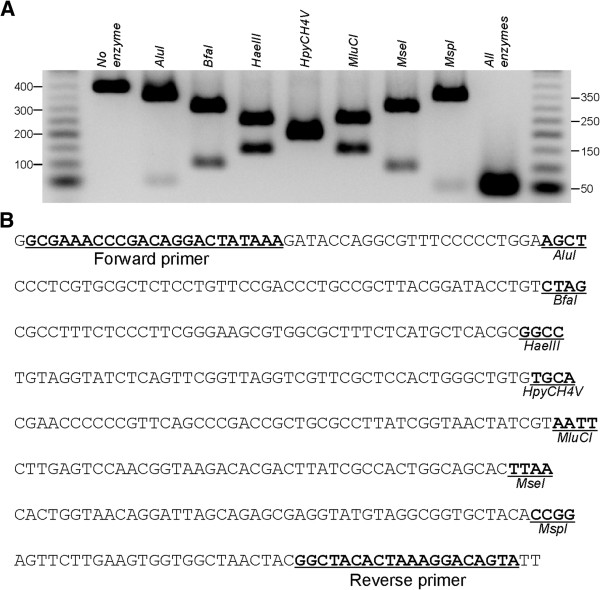
Restriction digestion assay to evaluate restriction enzyme efficiency and compatibility. (A) Restriction digestion of DNA bearing the seven restriction sites. (B) Sequence used for restriction reaction digestion. Underlined is the primer sequence used to amplify the synthetic insert and the restriction sites.
Conclusions
The increase in mass sequencing throughput allows for multiple enzyme combinations to expand RRBS CpG coverage. The use of two or three novel enzyme combinations improves the theoretical CpG coverage to almost half of the human genome. The increased coverage of low density CpG regions generates a significant amount of CpG-free fragments, which considerably increases the sequencing cost. We found that HaeIII or MspI HaeIII are enzymes that may be used as an alternative to MspI or MspI ApeKI.
Electronic supplementary material
Additional file 1: Table S1: Human near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S2. Rat near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S3. Mouse near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S4. Human combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. Table S5. Rat combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. Table S6. Mouse combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. (DOCX 55 KB)
Additional file 2: Figure S1: Fragment distribution of selected enzymes in Homo sapiens. (TIFF 2 MB)
Additional file 3: Figure S2: Fragment distribution of selected enzymes in Rattus novergicus. (TIFF 2 MB)
Additional file 4: Figure S3: Fragment distribution of selected enzymes in Mus musculus. (TIFF 2 MB)
Acknowledgements
This work was supported by grant MOP-111131 from the Canadian Institutes of Health Research and by a Canada Research Chair in Biochemical Pharmacology (to V.P.). The Research Institute is supported in part by a center grant from Fonds de la Recherche Quebec – Santé.
Abbreviations
- CGI
CpG islands
- NGS
Next generation sequencing
- RRBS
Reduced representation bisulfite sequencing.
Footnotes
Competing interests
The authors have nothing to disclose.
Authors’ contributions
DM performed the in silico analysis. DM and SL performed restriction digestion experiments. DM and VP designed research, analyzed data, and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Daniel B Martinez-Arguelles, Email: dan.martinez@mail.mcgill.ca.
Sunghoon Lee, Email: sunghoon.lee2@mail.mcgill.ca.
Vassilios Papadopoulos, Email: vassilios.papadopoulos@mcgill.ca.
References
- 1.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28(10):1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33(18):5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 5.Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48(3):226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YK, Jin S, Duan S, Lim YC, Ng DP, Lin XM, Yeo G, Ding C. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biol Proced Online. 2014;16(1):1. doi: 10.1186/1480-9222-16-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Xia Y, Li L, Gong D, Yao Y, Luo H, Lu H, Yi N, Wu H, Zhang X, Tao Q, Gao F. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics. 2013;14:11. doi: 10.1186/1471-2164-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee A, Stockwell PA, Rodger EJ, Morison IM. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res. 2012;40(10):e79. doi: 10.1093/nar/gks150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichiyanagi K. Inhibition of MspI cleavage activity by hydroxymethylation of the CpG site: a concern for DNA modification studies using restriction endonucleases. Epigenetics. 2012;7(2):131–136. doi: 10.4161/epi.7.2.18909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Human near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S2. Rat near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S3. Mouse near-neighbor analysis of 4 nt restriction sites most frequently found within 50 nt of a CpG. In bold are enzymes that may be used for RRBS, and which share similar restriction conditions. Underlined enzymes may need separate restriction reactions. Table S4. Human combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. Table S5. Rat combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. Table S6. Mouse combinatorial analysis of enzymes that may be used for RRBS. Calculations assumed a fragment size inclusion of 40–400 bp, a sequencing depth of 50 nt, and a 150 M read NGS throughput. (DOCX 55 KB)
Additional file 2: Figure S1: Fragment distribution of selected enzymes in Homo sapiens. (TIFF 2 MB)
Additional file 3: Figure S2: Fragment distribution of selected enzymes in Rattus novergicus. (TIFF 2 MB)
Additional file 4: Figure S3: Fragment distribution of selected enzymes in Mus musculus. (TIFF 2 MB)