

Highlights
-
•
Chromium(VI) impaired respiration and increased glycolytic flux in BEAS-2B cells.
-
•
Cr(VI)-exposed cells shifted to a more fermentative metabolism.
-
•
This metabolic shift was in line with a decreased β-F1-ATPase/GAPDH protein ratio.
-
•
Increased oxidative stress levels suggest impairment of antioxidant defenses.
Abbreviations: Cr(III), trivalent chromium; Cr(IV), tetravalent chromium; Cr(V), pentavalent chromium; Cr(VI), hexavalent chromium; DCF, 2′,7′-dichlorofluorescein; 2-DG, 2-deoxyglucose; 2,4-DNP, 2,4-dinitrophenol; EDTA, ethylenediaminetetracetic acid; ETC, mitochondrial electron transport chain; β-F1-ATPase, catalytic subunit (subunit β) of the mitochondrial H+-ATP synthase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IARC, International Agency for Research on Cancer; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; PBS, phosphate-buffered saline; PI, propidium iodide; ROS, reactive oxygen species; TCA, tricarboxylic acid
Keywords: Chromate lung cancer, Warburg effect, Aerobic glycolysis, Cellular respiration, Cellular bioenergetic index, Cellular energy status
Abstract
Previous studies on the impact of hexavalent chromium [Cr(VI)] on mammalian cell energetics revealed alterations suggestive of a shift to a more fermentative metabolism. Aiming at a more defined understanding of the metabolic effects of Cr(VI) and of their molecular basis, we assessed the impact of a mild Cr(VI) exposure on critical bioenergetic parameters (lactate production, oxygen consumption and intracellular ATP levels). Cells derived from normal human bronchial epithelium (BEAS-2B cell line), the main in vivo target of Cr(VI) carcinogenicity, were subjected for 48 h to 1 μM Cr(VI). We could confirm a shift to a more fermentative metabolism, resulting from the simultaneous inhibition of respiration and stimulation of glycolysis. This shift was accompanied by a decrease in the protein levels of the catalytic subunit (subunit β) of the mitochondrial H+-ATP synthase (β-F1-ATPase) and a concomitant marked increase in those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The corresponding alteration in the β-F1-ATPase/GAPDH protein ratio (viewed as a bioenergetic signature) upon Cr(VI) exposure was in agreement with the observed attenuation of cellular respiration and enhancement of glycolytic flux. Altogether, these results constitute a novel finding in terms of the molecular mechanisms of Cr(VI) effects.
1. Introduction
Heavy exposure to Cr(VI) compounds, extensively used in chemical, metallurgical and refractory industries, has been firmly associated with an increased risk of lung cancer and of a rare cancer of the sinonasal cavity [23,37]. The general population may also be at risk, as these occupational carcinogens are now widespread in the environment, due to landfill disposal of Cr(VI)-containing waste, automobile emissions and fuel combustion, amongst other causes [50]. Similar to all other cancers, Cr(VI)-induced lung cancer is likely associated with dynamic changes in the genome. Accordingly, much research effort has been devoted to the study of Cr(VI) genotoxicity. Interestingly, Cr(VI) itself does not react extensively with DNA, but its intracellular reduction generates a variety of DNA damaging species. Amongst these species are trivalent chromium [Cr(III)], the final product of this reduction [54], and strong oxidants such as tetravalent chromium [Cr(IV)], pentavalent chromium [Cr(V)] and, possibly, reactive oxygen species (ROS) [33]. Altogether, these reactive species induce a wide spectrum of potentially mutagenic DNA lesions (adducts, strand breaks, oxidized bases, abasic sites and crosslinks) and other genetic defects [49,50]. Significantly, in the TP53 gene, which encodes p53, a critical “guardian of the genome” [27], Cr(III) binds preferentially to -NGG- sequences [4]. As these are the sequences of three mutational TP53 hotspots in human lung cancer, it is conceivable that Cr(III)-DNA adducts might induce mutations in TP53 that, in turn, will facilitate the accumulation, along hundreds of generations, of further mutations in this and other genes needed for uncontrolled cellular proliferation.
Notwithstanding the fundamental role that mutations in established cancer genes play in carcinogenesis, it is becoming increasingly clear that defects in the mechanisms that govern normal cell proliferation, cell death and tissue homeostasis do not lead, per se, to tumor formation. Incipient cancer cells must also rewire their metabolism in order to meet the augmented demands for energy, reducing power and biosynthetic precursors associated with uncontrolled proliferation [20]. To this day, the best-characterized metabolic phenotype of cancer cells is a strong reliance on lactic fermentation for energy generation, despite the presence of an adequate oxygen supply [51]. First unveiled by Otto Warburg in the 1920s, this shift to “aerobic glycolysis” came to be known as the Warburg effect.
The exact molecular basis of the Warburg effect remains essentially unknown, but it has been reported that, in several common human cancers, it correlates closely with decreased levels of the mitochondrial marker β-F1-ATPase, compared to paired normal tissues [9,11,12,21,25], which suggests a loss of mitochondrial potential for energy generation. On the contrary, the same cancers exhibited frequently augmented levels of the glycolytic marker GAPDH [2,12,24,25,48], pointing to an increased glycolytic competence. Altogether, these changes translate into a lower β-F1-ATPase/GAPDH protein ratio, which has been defined a bioenergetic signature [43]. Very importantly, it was found that the in vivo glucose uptake of lung carcinomas correlated inversely with this ratio [29] and it was shown that, for some of these cancers, this bioenergetic signature might be used as a marker for cancer progression [11,12,24,28]. It was also suggested that H+-ATP synthase downregulation could be used as a predictive marker of tumor response to chemotherapy [46].
Impact of Cr(VI) exposure in mammalian cell bioenergetics remains largely unexplored. There is already evidence that it inhibits respiration and negatively affects the cellular energy status, and there are also some reports of changes in nutrient uptake and in aerobic glycolysis [1], but the establishment of Cr(VI)-induced metabolic trends and a clear understanding of their effective role in Cr(VI)-induced carcinogenesis requires much more additional work. In this study, we assessed the impact of a short-term (48 h) exposure to a low Cr(VI) concentration (1 μM) on the bioenergetic phenotype of human epithelial bronchial cells. Aiming at an elucidation of the underlying mechanisms of the changes that were observed, we also assessed the effects of this exposure on the cells’ bioenergetic signature, intracellular ATP levels and levels of oxidative stress.
2. Materials and methods
2.1. Cell culture
BEAS-2B cells, obtained from the European Collection of Animal Cell Cultures (Salisbury, UK; ECCAC no. 95102433) were used throughout this study. These cells were grown in a serum-free medium (Gibco® LHC-9 medium, from Invitrogen). Cultures were established in tissue culture-grade flasks or multiwell plates pre-coated with gelatine (from bovine skin, type B, Sigma–Aldrich) and maintained at 37 °C in a humidified atmosphere of 95% air/5% CO2. Cells were seeded at 4000 cells/cm2 and subcultured at 70–80% confluence (typically, 3–4 days after seeding). Under these culture conditions, the stationary phase of growth was never reached. Cultures used in the different experiments were in passages 11–19.
2.2. Cr(VI) exposures
In all experiments, cells were allowed to attach to the substrate for 24 h prior to the addition of Cr(VI). Final concentrations of Cr(VI), ranging between 0.1 and 2.0 μM, were achieved through addition to the growth medium of adequate volumes of filter-sterilized 5 or 50 μM K2Cr2O7 (Sigma–Aldrich) aqueous solutions. Control cultures, established and processed in parallel, received an equivalent amount of the vehicle (water).
2.3. Colony formation assay
Clonogenic potentials were determined using the colony formation assay, as described by Franken and collaborators [16]. Briefly, cells were seeded into 6-well plates (300 cells/well) and subjected to different Cr(VI) exposures. At least two cultures were prepared for each condition. Nine or ten days after seeding, colonies were fixed and stained with a solution containing 6.0% (v/v) glutaraldehyde (Sigma–Aldrich) and 0.5% (w/v) crystal violet (Sigma–Aldrich). Colonies were counted by direct observation, scoring all colonies that stained purple. For the calculation of plating efficiencies, defined as the ratio, in percentage, of number of colonies formed to number of cells seeded, the number of colonies in the replicate cultures was averaged.
2.4. Determination of protein contents
In those experiments where results were standardized to protein content, trypsinized cultures were washed with phosphate-buffered saline (PBS), resuspended in an adequate volume of lysis buffer [10 mM tris base (Sigma–Aldrich), pH 7.5; 130 mM NaCl; 1% (v/v) Triton X-100 (Merck); ethylenediaminetetracetic acid (EDTA)-free protease inhibitor cocktail (1 tablet per 0.95 mL buffer; Roche, Cat. No. 11 836 170 001)] and centrifuged at 11,000g and 4 °C for 15 min. Supernatants were transferred to new centrifuge tubes and their protein contents were determined by the Bradford method [6], using the Bio-Rad protein assay dye reagent concentrate (Bio-Rad) and bovine serum albumin as a standard.
2.5. Determination of oxygen consumption rates
Oxygen consumption rates (OCRs) were measured using the Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience). For these determinations, cells were seeded into the wells of a XF 24-well cell culture microplate (20,000 cells/well in 250 μL of medium). Eight replicate cultures were used for each condition. At the end of the Cr(VI) exposure, spent medium was replaced with 700 μL of fresh, Cr(VI)-free medium and cells were incubated at 37 °C for 60 min to allow for media temperature and pH to reach equilibrium. After obtaining the basal OCR, oligomycin (Sigma–Aldrich), 2,4-dinitrophenol (2,4-DNP, Sigma–Aldrich), rotenone (Sigma–Aldrich) and antimycin (Sigma–Aldrich) were sequentially added to each well (in 50 μL of LHC-9 medium) to final concentrations of 6 μM, 750 μM, 1 μM and 1 μM, respectively. OCRs under these different conditions were measured between additions. OCRs were expressed in pmol O2/min/μg protein.
2.6. Determination of lactate production
For these determinations, cells were seeded into 6-well plates (100,000 cells/well). Tetraplicate cultures were used for each condition. At the end of the Cr(VI) exposure, spent medium was replaced with fresh, Cr(VI)-free medium, with or without 6 μM oligomycin or 750 μM 2,4-DNP. Incubations with oligomycin lasted 4 h and those with 2,4-DNP 1 h. After the incubations, the decrease in lactate levels in the growth medium over a 4 h period was determined. To this end, 200 μL aliquots of growth medium were collected at times 0 and 4 h and treated with 6% (v/v) perchloric acid (Merck) to stop lactate dehydrogenase enzymatic activity. After 30 min on ice, the samples were centrifuged, for 5 min, at 11,000g and 4 °C. Supernatants, kept on ice, were neutralized with 20% (w/v) KOH (Merck) and centrifuged at 11,000g and 4 °C for 5 min to sediment the KClO4 salt. Lactate levels in the clear supernatants were determined using a lactate dehydrogenase (LDH)-based enzymatic assay. For standardization purposes, protein contents were determined as described in Section 2.4. The rate of lactate production was expressed in nmol lactate/h/μg protein.
2.7. Determination of intracellular ATP levels
Intracellular levels of ATP were determined enzymatically using the ATP Bioluminescence Assay Kit HS II (Roche), which is based on the ATP dependency of the light emitting oxidation of luciferin, catalyzed by luciferase [13]. Cultures were prepared in 6-well plates (seeding density of either 100,000 or 150,000 cells/well), using triplicate cultures for each condition. At the end of the Cr(VI) exposure, spent medium was replaced with fresh, Cr(VI)-free medium with or without 6 μM oligomycin, 750 μM 2,4-DNP or 100 μM 2-deoxyglucose (2-DG, Sigma–Aldrich). After a one-hour incubation, cells were collected by trypsinization and disrupted by addition of 400 μL of boiling ATP lysis buffer [100 mM tris base, pH 7.75; 4 mM EDTA]. After 2 min at 95 °C, extracts were centrifuged at 11,000g for 15 min. ATP solutions with concentrations ranging from 1.6 × 10–9 to 1.6 × 10–6 M were used as standards. Luminescence was measured using a FLUOstar OPTIMA plate luminometer (BMG Labtech). Intracellular ATP levels were expressed in nmol ATP/106 cells.
2.8. Western blot analysis
Proteins from cellular extracts were fractionated by SDS-PAGE. The fractionated proteins were transferred to a polyvinylidene fluoride (PVDF) membrane for immunoblot analysis. Mouse primary monoclonal antibodies were used for Hsp60 and GAPDH detection (Enzo Life Sciences and Abcam, respectively), whereas a polyclonal rabbit antibody was used for β-F1-ATPase detection [12]. Peroxidase-conjugated anti-mouse or anti-rabbit IgGs (Nordic Immunology) were used as secondary antibodies. The dilutions used were 1:20,000 for the primary antibodies and 1:5000 for the secondary antibodies. The blots were developed using the Novex® ECL chemiluminescent reagent kit (Invitrogen). Expression levels in control and Cr(VI)-exposed samples were normalized relative to those of the same proteins found in HCT116 cells extracted in parallel and assayed in the same gels and expressed relative to the mean value of unexposed cells.
2.9. Assessment of oxidative stress
Oxidative stress was assessed in cultures established in 6-well plates (100,000 cells/well). Tetraplicate cultures were used for each condition. After Cr(VI) exposure, control and Cr(VI)-exposed cultures were incubated for 1 h in fresh, Cr(VI)-free medium with or without 6 μM oligomycin. Cell pellets, obtained by trypsinization, were resuspended in 300 μL of a 5 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) solution (Invitrogen) and placed at 37 °C for 30 min (protected from light). The cellular suspensions were then centrifuged at 1500 rpm for 5 min and the resulting pellets were dispersed into 200 μL of a buffer containing 1.0% (v/v) fetal bovine serum (Gibco) and 0.1% (w/v) sodium azide in PBS. Propidium iodide (PI; 1 μL; Sigma–Aldrich) was added to each sample to detect dead cells. The fluorescence emitted by 2′,7′-dichlorofluorescein (DCF) and PI was detected by flow cytometry using a FACSCalibur System (BD Biosciences). The flow cytometer was equipped with two lasers, 488 nm (Blue) and 663 nm (Red), and four fluorescence detectors, FL1 (Blue, 530/30 nm), FL2 (Blue, 585/42 nm), FL3 (Blue, 670 nm LP) and FL4 (Red, 660/14 nm). DCF and PI emissions were collected through the FL1 and FL3 channels, respectively. Acquisition and analysis of gates were set using BD CellQuest Pro software. Briefly, live cells were identified based on PI dye exclusion and intact cells based on the FSC-H and SSC-H parameters. Then, DCF-positive cells were identified. At least 10,000 events were recorded in each determination.
2.10. Statistical analysis
Multiple variances (time and concentration) present in the clonogenic assays were statistically analysed with the two-way ANOVA model and the Bonferroni’s post hoc test. The results are represented as the mean ± standard error of the mean (SEM). Differences with p < 0.05 were considered statistically significant. The analysis was performed with the Graphpad Prism Software. All other results were statistically analysed with the unpaired Student’s t test, using the Excel software. The results are represented as the mean ± SEM. Differences with p < 0.05 were considered statistically significant.
3. Results
3.1. Short-term exposure to Cr(VI) in the low micromolar range had little impact on the clonogenic potential of BEAS-2B cells
Overtly cytotoxic exposure regimens are of questionable relevance in the context of carcinogenesis. To select an appropriate regimen for our evaluation of the impact of Cr(VI) on bioenergetics, we first assessed the cytotoxicity of various exposure regimens. The colony-forming assay was used to this end. As can be seen (Fig. 1), with the exception of the decreases in clonogenic potential produced by the highest Cr(VI) concentration tested (2 μM), all other decreases were rather small and devoid of statistical significance. Interestingly, after an initial decrease, the clonogenic potential of cells exposed to 0.5 μM Cr(VI) increased over time of exposure. Despite the absence of statistical significance, this increase in clonogenic potential over time for Cr(VI)-exposed cells is in line with a stimulation of proliferation suggested in two previous studies by our group with the same cell line and exposure regimen, but using different endpoints of toxicity (dehydrogenase activity [10] and trypan blue dye exclusion [15]. Cytotoxicity data reported by Caglieri and co-workers also suggested a stimulation of proliferation upon exposure to Cr(VI) in the low micromolar range [7]. Based on these results, an exposure regimen consisting of a 48 h exposure to 1 μM Cr(VI) was adopted.
Fig. 1.
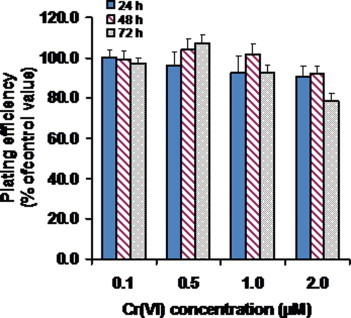
Short-term exposure (24–72 h) to Cr(VI) in the low micromolar range had little or no effect in the colony forming potential of BEAS-2B cells. Values represent the mean ± SEM of 3 independent experiments. In each experiment, at least duplicate cultures were prepared for each condition. Multiple comparisons by analysis of variance were done with two-way ANOVA model and the post hoc Bonferroni’s test confirmed that there were no significant differences (p > 0.05).
3.2. Cr(VI) compromised the respiratory capacity of BEAS-2B cells
To characterize the impact of the adopted Cr(VI) exposure regimen on cellular respiration, OCRs were determined for control and Cr(VI)-exposed cells. These determinations were carried out both under basal conditions and after sequentially exposing cells to four well-defined modulators of oxidative phosphorylation (OXPHOS): oligomycin (an inhibitor of the mitochondrial H+-ATP synthase), 2,4-DNP (which uncouples electron transport from ATP synthesis), rotenone (an inhibitor of complex I of the mitochondrial electron transport chain (ETC)) and antimycin (an inhibitor of complex III of the ETC). The real-time profiles obtained using the Seahorse XF24 Extracellular Flux Analyzer can be seen in Fig. 2A. Control and Cr(VI)-exposed cells responded identically to the three inhibitors tested, but OCRs were always slightly attenuated in Cr(VI)-exposed cells, as compared to the corresponding controls, an effect also observed under basal conditions. Differences in OCRs between control and Cr(VI)-exposed cells were much more pronounced under uncoupling conditions, i.e., when dissipation of the mitochondrial proton gradient maximized mitochondrial respiration. As can be appreciated in Fig. 2, while control cells responded to 2,4-DNP addition by a significant upregulation of mitochondrial respiration (by ca. 40%, compared to basal values), Cr(VI)-exposed cells exhibited a modest upregulation. Altogether, the results suggest that Cr(VI) compromised the respiratory capacity of BEAS-2B cells and that respiration of Cr(VI)-exposed cells under basal conditions was already close to its maximum.
Fig. 2.
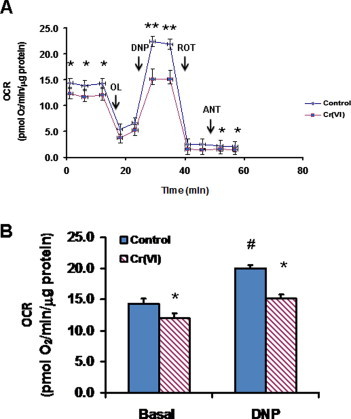
Short-term exposure (48 h) to 1 μM Cr(VI) compromised the respiratory capacity of BEAS-2B cells. (A) Bioenergetic profiles obtained using the XF24 Extracellular Flux Analyzer. The arrows represent the sequential addition of 6 μM oligomycin (OL), 750 μM 2,4-dinitrophenol (DNP), 1 μM rotenone (ROT) and 1 μM antimycin (ANT). (B) Oxygen consumption rates under basal and uncoupling conditions (i.e., after DNP addition) for control and Cr(VI)-exposed cells. Data represent the mean ± SEM for 8 determinations. Asterisks indicate p < 0.05 (*) or p < 0.01 (**), when compared with control values by Student’s t test; Cardinals indicate p < 0.05 (#) when compared with the respective basal value by Student’s t test.
3.3. Cr(VI) augmented the glycolytic capacity of BEAS-2B cells
Next, we assessed the impact of the same Cr(VI) exposure on aerobic glycolysis, by comparing the rates of lactate production by Cr(VI)-exposed cells with those of control cells. Determinations were conducted both under basal conditions and under conditions that impaired OXPHOS-mediated ATP synthesis (Fig. 3). Basal lactate production rates were slightly higher in Cr(VI)-exposed cells than in control cells. Oligomycin elicited a small stimulation of lactate production, which was essentially identical in both control and Cr(VI)-exposed cells. Once again, differences between control and Cr(VI)-exposed cells became more evident in the presence of 2,4-DNP. In fact, the upregulation of lactate production induced by this uncoupler in Cr(VI)-exposed cells was much stronger than that induced in control cells, which suggests an increased glycolytic capacity for Cr(VI)-exposed cells.
Fig. 3.
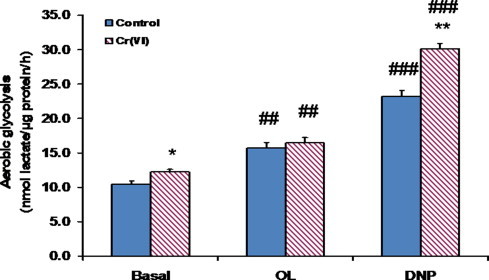
Short-term exposure (48 h) to 1 μM Cr(VI) augmented the glycolytic capacity of BEAS-2B cells under basal and uncoupling conditions. The results shown are the mean ± SEM of 4 determinations. Asterisks indicate p < 0.05 (*) or p < 0.01 (**), when compared with control values by Student’s t test; Cardinals indicate p < 0.01 (##) or p < 0.001 (###), when compared with the respective basal value by Student’s t test. OL, oligomycin; DNP, 2,4-dinitrophenol.
3.4. Intracellular ATP levels of BEAS-2B cells remained essentially unaltered upon Cr(VI) exposure
Intracellular ATP levels in control and Cr(VI)-exposed cells were determined under basal conditions and under conditions that impaired glycolytic or OXPHOS-mediated ATP synthesis (Fig. 4). Despite their attenuated OCRs, Cr(VI)-exposed cells exhibited ATP levels comparable to those of the corresponding controls under all four experimental conditions tested. The accompanying rise in aerobic glycolysis described in the previous section might have contributed to this compensatory mechanism. Inhibition of OXPHOS by oligomycin provoked a dramatic drop in ATP levels in both control and Cr(VI)-exposed cells, revealing that fermentation alone was unable to maintain basal ATP levels. This result indicates that BEAS-2B cells normally regenerate ATP mostly by electron transfer phosphorylation. An identically dramatic drop in intracellular ATP levels was observed upon incubation with 2-DG, a competitive inhibitor of hexokinase, the first enzyme of glycolysis. This observation shows that these cells were deriving most of their energy from the oxidation of glucose, and not from the oxidation of glutamine, a fuel molecule also present in the growth medium used. Although substantial, the decrease in intracellular ATP levels observed under uncoupling conditions was much less pronounced than those observed in the presence of oligomycin or 2-DG. The ability of 2,4-DNP to strongly upregulate glycolysis might have contributed to this result.
Fig. 4.
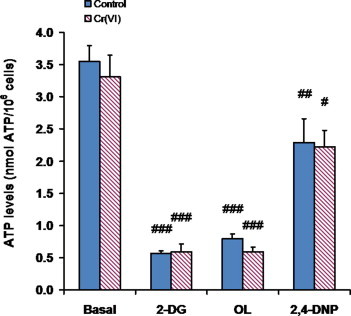
Intracellular ATP levels of BEAS-2B cells remained unaltered upon short-term exposure (48 h) to 1 μM Cr(VI), under all 4 experimental conditions tested. Values represent the means ± SEM of 3 independent experiments. Cardinals indicate p < 0.05 (#), p < 0.01 (##) or p < 0.001 (###), when compared with the respective basal value by Student’s t test. 2-DG, 2-deoxyglucose; OL, oligomycin; 2,4-DNP, 2,4-dinitrophenol.
3.5. Changes in the expression of GAPDH and β-F1-ATPase induced by Cr(VI) exposure translated into an altered bioenergetic signature
Our investigation into the mechanisms by which Cr(VI) altered the bioenergetic phenotype of BEAS-2B cells included the evaluation of the effects of this carcinogen on the protein levels of GAPDH and β-F1ATPase, which can be used as markers of glycolytic and OXPHOS activity, respectively. These determinations, which were carried out by Western blot analysis, showed that the levels of these two proteins were differentially affected by Cr(VI): whereas GAPDH levels were increased by nearly 50%, levels of β-F1ATPase were slightly decreased (Fig. 5). As a consequence, the β-F1-ATPase/GAPDH protein ratio, viewed as a cellular bioenergetic signature [43], was significantly diminished in Cr(VI)-exposed cells.
Fig. 5.
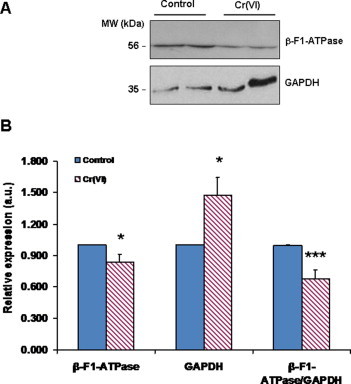
Short-term (48 h) exposure to 1 μM Cr(VI) altered the protein levels of glycolytic (GAPDH) and mitochondrial (β-F1-ATPase) markers. (A) Representative western blot of the expression of β-F1-ATPase and GAPDH in two different preparations of control and exposed cells. (B) Values represent the relative expression (i.e., compared to control cultures; mean ± SEM) of β-F1-ATPase (n = 14), GAPDH (n = 13) and β-F1-ATPase:GAPDH ratio. Asterisks indicate p < 0.05 (*) or p < 0.001 (***), when compared with control values by Student’s t test. β-F1-ATPase, catalytic subunit (subunit β) of the mitochondrial H+-ATP synthase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
3.6. Oligomycin treatment significantly increased levels of oxidative stress in Cr(VI)-exposed cells
In order to further investigate the mechanisms of Cr(VI)-induced changes in the bioenergetic phenotype of BEAS-2B cells, levels of oxidative stress were assessed, by flow cytometry and using the redox probe DCFH-DA, in control and Cr(VI)-exposed cells. These determinations were carried out both under basal conditions and in the presence of oligomycin (Fig. 6). Basal levels of oxidative stress were identical in control and Cr(VI)-exposed cells (the small increase observed upon Cr(VI) exposure was not statistically significant). However, whereas control cells were able to prevent an increase in oxidative stress when challenged by oligomycin treatment, Cr(VI)-exposed cells were not (Fig. 6B). This result might indicate a negative impact of Cr(VI) on the cellular antioxidant defenses.
Fig. 6.
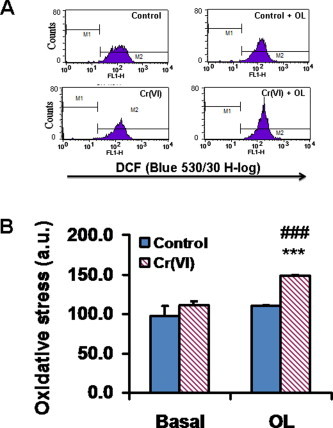
Short-term (48 h) exposure to Cr(VI) augmented oxidative stress levels in oligomycin (OL)-treated BEAS-2B cells. Values represent the mean ± SEM of four determinations. Asterisks indicate p < 0.001 (***), when compared with control values by Student’s t test; Cardinals indicate p < 0.001 (###) when compared with the respective basal value by Student’s t test.
4. Discussion
In 1990, based on extensive epidemiological, animal and other relevant studies, the International Agency for Research on Cancer (IARC) classified Cr(VI) compounds as encountered in the chromate production, chromate pigment production and chromium plating industries as carcinogenic to humans [23]. The observations made by our group upon exposure of human bronchial epithelial cells, the main targets of Cr(VI) toxicity, to low Cr(VI) concentrations are in line with this classification. These observations included changes in morphology and pattern of growth characteristic of an early phase of pre-malignant progression [10] and overexpression of genes commonly involved in malignant transformation (e.g., MYC) [40]. Yet, despite the combined efforts of many research groups, the molecular basis of Cr(VI) carcinogenicity remains elusive [49]. Namely, clarification is needed on the impact of Cr(VI) compounds on mammalian cell bioenergetics. In spite of various reports on changes in critical bioenergetic parameters induced by Cr(VI) exposure [1,49], specific Cr(VI)-induced bioenergetic phenotypes have not yet been firmly established. The main aim of the present study was to produce additional, biologically relevant data regarding this aspect.
The increased risk of chromate lung cancer is associated with the inhalation of Cr(VI) particles [5]. These particles adhere to the surface of bronchial epithelial cells and slowly release, in their close vicinity, soluble Cr(VI) oxyanions. Some of these ions, present essentially in the form of chromate, escape extracellular reduction and readily cross cell membranes [3,45]. Once in the intracellular milieu, they undergo reduction and various reactive species are formed that cause cellular damage [33,54].
To address the issue of biological relevance, we employed a cell line (BEAS-2B) that was derived from normal human bronchial epithelium. Although immortalized through viral infection, BEAS-2B cells retain features of human bronchial epithelial cells and only became tumorigenic (very weakly) at passages much higher than those used in this study [38,39]. In the present study, BEAS-2B cells were submitted to 1 μM Cr(VI) for 48 h, an exposure regimen that was not overtly cytotoxic to them, as demonstrated by us in this and previous studies [10,15], using three different endpoints of toxicity. Also, according to a study by Caglieri and collaborators, BEAS-2B cells exposed to this Cr(VI) concentration for both 8 and 24 h results exhibited an intracellular Cr content (ca. 50 μg/g of cells) [7] that compares well with the mean Cr content found by Tsunetta and co-workers in the peripheral lung tissue of chromate workers with lung cancer (36.7 μg/g wet weight) [47]. The impact of the chosen Cr(VI) exposure regimen on the bioenergetic phenotype of BEAS-2B cells was assessed in terms of cellular respiration, aerobic glycolysis and intracellular ATP levels. Determinations were carried out both under basal conditions and in the presence of well-defined bioenergetic effectors.
Bioenergetic analysis with the XF24 Extracellular Flux Analyzer demonstrated that a single insult with 1 μM Cr(VI) compromised the respiratory capacity of BEAS-2B cells. This effect was best appreciated under uncoupling conditions, which forced the ETC to operate at its maximal capacity. Inhibition of respiration occurred concurrently with a stimulation of aerobic glycolysis. Similar to its effect on respiration, the effect of Cr(VI) on aerobic glycolysis was rather modest under basal conditions or in the presence of oligomycin, but became more pronounced under uncoupling conditions, which collapsed the mitochondrial membrane potential (Δψm). It is of note that the stimulation of aerobic glycolysis produced by the uncoupler was significantly higher in Cr(VI)-exposed cells than in control cells. This observation suggests that Cr(VI) increased the maximal glycolytic capacity of BEAS-2B cells. Upregulation of aerobic glycolysis in response to Cr(VI) had been observed previously by our group in PC-12 cells (a cancer cell line) exposed for 6 h to 1–5 μM Cr(VI) [19] and in BEAS-2B cells over a 6-week exposure to 0.1 and 1.0 μM Cr(VI) [15]. In both cases, upregulation of aerobic glycolysis was accompanied by increases in glucose uptake rates.
The stronger reliance of cancer cells on a less efficient process of energy production is seemingly paradoxical, as these rapidly proliferating cells likely require more energy than their normal counterparts. Thus, the consistency of this bioenergetic phenotype among cancer cells strongly points to an impaired respiration, as proposed by Warburg, and/or to the acquisition of selective growth advantages not directly related to energy production. In spite of much speculation, these advantages remain essentially undefined. They might be related, for instance, to the generation of intermediates and reducing power for biosynthesis and/or to the acidification of the extracellular milieu [8,43]. Active biosynthesis is required to sustain proliferation and the acidification of the extracellular milieu, which accompanies lactate upregulation, has been shown to promote, in several model systems, neoangiogenesis, anchorage-independent growth, genetic instability, facilitating invasion. Interestingly, in these model systems, acidification occurred early in transformation [8]. Therefore, it is possible that the shift to a more fermentative metabolism observed in our study upon short-term incubation (48 h) of BEAS-2B cells to Cr(VI) constitutes an early step in Cr(VI)-induced lung cancer.
The ability of both control and Cr(VI)-exposed BEAS-2B cells to upregulate aerobic glycolysis in response to oligomycin-induced OXPHOS inhibition was very limited. As a consequence, their intracellular ATP levels decreased dramatically. On the contrary, these cells were able to more than double their rates of aerobic glycolysis when stressed with 2,4-DNP. Nonetheless, the upregulation of aerobic glycolysis was still insufficient to ensure basal ATP levels. Very pronounced decreases in ATP levels were also observed in the presence of 2-DG, which suggests that BEAS-2B cells are not able to use glutamine, present in the growth medium in levels analogous to those of glucose, as a major energy source. Lastly, differences in ATP levels between control and Cr(VI)-exposed cells were very modest under all four experimental conditions tested (the differences were never statistically significant).
Attenuation of respiration by Cr(VI) has been consistently reported in the literature, both in cultured cells and in isolated mitochondria [1]. However, the underlying mechanisms of this attenuation are poorly understood. It has been speculated that oxidation of NADH and NAD-linked substrates (e.g., glutamate and citrate) by Cr species might compromise substrate availability to the ETC [41,42]. It is also possible that processes of ATP generation are compromised due to the formation of stable coordination complexes between Cr(III) and ATP precursors and/or proteins involved in these processes [1]. There are, in fact, reports of changes in the activities of these enzymes upon Cr(VI) exposure, as well as changes in their intracellular levels and in the expression of the corresponding genes [1]. However, these changes were heavily dependent on the model systems employed, preventing the establishment of reliable trends. It is, nonetheless, worth mentioning that complex I of the ETC was found inhibited in six out of seven reported studies, and complex III was found inhibited in five out of six [14,17,31,32,34,52,53]. One of these studies assessed also the effects on aconitase, reporting the inhibition of this tricarboxylic acid (TCA) cycle enzyme [34]. By attenuating the TCA cycle, aconitase inhibition might slow down the production of NADH, the source of electrons to complex I, ultimately attenuating respiration. In fact, fluorocitrate, a specific inhibitor of aconitase, inhibited respiration [18].
The immunologic analysis carried out in the present study provided additional insight into the molecular basis of the observed Cr(VI)-induced bioenergetic effects. In this analysis, GAPDH and β-F1-ATPase were used as markers of glycolytic and OXPHOS activities, respectively. It is of note that β-F1-ATPase, which is the catalytic subunit of the H+-ATP synthase complex, is a rate-limiting component of OXPHOS. The results obtained are in line with the impaired respiration and augmented glycolytic potential just discussed: β-F1-ATPase proteins levels were decreased, whereas those of GAPDH were increased. Altogether, this dual effect resulted into a lower β-F1-ATPase/GAPDH protein ratio, similar to what has been described in common human cancers [2,12,25].
We next investigated the possible involvement of oxidative stress in the bioenergetic changes induced by Cr(VI) exposure, as the intracellular reduction of Cr(VI) is accompanied by the generation of strong oxidants, such as Cr(IV), Cr(V) and ROS [33]. We observed that the impact of Cr(VI) exposure on oxidative stress levels depended on the experimental conditions. Under basal conditions, these levels remained basically unaltered, indicating that the antioxidant defense system of BEAS-2B cells was able to cope with any upregulation of Cr(IV), Cr(V) and/or ROS (the probe used is not able to discriminate between these species [26]) that the Cr(VI) exposure regimen adopted might have produced. On the contrary, levels of oxidative stress were much increased when cells were also treated with oligomycin. In the presence of this specific inhibitor of the ATP synthase, which blocks its proton channel, protons tend to accumulate in the mitochondrial inter-membrane space, triggering an increase of Δψm [44]. In turn, increases in Δψm are associated with ROS formation [22]. Whereas control cells were able to maintain their levels of oxidative stress when treated with oligomycin, Cr(VI)-exposed cells did not, exhibiting much increased levels of oxidative stress in the presence of this inhibitor. This inability of Cr(VI)-exposed cells to counterbalance the increased production of ROS likely induced by oligomycin suggests that Cr(VI) compromises the antioxidant defenses of BEAS-2B cells. This is in line with previous reports of Cr(VI) interference with some components of the antioxidant defense system of mammalian cells [1], namely with the thioredoxin system [35,36], which plays a major role in the maintenance of the intracellular thiol-disulfide redox balance [30].
5. Conclusions
The results obtained in this study provided mechanistic insights into the metabolic reprogramming of lung cancer cells to an enhanced aerobic glycolysis. Specifically, our study revealed proteomic changes in Cr(VI)-exposed cells (decreased β-F1-ATPase levels and augmented GAPDH levels) that might have translated into a lower mitochondrial potential for energy generation and an increased glycolytic competence. Our results also suggest that Cr(VI) interferes with the cellular antioxidant system.
Acknowledgements
This work was supported by grants from MICINN (BFU2010-18903) and Comunidad de Madrid (S2011/BMD-2402) (from Spain, to J.M.C.), and CIMAGO (CIMAGO 26/07) and Fundação para a Ciência e a Tecnologia (FCT) (PEst-OE/QUI/UI0070/2011) (from Portugal, to A.M.U. and Unidade de Química Física Molecular, respectively).
Contributor Information
Joana F. Cerveira, Email: joana.cerveira@cancer.org.uk.
María Sánchez-Aragó, Email: msanchez@cbm.uam.es.
Ana M. Urbano, Email: amurbano@ci.uc.pt.
José M. Cuezva, Email: jmcuezva@cbm.uam.es.
References
- 1.Abreu P.L., Ferreira L.M.R., Urbano A.M. Impact of hexavalent chromium on mammalian cell bioenergetics: phenotypic changes, molecular basis and potential relevance to chromate-induced lung cancer. Biometals. 2014;27:409–443. doi: 10.1007/s10534-014-9726-7. [DOI] [PubMed] [Google Scholar]
- 2.Aldea M., Clofent J., de Arenas C.N., Chamorro M., Velasco M., Berrendero J.R., Navarro C., Cuezva J.M. Reverse phase protein microarrays quantify and validate the bioenergetic signature as biomarker in colorectal cancer. Cancer Lett. 2011;311:210–218. doi: 10.1016/j.canlet.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 3.Alexander J., Aaseth J. Uptake of chromate in human red-blood-cells and isolated rat-liver cells – the role of the anion carrier. Analyst. 1995;120:931–933. doi: 10.1039/an9952000931. [DOI] [PubMed] [Google Scholar]
- 4.Arakawa H., Wu F., Costa M., Rom W., Tang M.S. Sequence specificity of Cr(III)-DNA adduct formation in the p53 gene: NGG sequences are preferential adduct-forming sites. Carcinogenesis. 2006;27:639–645. doi: 10.1093/carcin/bgi249. [DOI] [PubMed] [Google Scholar]
- 5.Barceloux D.G. Chromium. J. Toxicol. Clin. Toxicol. 1999;37:173–194. doi: 10.1081/clt-100102418. [DOI] [PubMed] [Google Scholar]
- 6.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Caglieri A., Goldoni M., De Palma G., Mozzoni P., Gemma S., Vichi S., Testai E., Panico F., Corradi M., Tagliaferri S., Costa L.G. Exposure to low levels of hexavalent chromium: target doses and comparative effects on two human pulmonary cell lines. Acta Biomed. 2008;79(Suppl. 1):104–115. [PubMed] [Google Scholar]
- 8.Cardone R.A., Casavola V., Reshkin S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 9.Chen J., Kähne T., Röcken C., Götze T., Yu J., Sung J.J.Y., Chen M., Hu P., Malfertheiner P., Ebert M.P.A. Proteome analysis of gastric cancer metastasis by two-dimensional gel electrophoresis and matrix assisted laser desorption/ionization-mass spectrometry for identification of metastasis-related proteins. J. Proteome Res. 2004;3:1009–1016. doi: 10.1021/pr049916l. [DOI] [PubMed] [Google Scholar]
- 10.Costa A.N., Moreno V., Prieto M.J., Urbano A.M., Alpoim M.C. Induction of morphological changes in BEAS-2B human bronchial epithelial cells following chronic sub-cytotoxic and mildly cytotoxic hexavalent chromium exposures. Mol. Carcinog. 2010;49:582–591. doi: 10.1002/mc.20624. [DOI] [PubMed] [Google Scholar]
- 11.Cuezva J.M., Chen G., Alonso A.s.M., Isidoro A., Misek D.E., Hanash S.M., Beer D.G. The bioenergetic signature of lung adenocarcinomas is a molecular marker of cancer diagnosis and prognosis. Carcinogenesis. 2004;25:1157–1163. doi: 10.1093/carcin/bgh113. [DOI] [PubMed] [Google Scholar]
- 12.Cuezva J.M., Krajewska M., de Heredia M.L., Krajewski S., Santamaría G., Kim H., Zapata J.M., Marusawa H., Chamorro M., Reed J.C. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62:6674–6681. [PubMed] [Google Scholar]
- 13.Deluca M., McElroy W.D., Marlene A.D. Purification and properties of firefly luciferase. Methods Enzymol. 1978;57:3–15. [Google Scholar]
- 14.Fernandes M.A.S., Santos M.S., Alpoim M.C., Madeira V.M.C., Vicente J.A.F. Chromium(VI) interaction with plant and animal mitochondrial bioenergetics: a comparative study. J. Biochem. Mol. Toxicol. 2002;16:53–63. doi: 10.1002/jbt.10025. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira L.M.R., Guiomar A.J., Santos M.S., Alpoim M.C., Urbano A.M. Impact of carcinogenic chromium(VI) on the energy metabolism of human bronchial epithelial cells. Acta Med. Port. 2011;(Suppl. 1):P45. [Google Scholar]
- 16.Franken N.A.P., Rodermond H.M., Stap J., Haveman J., van Bree C. Clonogenic assay of cells in vitro. Nat. Protocols. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Nino W.R., Tapia E., Zazueta C., Zatarain-Barron Z.L., Hernandez-Pando R., Vega-Garcia C.C., Pedraza-Chaverri J. Curcumin pretreatment prevents potassium dichromate-induced hepatotoxicity, oxidative stress, decreased respiratory complex I activity, and membrane permeability transition pore opening. Evid. Based Complement. Alternat. Med. 2013;2013:424692. doi: 10.1155/2013/424692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner P.R., Nguyen D.D., White C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonçalves M.J., Santos A.C.C., Coelho P., Costa A.N., Rodrigues C.F.D., Santos M.S., Guiomar A.J., Alpoim M.C., Urbano A.M. Changes in glucose uptake rate and in the energy status of PC-12 cells acutely exposed to hexavalent chromium, an established human carcinogen. Toxicol. Environ. Chem. 2011;93:1202–1211. [Google Scholar]
- 20.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 21.He Q.-Y., Chen J., Kung H.-F., Yuen A.P.-W., Chiu J.-F. Identification of tumor-associated proteins in oral tongue squamous cell carcinoma by proteomics. Proteomics. 2004;4:271–278. doi: 10.1002/pmic.200300550. [DOI] [PubMed] [Google Scholar]
- 22.Hüttemann M., Lee I., Pecinova A., Pecina P., Przyklenk K., Doan J. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008;40:445–456. doi: 10.1007/s10863-008-9169-3. [DOI] [PubMed] [Google Scholar]
- 23.IARC . IARC Scientific Publications; Lyon: 1990. Chromium, Nickel and Welding. [Google Scholar]
- 24.Isidoro A., Casado E., Redondo A., Acebo P., Espinosa E., Alonso A.M., Cejas P., Hardisson D., Fresno Vara J.A., Belda-Iniesta C., González-Barón M., Cuezva J.M. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis. 2005;26:2095–2104. doi: 10.1093/carcin/bgi188. [DOI] [PubMed] [Google Scholar]
- 25.Isidoro A., Martinez M., Fernandez P.L., Ortega A.D., Santamaria G., Chamorro M., Reed J.C., Cuezva J.M. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem. J. 2004;378:17–20. doi: 10.1042/BJ20031541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalyanaraman B., Darley-Usmar V., Davies K.J.A., Dennery P.A., Forman H.J., Grisham M.B., Mann G.E., Moore K., Roberts Ii L.J., Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lane D.P. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 28.Lin P.-C., Lin J.-K., Yang S.-H., Wang H.-S., Li A.-Y., Chang S.-C. Expression of beta-F1-ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: clinical data analysis and evidence from an in vitro study. Int. J. Colorectal Dis. 2008;23:1223–1232. doi: 10.1007/s00384-008-0539-4. [DOI] [PubMed] [Google Scholar]
- 29.López-Ríos F., Sánchez-Aragó M., García-García E., Ortega Á.D., Berrendero J.R., Pozo-Rodríguez F., López-Encuentra Á., Ballestín C., Cuezva J.M. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–9017. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 30.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 31.Messer R.L.W., Doeller J.E., Kraus D.W., Lucas L.C. An investigation of fibroblast mitochondria enzyme activity and respiration in response to metallic ions released from dental alloys. J. Biomed. Mater. Res. 2000;50:598–604. doi: 10.1002/(sici)1097-4636(20000615)50:4<598::aid-jbm16>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 32.Molina-Jijon E., Tapia E., Zazueta C., El Hafidi M., Zatarain-Barron Z.L., Hernandez-Pando R., Medina-Campos O.N., Zarco-Marquez G., Torres I., Pedraza-Chaverri J. Curcumin prevents Cr(VI)-induced renal oxidant damage by a mitochondrial pathway. Free Radic. Biol. Med. 2011;51:1543–1557. doi: 10.1016/j.freeradbiomed.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 33.Myers C.R. The effects of chromium(VI) on the thioredoxin system: implications for redox regulation. Free Radic. Biol. Med. 2012;52:2091–2107. doi: 10.1016/j.freeradbiomed.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers C.R., Antholine W.E., Myers J.M. The pro-oxidant chromium(VI) inhibits mitochondrial complex I, complex II, and aconitase in the bronchial epithelium EPR markers for Fe-S proteins. Free Radic. Biol. Med. 2010;49:1903–1915. doi: 10.1016/j.freeradbiomed.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers J.M., Antholine W.E., Myers C.R. Hexavalent chromium causes the oxidation of thioredoxin in human bronchial epithelial cells. Toxicology. 2008;246:222–233. doi: 10.1016/j.tox.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers J.M., Myers C.R. The effects of hexavalent chromium on thioredoxin reductase and peroxiredoxins in human bronchial epithelial cells. Free Radic. Biol. Med. 2009;47:1477–1485. doi: 10.1016/j.freeradbiomed.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.NTP, 2011. 12th Report on Carcinogens – Chromium Hexavalent Compounds, 11th ed. U.S. Department of Health and Human Services, Public Health Service.
- 38.Reddel R.R., Ke Y., Gerwin B.I., McMenamin M.G., Lechner J.F., Su R.T., Brash D.E., Park J.B., Rhim J.S., Harris C.C. Transformation of human bronchial epithelial-cells by infection with Sv40 or adenovirus-12 Sv40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing Sv40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- 39.Reddel R.R., Salghetti S.E., Willey J.C., Ohnuki Y., Ke Y., Gerwin B.I., Lechner J.F., Harris C.C. Development of tumorigenicity in Simian Virus-40-immortalized human bronchial epithelial-cell lines. Cancer Res. 1993;53:985–991. [PubMed] [Google Scholar]
- 40.Rodrigues C.F.D., Urbano A.M., Matoso E., Carreira I., Almeida A., Santos P., Botelho F., Carvalho L., Alves M., Monteiro C., Costa A.N., Moreno V., Alpoim M.C. Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009;670:42–52. doi: 10.1016/j.mrfmmm.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Ryberg D., Alexander J. Inhibitory action of hexavalent chromium (Cr(VI)) on the mitochondrial respiration and a possible coupling to the reduction of Cr(VI) Biochem. Pharmacol. 1984;33:2461–2466. doi: 10.1016/0006-2952(84)90718-4. [DOI] [PubMed] [Google Scholar]
- 42.Ryberg D., Alexander J. Mechanisms of chromium toxicity in mitochondria. Chem. Biol. Interact. 1990;75:141–151. doi: 10.1016/0009-2797(90)90114-3. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Arago M., Formentini L., Cuezva J.M. Mitochondria-mediated energy adaption in cancer: the H+-ATP synthase-geared switch of metabolism in human tumors. Antioxid. Redox Signal. 2013;19:285–298. doi: 10.1089/ars.2012.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sánchez-Cenizo L., Formentini L., Aldea M., Ortega Ã.l.D., García-Huerta P., Sánchez-Aragó M., Cuezva J.M. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a warburg phenotype. J. Biol. Chem. 2010;285:25308–25313. doi: 10.1074/jbc.M110.146480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sehlmeyer U., Hechtenberg S., Klyszcz H., Beyersmann D. Accumulation of chromium in Chinese hamster V79-cells and nuclei. Arch. Toxicol. 1990;64:506–508. doi: 10.1007/BF01977636. [DOI] [PubMed] [Google Scholar]
- 46.Shin Y.-K., Yoo B.C., Chang H.J., Jeon E., Hong S.-H., Jung M.-S., Lim S.-J., Park J.-G. Down-regulation of mitochondrial F1F0-ATP synthase in human colon cancer cells with induced 5-fluorouracil resistance. Cancer Res. 2005;65:3162–3170. doi: 10.1158/0008-5472.CAN-04-3300. [DOI] [PubMed] [Google Scholar]
- 47.Tsuneta Y., Ohsaki Y., Kimura K., Mikami H., Abe S., Murao M. Chromium content of lungs of chromate workers with lung-cancer. Thorax. 1980;35:294–297. doi: 10.1136/thx.35.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unwin R.D., Craven R.A., Harnden P., Hanrahan S., Totty N., Knowles M., Eardley I., Selby P.J., Banks R.E. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics. 2003;3:1620–1632. doi: 10.1002/pmic.200300464. [DOI] [PubMed] [Google Scholar]
- 49.Urbano A.M., Ferreira L.M.R., Alpoim M.C. Molecular and cellular mechanisms of hexavalent chromium-induced lung cancer: an updated perspective. Curr. Drug Metab. 2012;13:284–305. doi: 10.2174/138920012799320464. [DOI] [PubMed] [Google Scholar]
- 50.Urbano A.M., Rodrigues C.F.D., Alpoim M.C. Hexavalent chromium exposure, genomic instability and lung cancer. Gene Ther. Mol. Biol. 2008;12B:219–238. [Google Scholar]
- 51.Warburg O. Constable & Co. Ltd.; London: 1930. The Metabolism of Tumors. [Google Scholar]
- 52.Xiao F., Feng X., Zeng M., Guan L., Hu Q., Zhong C. Hexavalent chromium induces energy metabolism disturbance and p53-dependent cell cycle arrest via reactive oxygen species in L-02 hepatocytes. Mol. Cell. Biochem. 2012;371:65–76. doi: 10.1007/s11010-012-1423-7. [DOI] [PubMed] [Google Scholar]
- 53.Xiao F., Li Y.H., Dai L., Deng Y.Y., Zou Y., Li P., Yang Y., Zhong C.G. Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent caspase-3 activation in L-02 hepatocytes. Int. J. Mol. Med. 2012;30:629–635. doi: 10.3892/ijmm.2012.1031. [DOI] [PubMed] [Google Scholar]
- 54.Zhitkovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium (VI) Chem. Res. Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]