

Abstract
Mice bearing germ line mutations of p53 develop sarcomas at a significant rate. Since they are susceptible to a variety of other malignancies, they are not ideally suited to the study of sarcomas. To test the possibility that targeted mutation of tumor suppressor genes in early mesenchymal cells would induce formation of sarcomas, the Prx1-cre transgenic mouse was crossed to mice-bearing floxed alleles of p53 and Rb. Mice with homozygous deletion of p53 (Prx1-cre p53lox/lox) developed sarcomas in the extremities at a mean time of 50 weeks. Osteosarcomas (OS) were the most common type of sarcoma (61%) followed by poorly differentiated soft tissue sarcomas (PDSTS) (32%). Homozygous deletion of p53 produced sarcomas significantly more rapidly than heterozygous deletion, which resulted in sarcoma formation after a mean of 96 weeks. Mice with homozygous Rb mutation (Prx1-cre Rblox/lox) developed normally and had no ostensible defects in the limbs. In contrast to p53, targeted deletion of Rb did not produce sarcomas in the limbs. However, simultaneous deletion of Rb and p53 accelerated the time to sarcoma formation, and a greater percentage of PDSTS were found. Deletion of p53 in committed osteoblasts by the Col1a1-cre transgenic mouse bearing an osteoblast-specific enhancer resulted in a high percentage of OS. These findings suggest that deletion of p53 in mesenchymal cells that give rise to osteoblasts is a powerful initiator of OS. Deletion of Rb does not initiate sarcoma formation in mice, but it accelerates formation of both soft tissue sarcomas and OS.
Introduction
The sequence of genetic events leading to the formation of sarcomas has not been well established. Although many genes have been implicated, it is not clear which mutations initiate the process and which mutations enable the disease to progress. It is also not known what cell types give rise to particular sarcomas and whether certain stages of differentiation are especially prone to formation of certain types of sarcomas.
Previous models of sarcomas based upon tumor-derived cell lines and xenografts have yielded important discoveries (1–4). However, a disadvantage of this approach is that the tumor cell lines have already acquired a large number of genetic changes. This places constraints upon the ability to study the genetic etiology of the disease, especially with regard to the early key mutations that give rise to the tumors. Mice bearing germ line deletion mutations of p53 develop osteosarcoma (OS) and soft tissue sarcomas at a significant rate (5,6). This parallels the relatively high rate of sarcomas in patients with the Li-Fraumeni syndrome (7). However, mice with global deletion of p53 suffer from two drawbacks with regard to the study of sarcomas. First, the sarcomas typically develop late in the life of heterozygous mice, well after 12 months. Second, the homozygous mutant mice die early, most commonly as a result of lymphomas and very few sarcomas are observed prior to the onset of lymphomas.
In addition to p53, the Rb gene is another important tumor suppressor gene (8–10). In humans, patients with hereditary retinoblastoma have a high incidence of OS, thereby raising the possibility that Rb may be involved in the initiation of OS (11). The study of Rb in mice has been hampered somewhat by the fact that mice bearing homozygous null mutations of Rb are not viable. It is not known whether homozygous deletion of Rb in the extremities might give rise to sarcomas. Mice bearing heterozygous mutation of Rb develop pituitary tumors but not sarcomas (12).
Tissue-specific targeted disruption of both the p53 and Rb genes may be achieved by Cre-Lox recombination of conditional alleles of the genes (13). The Prx1-cre transgenic mouse is an attractive mouse to use for sarcoma studies because it expresses Cre recombinase in the early mesenchymal tissues of embryonic limb buds (14). The primitive, undifferentiated mesenchymal cell may be a good candidate for the cell of origin of certain sarcomas. Since many sarcomas recapitulate features of mesodermally derived tissues, it is reasonable that a pluripotential mesenchymal cell may give rise to a variety of sarcomas. The Prx1-cre mouse provides a possible means of creating mutations in undifferentiated mesenchymal cells as opposed to more differentiated cells, such as osteoblasts, which may be more restricted in the types of tumors they can form. It is an attractive mouse to use since it spares the vital internal organs and theoretically enables mutations of genes to be studied that could be lethal in central organs. One of our primary goals was to test the ability of the Prx1-cre mouse to generate sarcomas and to determine the spectrum of tumors that would arise.
In this study, it was hypothesized that sarcomas could develop after targeted deletion of p53 and Rb in the mesenchymal cells of the limb bud by Prx1-cre. We show that a strategy employing the Prx1-cre transgenic mouse is capable of generating sarcomas in a high proportion of animals. Homozygous deletion of p53 produces sarcomas consistently, but homozygous deletion of Rb does not initiate formation of sarcomas in mice.
Materials and methods
Reagents
Tris, glycine, NaCl, sodium dodecyl sulfide and bovine serum albumin were purchased from Sigma–Aldrich (St Louis, MO). Rabbit polyclonal antibody to Runx2, c-Myc, Mdm-2 and p53 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against Col1 type 1 was obtained from Abcam (Cambridge, MA). Antibody against β-actins was purchased from Sigma–Aldrich.
Mice
Mice bearing floxed alleles of the Rb and p53 gene have been previously described (13). The p53lox mice carry loxP sites flanking exons 2–10 of the mouse p53 gene, and the Rblox mice carry loxP sites around exon 19. The Prx1-cre transgenic mouse has been characterized (15). This mouse was generated by a DNA construct that incorporated a 2.4 kb promoter fragment from the Prx1 gene to control expression of Cre recombinase. The Prx1-cre transgenic mouse was selected because in previous experiments (P.P.Lin, M.K.Pandey, unpublished data), we ascertained that Prx1 does affect most of the mesenchymally derived cells and this is consistent with the published reports using lacZ reporter genes that show expression of Prx1 in the mesodermally derived tissues of the limb bud (15). The Col1a1-cre transgenic mouse bearing an osteoblast-specific enhancer element has also been described (16). The Rosa26-lacZ reporter mouse (R26R) was used to confirm tissue expression of Cre recombinase (17). All mice were crossed to and maintained in a C57/Bl6 background.
Genotyping of mice
The genotypes of mice were determined by extracting DNA from standard tail preparations. The primers for verifying the genotype of the conditional alleles p53lox and Rblox have previously been published (13).
Embryo analysis for lacZ expression
Embryos at E12.5–16.5 were isolated after crossing Prx1-cre mice to R26R mice. The embryos were rinsed in phosphate-buffered saline and briefly fixed for 30 min with glutaraldehyde before staining overnight for lacZ. Seven micron thick sections were made and lightly counterstained with nuclear fast red.
Radiographs
Mice bearing tumors were analyzed by whole-body X-ray using a Faxitron machine to determine the sites of skeletal involvement.
Histopathology
After standard necropsy, the tissues and organs were fixed in 10% buffered formalin. Osseous tissues were decalcified in standard acidic decalcifying solution prior to preparation of slides. Seven micron thick sections were prepared and stained with hematoxylin and eosin.
Western blot analysis
To detect various proteins, various tissues were extracted by 0.05 ml lysis buffer containing 20 mmol/l N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4), 2 mmol/l ethylenediaminetetraacetic acid, 250 mmol/l NaCl, 0.1% NP-40, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mmol/l phenylmethylsulfonyl fluoride, 0.5 μg/ml benzamidine, 1 mmol/l dithiothreitol and 1 mmol/l sodium vanadate by incubation for 30 min on ice. The lysate was centrifuged and the supernatant was collected. Whole-cell protein (30 μg) was resolved on 10–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred onto a nitrocellulose membrane, blotted with respective antibodies and then detected by chemiluminescence (Amersham, Piscataway, NJ).
Statistical analysis
Statistical analysis was performed with the aid of the SPSS 15.0 computer software program. Statistical significance was defined as P < 0.05. The survival of animals was analyzed by Kaplan–Meier curves, and the log rank test was used to determine whether there was a difference in survival.
Results
Functional assessment of Cre recombinase expression
The expression pattern of Cre recombinase in the Prx1-cre and Col1a1-cre transgenic mice was confirmed by crossing to the R26R reporter strain, which carries the lacZ gene. For the Prx1-cre mice, strong staining for lacZ was observed throughout the mesodermal tissues of the limb bud, including the condensing mesenchyme giving rise to the long bones (Figure 1A). No staining was observed in dermal and epidermal tissues. For the Col1a1-cre mice, lacZ staining was observed in the skeletal tissues, including the cranium, facial bones, spine and limbs (Figure 1B).
Fig. 1.
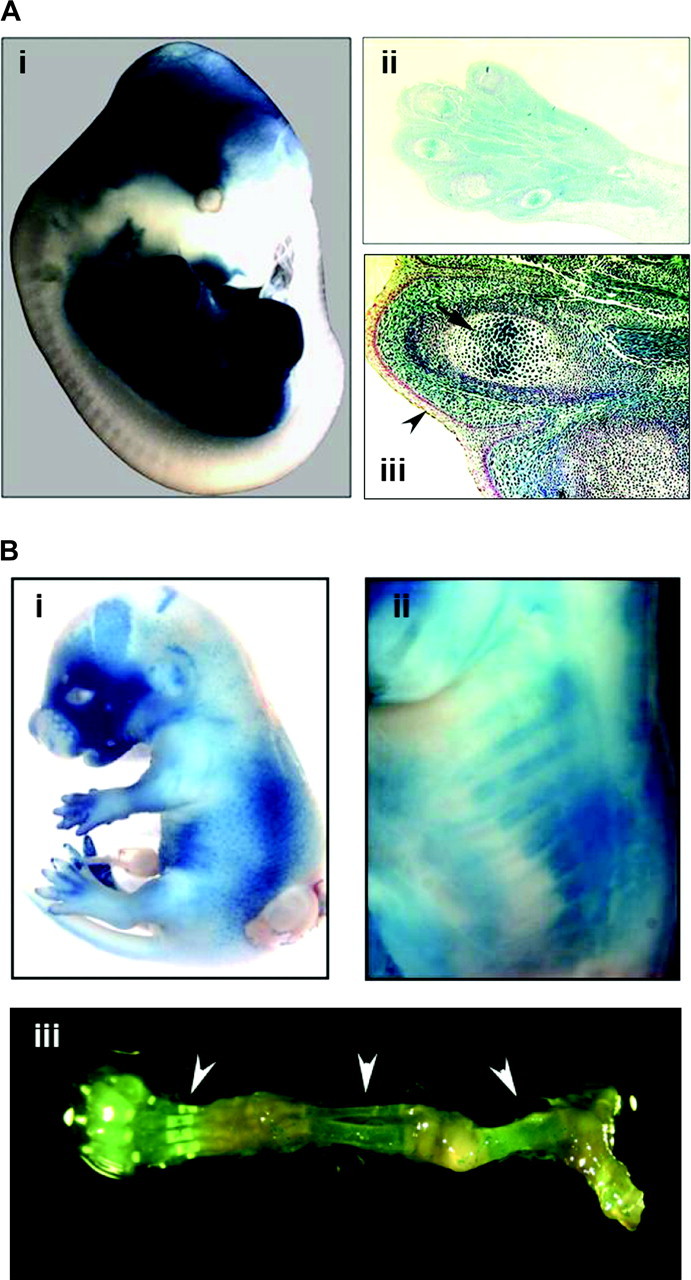
Assessment of Cre recombinase activity. (A) The Prx1-cre mouse was crossed to the Rosa26-lacZ reporter (R26R) mouse. (i) Whole-embryo staining at E13.5 for lacZ demonstrates strong staining in the limbs. Some staining is also present in the cranium, which corresponds to the normal expression pattern for the Prx1 gene. (ii) Histological examination of the upper forelimb shows lacZ staining throughout the mesodermal tissues, including the muscles, connective tissues and bones (×10 magnification). (iii) There is absence of staining of the dermal and epidermal tissues (arrowhead), but strong staining in the germinal centers of bones (arrow). Magnification is ×40. (B) LacZ staining was performed on E15.5 embryos for Col1A1-cre mice that were crossed to R26R mice. Positive staining was noted especially in the facial bones (i) and ribs (ii), which became evident after removal of skin. LacZ staining is also visible in the skeletal tissues (arrowheads) of the lower extremity of a 3 week old mouse, which has been dissected free of muscle and soft tissue (iii).
Generation of cohorts
Six cohorts of mice were generated with the following genotypes: (i) Prx1-cre Rblox/+; (ii) Prx1-cre Rblox/lox; (iii) Prx1-cre p53lox/+; (iv) Prx1-cre p53lox/lox; (v) Prx1-cre p53lox/lox Rblox/lox and (vi) Col1a1-cre p53lox/lox (Table I). There were no overt developmental abnormalities in any of the cohorts, and all the mice had a normal appearance. The limb lengths were all of normal length, and X-rays of the skeleton revealed no discernable anomalies (data not shown).
Table I.
Mouse cohorts
| Cohort | No. of mice | Sarcomas (%) | Other tumors (%) |
| Prx1-cre Rblox/+ | 10 | 0 (0%) | 0 (0%) |
| Prx1-cre Rblox/lox | 22 | 1 (5%) | 5 (23%) |
| Prx1-cre p53lox/+ | 22 | 7 (32%) | 5 (23%) |
| Prx1-cre p53lox/lox | 33 | 29 (88%) | 2 (6%) |
| Prx1-cre p53lox/lox Rblox/lox | 16 | 12 (75%) | 2 (13%) |
| Col1a1-cre p53lox/lox | 23 | 21 (91%) | 0 (0%) |
Deletion of p53 in early mesenchymal cells of the limb bud induced sarcoma formation
Conditional deletion of p53 in the early mesenchymal cells of the embryonic limb bud was achieved by crossing the Prx1-cre mouse to p53lox/lox mice. For the mice with a heterozygous deletion of p53 (Prx1-cre p53lox/+), OS arose in 22% of the mice, and soft tissue sarcomas developed in 17% of the mice. Other tumor types were observed as well, and these included thymic lymphoma, lung carcinoma, squamous cell carcinoma of the skin and mammary carcinoma (one case each).
Homozygous deletion of p53 by Prx1-cre resulted in a higher proportion of tumors being sarcomas (88%, Table I) and an acceleration of time to develop tumors. The mean time to tumor formation was 96 weeks for heterozygous deletion of p53 (Prx1-cre p53lox/+) and 50 weeks for homozygous deletion of p53 (Prx1-cre p53lox/lox, P = 0.008, Figure 2A). For the mice with homozygous deletion of p53, the most common tumor that formed was OS (Figures 2C and 3). The most frequent site of involvement was the femur (39%) followed by the tibia (22%), humerus (13%), pelvis (11%), ribs (7%), spine (4%) and scapula (4%). Multiple bone lesions were found in 17% of the mice with OS. In these mice, it was not possible to determine whether the lesions represented multiple primary tumors or metastases. Metastatic disease was identified in 24% of animals at necropsy, with the lungs being the most common site of involvement.
Fig. 2.
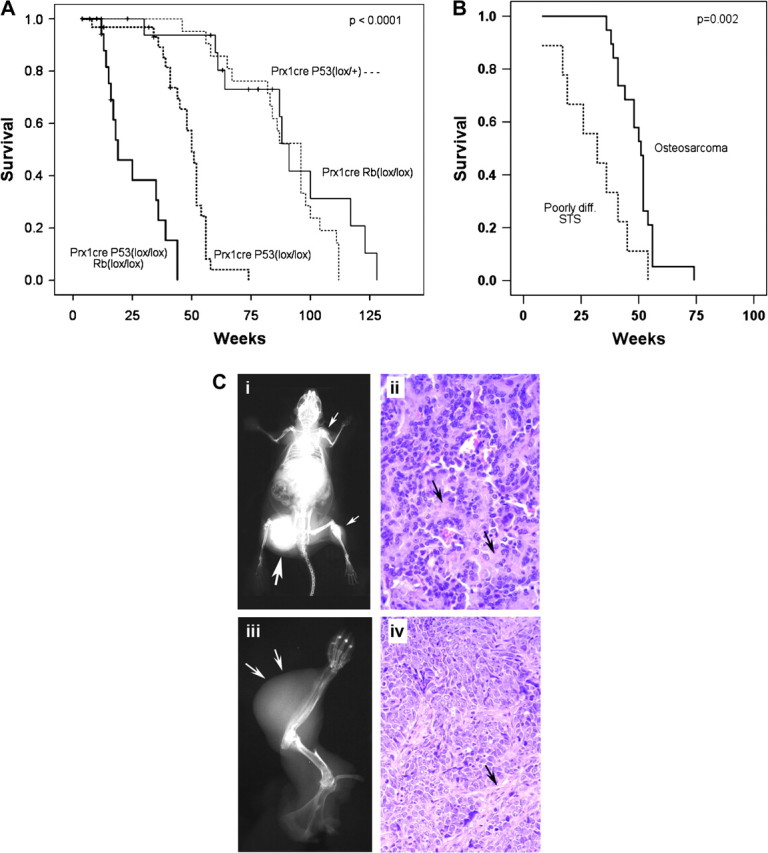
Homozygous deletion of p53 in mesenchymal cells of limb buds enhanced sarcoma formation. (A) Overall survival is shown by Kaplan–Meier analysis for mice with Prx1-cre-mediated conditional deletion of p53. The homozygous Prx1-cre p53lox/lox deletion developed tumors at a mean of 50 weeks, which was significantly earlier than the heterozygous Prx1-cre p53lox/+ mice, which developed tumors at a mean of 96 months (P = 0.008). The survival of mice with simultaneous homozygous deletion of both p53 and Rb (Prx1-cre p53lox/lox Rblox/lox) was significantly shorter than all other cohorts, with a mean time to tumor formation of 19 weeks (P < 0.0001). (B) When a subset analysis was performed for mice bearing the Prx1-cre p53lox/lox genotype, PDSTS were found to develop significantly faster than OS, with a mean time to tumor formation of 32 versus 51 weeks, respectively (P = 0.47). (C) The most common tumors generated by Prx1-cre-mediated deletion of p53 were OS (i and ii) and PDSTS (iii and iv). (i) Whole-body X-ray of a mouse with OS bearing the genotype Prx1-cre p53lox/lox demonstrates multiple osteoblastic lesions throughout the skeleton (arrows). The largest lesion is present in the femur (large arrow), which probably represents the earliest tumor to form. The other tumors may be metastatic lesions or separate primary tumors. A metastatic tumor is visible in the lower left lung. (ii) Hematoxylin and eosin (H&E) stain of a histological section of the OS arising from the right femur demonstrates abundant pink osteoid (arrows) and plump spindle-shaped sarcoma cells. (iii) A poorly differentiated soft tissue sarcoma developed in the forelimb of a mouse with the genotype Prx1-cre p53lox/lox (arrows). (iv) H&E stain shows pleomorphic spindle-shaped and compact cells without the osteoid formation that is characteristic of OS. A pink fibrovascular stroma is present (arrow), which should not be mistaken for osteoid. The photographs were taken at the magnification of ×40.
Fig. 3.

Effect of Rb deletion upon the distribution of tumor types for Prx1-cre-mediated deletion of p53. (A) Homozygous deletion of p53 alone in primitive mesenchymal cells of the limb bud by Prx1-cre resulted primarily in OS (61%) as the major tumor type, with soft tissue sarcomas representing a smaller percentage (32%) of tumors. One case of lymphosarcoma and lymphoma each were seen in this cohort. (B) Homozygous deletion of both p53 and Rb by Prx1-cre resulted in predominantly soft tissue sarcomas (57%) and a smaller fraction of OS (29%). Lymphomas were seen to a greater degree (14%).
Poorly differentiated soft tissue sarcomas (PDSTS) were the second most common tumor type, comprising 32% of the tumors (Figure 3A). These tumors, interestingly, developed at a significantly faster rate than the OS (Figure 2B). More differentiated or specific types of soft tissue sarcoma, such as rhabdomyosarcoma, were rarely seen. Lymphomas were also not frequently observed. One mouse bearing the Prx1-cre p53lox/lox genotype developed primary lymphoma of bone (lymphosarcoma) in the femur (Figure 3A). Another mouse had a lymphoma that was discovered in the spleen on necropsy. This mouse also had a simultaneous OS of the tibia and was the only mouse to have two malignancies of different histology.
Deletion of Rb did not induce formation of sarcomas
In contrast to p53 deletion, disruption of Rb by Prx1-cre in mesenchymal cells without other mutations did not produce sarcomas in the extremities. Heterozygous conditional deletion of Rb (Prx1-cre Rblox/+) did not result in any tumors. Homozygous conditional deletion of Rb (Prx1-cre Rblox/lox) did produce a few tumors by 96 weeks (Figure 2A, Table I), but none of these were in the limbs. There was one sarcoma, which developed intra-abdominally. The remaining tumors consisted of three thyroid carcinomas and one bronchioalveolar carcinoma of the lung (Table I).
Simultaneous deletion of Rb and p53 accelerates formation of sarcomas
Deletion of Rb together with p53 had two discernible effects. First, there was a significant reduction in the amount of time to the formation of sarcomas. The mean time to tumor formation was 50 weeks for p53 deletion and 19 weeks for simultaneous Rb and p53 deletion (P < 0.0001, Figure 2A). Second, deletion of Rb favored the formation of poorly differentiated sarcomas over OS. There was essentially a reversal of the ratios of these tumor types, with PDSTS comprising 57% of tumors and OS 29% of tumors (Figure 3B).
OS formation in committed osteoblasts
To determine whether sarcomas could be induced in cells committed to the osteoblast lineage, the Col1a1-cre transgenic mouse was used to mediate homozygous p53 deletion in osteoblasts. Unlike Prx1-cre, which is expressed in the early primitive mesenchyme of the limb bud, Col1a1-cre is expressed in osteoblasts since the promoter contains an osteoblast-specific enhancer. A high percentage of animals (85%) developed OS. In 15% of cases, the tumors were undifferentiated sarcomas. The mean time to tumor formation was 45 weeks for the Col1a1-cre p53lox/lox mice, which was less than the mean time of 50 weeks for Prx1-cre p53lox/lox mice (P = 0.003, Figure 4). However, when non-OS histologies were excluded, the mean time for OS formation was not statistically different for Col1a1-cre p53lox/lox mice and Prx1-cre p53lox/lox mice (Figure 4B).
Fig. 4.

Induction of OS by osteoblast-specific homozygous deletion of p53. (A) The overall survival of mice with an osteoblast-specific homozygous deletion of p53 induced by Col1a1-cre was slightly shorter than the survival of mice bearing a homozygous deletion of p53 in primitive mesenchymal cells induced by Prx1-cre. The mean time to tumor formation was 45 weeks compared with 50 weeks, respectively (P = 0.003). (B) When the survival of mice was restricted to those bearing OS, the mice with Col1a1-cre p53lox/lox genotype developed tumors at the same rate as those with Prx1-cre p53lox/lox genotype, with a mean time to tumor formation of 48 versus 51 weeks, respectively (P = 0.47). (C) Homozygous deletion of p53 in osteoblasts by Col1a1-cre resulted in primarily OS (85%) with a small fraction of undifferentiated sarcomas (15%).
Comparison of OS with poorly differentiated sarcomas
In addition to histological differences in morphology, the OS that were generated in this model differed from the PDSTS in the expression of several key genes, including several related to osteogenesis (18). We found that Mdm2 expression levels were upregulated in both PDSTS and OS, but especially for the former (Figure 5). In contrast, the levels of p53 were low in all samples. We observed that levels of Runx2 and c-Myc were also upregulated in PDSTS but not in OS samples. Expression of Col1a1 (collagen type 1, alpha 1) was found to be elevated in OS samples but not in PDSTS (Figure 5).
Fig. 5.
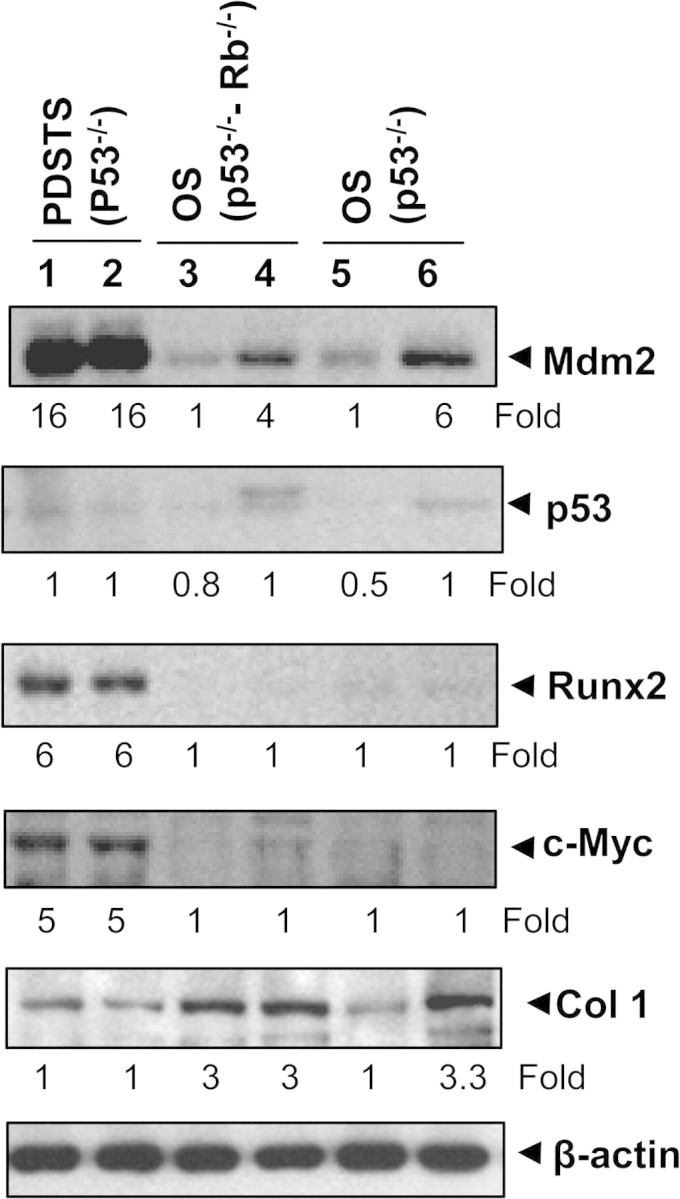
Analysis of gene products in tumor specimens. Western blot analysis showed expressions of Mdm2, p53, Runx2, c-Myc and Col1a1 in various tumor specimens. Lanes 1–2, PDSTS from two different mice with the genotype Prx1-cre p53lox/lox; lanes 3–4, OS from two different mice with the genotype Prx1-cre p53lox/lox Rblox/lox and lanes 5–6, OS from two different mice with the genotype Col1a1-cre p53lox/lox. Values under the blot indicate the fold changes.
Discussion
The results of the present study indicate that homozygous deletion of p53 in the primitive mesenchymal cells of the limb bud is a powerful initiator of sarcoma formation. Nearly, all mice with such mutations develop sarcomas. This supports the idea that pluripotential mesenchymal cells can function as precursor cells for sarcomas. A variety of sarcomas were observed, suggesting that differences in the subsequent mutations can have an important role in determining the ultimate type of the sarcoma that forms.
It was not surprising that undifferentiated soft tissue sarcomas were relatively common in the present model. These tumors accounted for approximately one-third of the malignancies that arose after Prx1-cre-mediated deletion of p53. Since the mutation occurred in the primitive mesenchymal cells, it seems plausible that persistence of the cells and failure to differentiate further would lead to the formation of undifferentiated sarcomas.
An intriguing finding was that OS comprised over half of the tumors in the Prx1-cre p53lox/lox cohort. Since deletion of p53 occurred in the undifferentiated mesenchymal cells, which had not yet differentiated into osteoblasts, this raises an important question. Does acquisition of key mutations by undifferentiated mesenchymal cells confer the OS phenotype? Alternatively, does the transformation to OS occur after the mesenchymal cell differentiates into an osteoblast? Since complete abrogation of p53 activity does not impair development of the limb, including the bones, it is possible that osteoblasts lacking p53 may continue to divide and function normally until enough mutations accumulate to create a malignant cell.
The results of our study indicate that the latter possibility can certainly occur. When homozygous deletion of p53 was induced in osteoblasts by Col1a1-cre, a high percentage of OS was obtained. Furthermore, the OS develop at nearly the identical rate as those that arise from Prx1-cre-mediated homozygous deletion of p53. The finding suggests that transformation to OS occurs after cells become committed to the osteoblast lineage and that the rate-limiting mutations for OS formation occur in the osteoblast stage.
Our results are consistent with the recently published studies of Lengner et al. (19), who also used a Col1a1-cre mouse with osteoblast specificity to delete p53 and generate OS. In two other studies, an osteoblast-specific promoter from the osterix gene was used to express Cre recombinase in transgenic mice and mediate conditional deletion of p53 and Rb in osteoblasts (20,21). OS were produced with complete penetrance in both studies. The deletion of Rb together with p53 was found to accelerate the formation of tumors and increase metastases from tumors.
This of course does not necessarily rule out the other possibility, namely that a key mutation (or mutations) of a pluripotential mesenchymal cell converts it directly to OS without the cell first having to differentiate into an osteoblast. Clear experimental data testing this concept were sparse. It is relevant to note that the study of Berman et al. (20) showed that Sca-1-positive mesenchymal stem cells bearing p53 and Rb mutations were capable of forming tumors bearing some osteoblastic markers when injected into immunocompromised mice. However, it was not clear whether the cells gradually acquired osteoblastic features or became OS-like instantaneously.
It is perhaps possible within our experimental design to test this idea more critically in the future. In contrast to the previous authors, we employed the Prx1-cre transgenic mouse, which affects an early undifferentiated cell. Further genetic manipulations of these cells, for example by crossing to floxed alleles of specific genes, might enable mutations to be tested for their ability to create other types of sarcomas. This is both an advantage and disadvantage of utilizing the Prx1-cre mouse, as opposed to a more specific tissue-restricted cre transgenic mouse, such as the Osterix-cre mouse. A wider array of sarcomas could potentially be generated with Prx1-cre, and this mouse would be ideally suited to questions regarding mutations in undifferentiated mesenchymal cells. On the other hand, if one were interested in studying a specific differentiated tumor such as OS, a more specific Cre mouse such as Osterix-cre would be more appropriate.
For the undifferentiated sarcomas, our results suggest that there may be a block to osteoblastic differentiation. The mammalian Runx genes are Runt-related transcription factors that regulate cellular differentiation and cell cycle progression. Runx2 is a critical inducer of osteoblast differentiation, and its expression induces mesenchymal stem cells to begin the progression toward the osteoblastic lineage. Runx2 also stimulates chondrocyte hypertrophy and contributes to endothelial cell migration and vascular invasion of developing bones (22). Interestingly, the level of Runx2 was found to be especially high in the poorly differentiated sarcomas of this study and elevated above the levels in OS. This may be somewhat counterintuitive since Runx2 is associated with osteoblast differentiation; however, it may be recalled that Runx2 is expressed in the pre-osteoblastic cell, and the level of expression is high in cells that have not yet acquired the full osteoblastic phenotype (23). The fact that certain tumors have a high level of expression of Runx2 without overt osteoblastic features on histology suggests that there may be a block to osteoblastic differentiation in these undifferentiated cells. Further work would be needed to determine what the mechanism of the block might be.
We found that there was an elevated expression of Mdm2 in OS and poorly differentiated sarcomas, but especially in the latter. The fact that Mdm2 was elevated despite deletion of p53 was intriguing and suggested a possible role for Mdm2 independent of p53 in some of these tumors. It may also be relevant that the studies of Lengner et al. (19) suggested that Mdm2 may be indirectly involved in the control of the expression of Runx2, and our results would be compatible with this idea.
The present model, which is based upon conditional mutation of tumor suppressor genes, appears to be attractive for the further study of early events in the process of sarcoma formation. The Prx1-cre and Col1a1-cre mice both produced tumors in the anatomic pattern expected from previous work and paralleled the pattern of lacZ staining in the R26R cross (15). For mice with homozygous p53 deletion (Prx1-cre p53lox/lox), the majority of tumors developed in the extremities. There were no carcinomas and there was only one centrally located thymic lymphoma. In the cohort involving heterozygous deletion of p53 (Prx1-cre p53lox/+), a few central tumors were observed at late age. This may be due to low-level expression of Prx1-cre in non-extremity tissues. It has been shown that Prx1 is expressed in certain adult tissues of mesodermal derivation, including heart, uterus and derivatives of the branchial arches (14,24,25). In the Col1a1-cre p53lox/lox cohort, a few cases of undifferentiated sarcomas were seen. While it is possible that the osteoblastic cells dedifferentiated in these tumors, it may also be possible that there was a low level of Cre expression in non-osteoblastic cells.
Our results confirmed the idea that sarcomas would form earlier and at a higher frequency in homozygous p53 deletion compared with heterozygous p53 deletion. This is consistent with the classic two-hit hypothesis of Knudson (26) for tumor suppressor genes. Although the observation was expected, it could not be shown in the early studies employing mice with global homozygous null deletion of p53 since they died of other causes such as lymphoma at an early age (5,6). Our findings support the use of sarcoma models based upon conditional homozygous deletion of p53, which reduces the time to generate sarcomas and increases the frequency of sarcomas.
Mutation of the Rb gene does not appear to be a probable mechanism for initiating the formation of sarcomas in mice. We did not find sarcomas when Prx1-cre was used to induce Rb mutation in mesenchymal cells. Similarly, Walkley et al. (21) did not observe OS when Rb was deleted by Osterix-cre. This is in distinct contrast to the role of Rb in the generation of pituitary tumors and medulloblastomas (12,13). It is also different from the situation in humans, who have a certain predisposition toward formation of OS in the hereditary retinoblastoma syndrome. Certain species differences may modulate the relative role of the Rb gene in the pathogenesis of sarcoma formation. Furthermore, there may be an effect of genetic background, and our experiments were limited to C57 strains. Nevertheless, it is clear that for mice, mutation of p53 is a much more powerful initiating event for sarcomas than mutation of Rb.
Mutation of the Rb gene does have two important effects upon sarcoma formation in mice. First, it accelerates the time to tumor formation from 50 to 19 weeks. A similar degree of acceleration was observed in the formation of OS by previous authors (20,21). This may have important considerations for therapeutic murine studies that rely upon the rapid generation of tumors. However, we did note that reproductive fertility of mice bearing the genotype Prx1-cre p53lox/lox Rblox/lox seemed markedly diminished, and this made it difficult to generate mice for the cohort. The effect may be specific for the Prx1-cre transgenic mouse since we have been able to generate p53/Rb double mutations more readily with other cre transgenic mice (P.P.Lin, M.K.Pandey, unpublished data). The other major effect of Rb mutation is that the spectrum of tumors is shifted more toward undifferentiated sarcomas. Although OS were observed, they no longer represented the predominant histological tumor type. The finding suggests that the mutation of Rb may have effects on cell differentiation that help determine tumor type. It is possible that Rb mutation may play a role in blocking osteoblastic differentiation, and some recent studies have shown an effect of Rb mutation on osteoblastic differentiation (27,28), but more work would be needed to address this question.
In conclusion, conditional homozygous deletion of p53 in mice bearing the genotype Prx1-cre p53lox/lox causes sarcomas to develop in a consistent manner. Mutation of p53 in primitive mesenchymal cells appears to be one general mechanism for the initiation of sarcomas. This is in distinct contrast to the Rb gene, which was not found to be effective at inducing sarcoma formation. Deletion of Rb did have some important effects, including acceleration of the time to formation of sarcomas and an increase in the number of undifferentiated soft tissue sarcomas as opposed to OS. Homozygous deletion of p53 in committed osteoblasts led to a high percentage of animals developing OS, suggesting that osteoblasts can act as the primary cell of origin for OS.
Funding
Texas ARP/ATP Program; Edward B. Heard Fund; Orthopaedic Research and Education Foundation.
Acknowledgments
The authors would like to thank James Martin for the Prx1-cre transgenic mouse and Anton Berns for the mice bearing the conditional alleles p53lox/lox and Rblox/lox. Histopathological preparation and analysis was performed in part by Carolyn Van Pelt. The authors are grateful to Terri Robinson for assistance with preparation of the manuscript.
Glossary
Abbreviation
- OS
osteosarcomas
References
- 1.Kirsch DG, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 2.Sommer G, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc. Natl Acad. Sci. USA. 2003;100:6706–6711. doi: 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubin BP, et al. A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E. Cancer Res. 2005;65:6631–6639. doi: 10.1158/0008-5472.CAN-05-0891. [DOI] [PubMed] [Google Scholar]
- 4.Nakai N, et al. A mouse model of a human multiple GIST family with KIT-Asp820Tyr mutation generated by a knock-in strategy. J. Pathol. 2008;214:302–311. doi: 10.1002/path.2296. [DOI] [PubMed] [Google Scholar]
- 5.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 6.Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin. Cancer Biol. 1996;7:269–278. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 7.Li FP, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 8.Jacks T, et al. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [see comments] [DOI] [PubMed] [Google Scholar]
- 9.Lee EY, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [see comment] [DOI] [PubMed] [Google Scholar]
- 10.Clarke AR, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [see comment] [DOI] [PubMed] [Google Scholar]
- 11.Draper GJ, et al. Second primary neoplasms in patients with retinoblastoma. Br. J. Cancer. 1986;53:661–671. doi: 10.1038/bjc.1986.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu N, et al. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–1027. [PubMed] [Google Scholar]
- 13.Marino S, et al. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 14.Martin JF, et al. Identification of a prx1 limb enhancer. Genesis J. Genet. Dev. 2000;26:225–229. [PubMed] [Google Scholar]
- 15.Logan M, et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis J. Genet. Dev. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 16.Nakanishi R, et al. Osteoblast-targeted expression of Sfrp4 in mice results in low bone mass. J. Bone Miner. Res. 2008;23:271–277. doi: 10.1359/jbmr.071007. [DOI] [PubMed] [Google Scholar]
- 17.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 18.Zamurovic N, et al. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J. Biol. Chem. 2004;279:37704–37715. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- 19.Lengner CJ, et al. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J. Cell Biol. 2006;172:909–921. doi: 10.1083/jcb.200508130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman SD, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc. Natl Acad. Sci. USA. 2008;105:11851–11856. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walkley CR, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008;22:1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein GS, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23:4315–4329. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 23.Galindo M, et al. The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J. Biol. Chem. 2005;280:20274–20285. doi: 10.1074/jbc.M413665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cserjesi P, et al. MHox: a mesodermally restricted homeodomain protein that binds an essential site in the muscle creatine kinase enhancer. Development. 1992;115:1087–1101. doi: 10.1242/dev.115.4.1087. [DOI] [PubMed] [Google Scholar]
- 25.Leussink B, et al. Expression patterns of the paired-related homeobox genes MHox/Prx1 and S8/Prx2 suggest roles in development of the heart and the forebrain. Mech. Dev. 1995;52:51–64. doi: 10.1016/0925-4773(95)00389-i. [DOI] [PubMed] [Google Scholar]
- 26.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berman SD, et al. The retinoblastoma protein tumor suppressor is important for appropriate osteoblast differentiation and bone development. Mol. Cancer Res. 2008;6:1440–1451. doi: 10.1158/1541-7786.MCR-08-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gutierrez GM, et al. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1-/- mice. Proc. Natl Acad. Sci. USA. 2008;105:18402–18407. doi: 10.1073/pnas.0805925105. [DOI] [PMC free article] [PubMed] [Google Scholar]