

Abstract
Mammalian target of rapamycin (mTOR) is a kinase protein involved in PI3K/AKT signaling with a central role in the processes of cell growth, survival and angiogenesis. Frequent mutations of this pathway make upstream and downstream components novel targets for tailored therapy design. Two mTOR inhibitors – everolimus and temsirolimus - enable an increase in overall survival (OS) or progression-free survival (PFS) time in a treatment of renal cancer. Despite recent advances in renal cancer treatment, resistance to targeted therapy is common. Understanding of molecular mechanisms is the basis of drug resistance which can facilitate prediction of success or failure in combinational or sequential targeted therapy. The article provides current knowledge on the mTOR signaling network and gives insight into the mechanisms of resistance to mTOR inhibitors from the complex perspective of RCC biology. The mechanisms of resistance developed not only by cancer cells, but also by interactions with tumor microenvironment are analyzed to emphasize the role of angiogenesis in ccRCC pathogenesis. As recent studies have shown the role of PI3K/AKT-mTOR pathway in proliferation and differentiation of cancer stem cells, we discuss cancer stem cell hypothesis and its possible contribution to ccRCC resistance. In the context of drug resistance, we also elaborate on a new approach considering ccRCC as a metabolic disease. In conclusion we speculate on future developments in agents targeting the mTOR pathway taking into consideration the singular biology of ccRCC.
Keywords: Anti-angiogenic therapy, cancer stem cells, clear cell renal cell carcinoma, drug resistance, dual mTOR inhibitors, everolimus, temsirolimus, tumor microenvironment
Introduction
Renal cell carcinoma (RCC) is the most common type of kidney cancer. As it is not a single entity, but rather a group of histologically, clinically and genetically unique cancers, we focus mainly on clear cell renal cell carcinoma (ccRCC), which is its most prevalent form. ccRCC is among the most resistant tumors to traditional chemotherapy, radiotherapy, and hormone therapy, because of its primary drug resistance driven by multi drug resistance mechanism [1]. Moreover, in most cases, it lacks the genetic hallmarks of solid tumors, such as TP53 and KRAS mutations [2]. Therefore, the classical approaches and standard paradigms of cancer therapy are irrelevant.
Recent achievements in translational research, enabled by advances in genomic biology, have changed the therapeutic perspective for patients with ccRCC. The recognition of the role of hypoxia inducible factor - 1 alpha (HIF 1α) in the pathogenesis of RCC [3], has led to development of angiogenesis inhibitors [2]. HIF 1α is a transcription factor expressed in response to changes in oxygen conditions and its role is to regulate expression of vascular endothelial growth factor (VEGF) - the main mediator of angiogenesis in ccRCC [4]. Angiogenesis inhibitors are unique anticancer agents as they prevent the formation of blood vessels rather than proliferation of tumor cells. To date, there are several registered and clinically evaluated angiogenesis inhibitors exhibiting different mechanisms of action: monoclonal antibody blocking directly VEGF ligand (bevacizumab [5]), tyrosine kinase inhibitors blocking VEGF receptors (sorafenib [6], sunitinib [7], pazopanib [8], axitinib[9]) and inhibitors of the intracellular mammalian target of rapamycin (mTOR) kinase (temsirolimus [10] and everolimus [11]).
In this review we focus on mTOR inhibitors. Targeting the mTOR pathway may be crucial as its activation leads to constitutive HIF-1α expression [12]. Moreover the rationale for the development of mTOR inhibitors have been supported by their potential to inhibit both tumor cell proliferation and angiogenesis, as mTOR signaling pathway is hyperactivated both in cancer and epithelial cells [13]. Recently, in a comprehensive molecular characterization of 400 RCC tumor samples using different genomic platforms, 19 significantly mutated genes were identified in addition to showing a deregulated PIK3/AKT pathway [14]. Here we focus only on ccRCC, however mTOR inhibitors also could be used in chromophobe RCC [15].
Despite solid rationale for developing mTOR inhibitors, existing clinical data have unsatisfactory preclinical results, even with combination therapy [16]. Temsirolimus improves overall survival (OS), but it refers only to minority of selected patients (first-line therapy for patients with poor-risk features [17]). The clinical utility of everolimus in the refractory setting characterizes 36% of 6-month progression free survival (PFS) rate and 31% of 3-month PFS rate [18]. Because responses to the current mTOR agents are not long lasting and most of the patients experience progression of the disease while on treatment [19], identification of the mechanisms of resistance to mTOR inhibitors in ccRCC is critical to improve clinical outcomes.
ccRCC constitutes a comprehensive model to study mechanisms of drug resistance, but its unique features and biological complexity pose challenges to design effective classical and angiogenic therapy. Efforts are now in place to develop compounds inhibiting mTOR and the its upstream signaling pathways. The upstream targets were elucidated after identification of drug resistance mechanisms connected with mTOR signaling. While the exact mechanisms for resistance have yet to be uncovered, several possibilities have already been proposed [19].
In this review, we provide insights into the complex mechanisms of mTOR inhibitors resistance in the context of ccRCC biology. The first part of the article brings together current data on signaling pathway of mTOR and mechanisms of resistance to mTOR inhibitors in ccRCC. In the second part, we analyze potential resistance mechanisms related to cancer niche emphasizing role of angiogenesis in the process of ccRCC development, and provide characterization of cancer stem cells and its possible contribution to cancer resistance. We also elaborate on novel approaches showing ccRCC as a metabolic disease in the context of drug resistance. We finally speculate on future developments of agents targeting mTOR pathway while taking into consideration the singular biology of ccRCC.
Molecular basis of mTOR signaling network and insight in the mechanisms of resistance to mTOR inhibitors
The process of activation and modulation of the mTOR pathway together with its downstream effects involves a series of molecular interactions linking growth factor receptors and intracellular regulatory factors in the processes of angiogenesis, tumorigenesis and cellular metabolism. Proteins upstream and downstream of mTOR are the nodes of a complex signaling network, but the linkage between many of them remains to be discovered to fully understand interactions leading to drug resistance. In this part of the article we summarize current knowledge on selected pathways directly connected to elucidated mechanisms which has been shown to potentially contribute to resistance to mTOR inhibitors in ccRCC cells. We feature the signaling network derived from the data (see Fig. 1) to provide a clear point of reference through analysis of several mechanisms reviewed here.
Fig. (1).
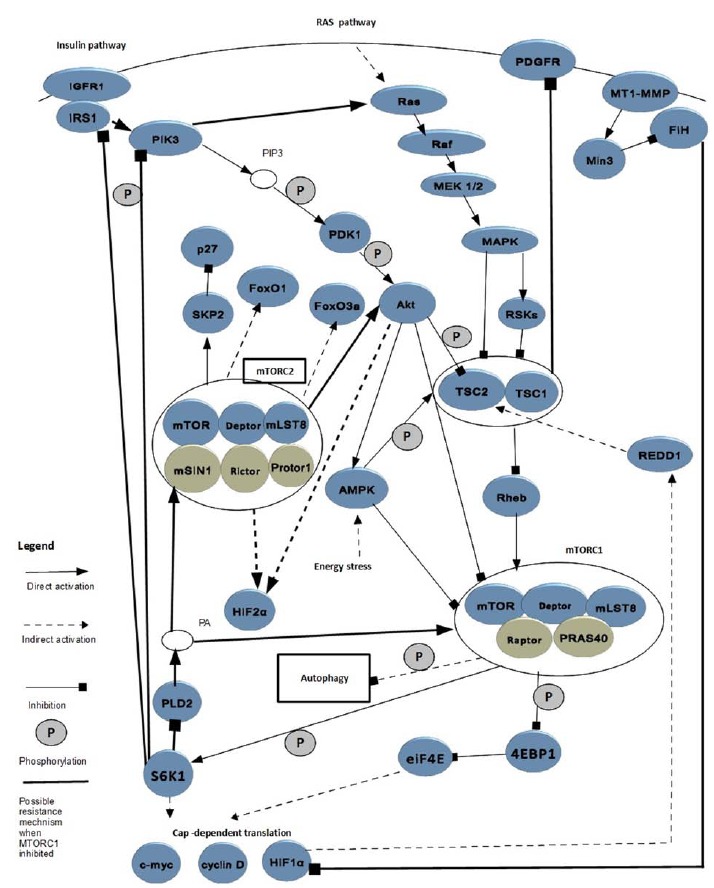
Partial mTOR signaling network with pathways of potential contribution to mTOR inhibitors resistance.
The basis to understanding mechanisms of resistance is to recognize the difference between two mTOR complexes. mTOR is a 289 kDa serine/threonine kinase and is classified as a member of the large phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family [20]. However, there are two different mTOR complexes, mTORC1 and mTORC2. Both of them consist of: mTOR, DEP-domain-containing mTOR-interacting protein (Deptor) and mammalian lethal with Sec13 protein 8 (mLST8) [21]. mTORC1 comprises 2 distinct components: regulatory associated protein of mTOR (Raptor) and proline-rich AKT substrate 40 kDa (PRAS40); whereas mTORC2 consists of 3 other proteins: mammalian stress-activated protein kinase interacting protein (mSIN1) and protein observed with Rictor-1 (Protor-1) [21]. It is important to notice that mTORC1 is sensitive to mTOR inhibitors, in contrary to mTORC2 which is largely resistant [19].
It is also useful to distinguish two basic signal cascades leading to mTOR1 activation. The first one occurs via the insulin - and the second one via Ras – pathway [21]. The first stage of the insulin signaling cascade involves binding of insulin to its receptor which displays tyrosine kinase activity towards insulin receptor substrate 1 (IRS1) [22]. When IRS1 is recruited and activated, the signal is transduced via the activation of phosphatidylinositide 3-kinase (PI3K), which subsequently activates phosphoinositide-dependent kinase-1 (PDK1) and then Akt [23]. The second pathway, which starts from Ras activation, involves signal transduction via Raf and then MEK 1/2 to mitogen-activated protein kinase (MAPK) and ribosomal s6 kinases (RSKs) [24, 25]. The stimulation of these two pathways by growth factors increases the phosphorylation of tuberin (TSC2) and this way inactivate hamartin-tuberin complex. In the insulin pathway phosphorylation is driven by AKT [23] and in the RAS pathway signal transduction is mediated by MAPK also known as extracellular signal-regulated kinase 1/2 (ERK1/2) [24]. Inactivation of TSC1/2 leads to mTORC1 activation [21]. More precisely, mTORC1 is under control of TSC1/2 complex [25] through its GTPase-activating protein activity towards the G-protein Ras homologue enriched in brain (Rheb) [25]. When TSC1 is inactivated, Rheb level is increased and activation of mTOR pathway occurs [29]. Therefore, TSC1 is perceived as a critical negative regulator of mTORC1 [25]. Regardless this core pathway, the AKT is able to activate mTORC1 in a TSC1/2 nondependent manner. The mechanism involves prolinerich Akt substrate 40 (PRAS40), which regulates mTORC1 by functioning as a direct inhibitor of substrate binding [30] .
There are two main downstream targets of mTORC1 activity: the eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the p70 ribosomal S6 kinase 1 (S6K1). The phosphorylation of 4E-BP1 prevents its binding to eIF4E, enabling eIF4E to promote cap-dependent translation [30]. Translation is also stimulated by S6K1 [31]. These processes lead to production of HIF 1α and cell-cycle regulators: c-myc and cyclin D1 [32]. In ccRCC, the von Hippel-Lindau gene (VHL) is frequently inactivated leading to constitutive activation of HIF-1 α and/or HIF-2α, which may promote up-regulation of REDD1 (regulated in development and DNA damage response 1)) and this way inhibit mTORC1. However, paradoxically, mTORC1 is frequently activated in ccRCC [33]. The relative therapeutic significance of HIF1α regulation via mTORC1 versus alternative downstream effects should be further investigated in ccRCC [32].
Important to notice are crosstalks between elements of PI3K/AKT axis which leads to mTORC1 activation with the second mTOR complex. Full activation of AKT requires its phosphorylation not only by PDK1, but also by mTORC2 [34]. Recently, additional genetic evidence has been provided that PI3K signaling activates mTORC2 [35]. Thus, interactions between mTOR complexes seem to be bidirectional.
With understanding of the basic components of this complex network, it is easier to analyze the mechanism of action of currently used mTOR inhibitors in RCC in the context of drug resistance. mTOR inhibitors form a complex with the FK binding protein (FKBP) target Raptor and prevent activation of mTORC1 [32]. Because inhibition of mTORC2 activity requires significantly higher doses than mTORC1, mTOR inhibitors are perceived as selective allosteric inhibitors of mTORC1 [19]. Although it has been suggested that the effects of mTOR inhibition are enhanced in VHL −/− cells, other data indicate that mTOR inhibitors are more effective in non-ccRCC implying that direct anti-tumor rather than anti-angiogenic effects might be the dominant component of mTOR inhibitor activity [32]. In the next subsections, we describe in detail several mechanisms of resistance to mTOR inhibitors connected with specific molecular pathways and phenomena.
Feedback Loops Activation
The loss of negative feedback loops may be an important mechanism of resistance to mTOR inhibitors [36]. Several feedback loops in which activation of mTORC1 strongly suppresses the PI3K-AKT axis have been found. For example, the negative feedback loop in which S6K1 promotes the phosphorylation of IRS1 and this way reduces its stability [37]. S6K1 also activates the feedback loop, which inactivates phospholipase D2 (PLD2 and this way decreases the phosphatidic acid (PA) level [38], small molecule compound which is necessary for mTORC1 and mTORC2 association [39]. Another pathway is mTORC1-MAPK feedback loop depending on S6K/PI3-K/Ras signaling. In accordance with the claim that the crosstalk between Ras and insulin pathway are to be elucidated, it has been shown that inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The most probable explanation is that mTORC1 inhibition increases IRS-1 and subsequently PI3K activity toward Ras and MAPK, therefore promoting both AKT activation and MAPK phosphorylation in what constitutes a dual feedback mechanism [36].
MAPK Up-regulation and Protective Autophagy
Recently it has been shown that MAPK undergoes ubiquitin-proteasome degradation through a prolyl hydroxylation-dependent mechanism in a process mediated by VHL [40]. They concluded that MAPK is a novel target of VHL [40]. Not only activation of feedback loops but also VHL mutation itself may up-regulate MAPK. This seems to be especially important when speculating on mechanisms of mTOR inhibitors resistance as it has been shown that tumor specimens from both patients and animal models treated with everolimus can be characterized with increased activation of MAPK, the protein, which is implicated in proliferation and angiogenesis [36]. Furthermore, high levels of MAPK correlate with advanced disease in ccRCC [40].
Interesting conclusions comes also with the research on β-Elemene - natural sesquiterpene with potential anti-tumor activity [41]. It has been observed that the compound inhibits the viability of RCC cells in a dose- and time-dependent manner via induction of apoptosis as well as protective autophagy [42]. Further experiments have shown that β-Elemene inhibits the MAPK/ERK as well as PI3K/Akt/ mTOR signaling pathways, and combined treatment with autophagy inhibitors increases the anti-tumor activity [42]. Therefore, inhibition of autophagy might be a useful strategy to enhance the anti-tumor effect on RCC cells and overcome mTOR resistance. In other studies it has been shown that mTORC1 inhibition increases autophagy [43]. However it has been concluded that mTORC1 controls autophagy through an unknown mechanism that is essentially insensitive to inhibition by mTOR inhibitors [44]. Whether resistance to mTOR inhibitors connects both pathways of autophagy and MAPK signaling remains to be determined.
Driving Resistance by mTORC2 Open Gate
There is one noticeable vulnerability in the classical mTOR inhibitors mechanism of action related to its selectivity to mTORC1. Data suggest that when inhibition of mTORC1 up-regulates PI3K and AKT [26], resistance can be potentially driven by the activation of mTORC2 [32]. mTORC2 activates AKT and HIF2α [45] and thus the anti-tumor effect achieved with administration of mTORC1 inhibitors is reduced [32].
To understand its profound reason is worth to notice that although HIF1α is dependent on both mTORC1 and mTORC2, the expression of HIF-2α is dependent only on mTORC2 activity [45]. Simultaneously, it has been shown that a substantial fraction of VHL−/− RCCs express only HIF-2α [46]. Moreover, the conclusion that the expression of HIF2α alpha is dependent on Akt2 and HIF1α on Akt3 was drawn from experimental research [45]. Because HIF-2α may be critical for RCC pathogenesis, the authors of this study emphasized the importance of targeting mTORC2 and perhaps Akt2 in RCC [45]. The inability of selective mTORC1 inhibitors to decrease HIF-2α expression is a possible mechanism for resistance.
More insight into the mechanisms of mTOR resistance provides the research of both upstream (AKT and MAPK) and downstream (S6K) nodes of the mTOR network in resistant and sensitive RCC cell line compared before and after treatment with temsirolimus [47]. It has been shown that phosphorylation of S6K in the non-resistant cell line was significantly inhibited, whereas there was no expression of phosphorylated S6K in resistant cell line before and after temsirolimus exposure [47]. The second observation was that both AKT and MAPK were constitutively phosphorylated even after temsirolimus treatment in both cell lines [47]. There was no difference in sensitivity of both cell lines to a dual inhibitor of mTORC1/2, but it was found that sensitivity to temsirolimus in resistant and non-resistant cell line could be achieved by additional treatment with specific inhibitors of the AKT- and MAPK-signaling pathways [47]. The authors concluded that activation of signal transduction pathways via mTORC2, but not via mTORC1, may have an important role in the acquisition of a resistant phenotype to temsirolimus in RCC [47]. Moreover another study has revealed effectiveness of the dual mTORC1/2 inhibitor which could be related to inhibition of autophagy in contrary to temsirolimus effects alone [46]. This suggests that modulation of autophagy is under the control of mTORC2.
In addition, a role for mTORC2 in the regulation of S-phase kinase-associated protein 2 (SKP-2) has been demonstrated. It has been elucidated that mTORC2 signaling decreases the level of cyclin-dependent kinase inhibitor - p27 through the increase in the expression of SKP-2.
As p27 has a prognostic value for RCC, involvement of mTORC2 signaling in the regulation of the SKP-2/p27 axis indicates next signaling node altered in RCC [35] which might constitute an additional strategy developed by cancer cells to circumvent mTORC1 inhibitor effects.
Differences in Sensitivity of Executory Proteins
The mTOR inhibitors exert not equal effect on the primary executors of mTORC1, S6K, and 4E-BP1 [31]. It has been shown that while the phosphorylation of S6K is extremely sensitive to inhibition and durable during treatment, suppression of 4E-BP1 phosphorylation in some cells is reversed within 6 hours of drug exposure despite initial inhibition (1-3 h) [31]. This reemerged 4E-BP1 phosphorylation is resistant to mTOR inhibition, the process still requires mTORC1 activity. Moreover, the ability to stimulate cap-dependent translation varies in different cell types [31]. These data taken together explains how cap-dependent translation can be maintained when mTORC1 is inhibited [31] and indicate a possible mechanism of resistance to mTOR inhibitors.
PLD2 Overexpression
Both PLD2 and its metabolite PA have been implicated in the regulation of mTOR [48]. It has been reported that suppression of PLD prevents phosphorylation of the mTORC1 substrate S6K and the mTORC2-dependent phosphorylation of PRAS40 [39]. PA is necessary for the association of mTORC1 and mTORC2 [39]. Inhibition of PA generation significantly increases the sensitivity of mTORC2 to rapamycin. RCC has been shown to overexpress PLD2 [38], but whether the differential expression of PD2 can predict response to mTOR inhibitors remains to be investigated [39].
Increased Survivin Expression
Survivin, a member of the inhibitor of apoptosis family, may have a role in resistance to mTOR inhibitors. To determine whether survivin expression modulates apoptosis induced by temsirolimus, RNA silencing of its expression was performed in two RCC cell lines (786-O and Caki-1). It has been shown that knockdown of survivin enhanced the anti-cancer activity of temsirolimus [49]. Survivin expression may be related to resistance mechanisms upstream mTORC1. One of the latest studies supports this claim showing that a novel histone deacetylase inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and X-linked inhibitor of apoptosis protein (XIAP) in RCC [50]. The combination of drugs activated caspase-3, -8 and -9 and induced intracellular reactive oxygen species, however Bcl-2 family members were not altered [50].
Activation of Potential Suppressor Gens
Interestingly, the results of sequencing analyses of 77 ccRCC samples for mutations in TSC1, TSC2 and REDD1 demonstrated that the TSC1 gene, and potentially REDD1 could be novel suppressor genes in ccRCC [33]. Whereas mTORC1 is inhibited by REDD1 in some tumors, in others, mechanisms have evolved to uncouple mTORC1 from REDD1 inhibition. One such mechanism is based on the disassociation of the TSC1/TSC2 complex. The authors have indicated there is a possibility that inactivation of a single TSC1 allele may activate mTORC1 and triggers its unresponsiveness to REDD1. Then modest depletion of TSC2 may be sufficient to prevent REDD1-induced mTORC1 inhibition [33]
However, the frequency of mutations in TSC1 has proven to be low [33, 51]. Therefore other mechanisms preventing mTORC1 inhibition by REDD1 are to be determined. Recently, mutations in the MTOR gene itself were identified in ccRCC [52]. In the mutants mTORC1 is highly activated, but MTOR mutations are also rare [33]. Understanding how ccRCCs become refractory to REDD1-induced mTORC1 inhibition should shed light into the potential mechanism of resistance for mTOR inhibitors in ccRCC [33].
Moreover, there is an interesting observation contributing to understanding mechanisms of resistance that ccRCC cells with intact VHL, as well as with VHL -/- expressing HIF-1/2α, exhibited enhanced Akt/mTOR and MAPK signaling. In contrast, cells with VHL -/- expressing only HIF-2 displayed elevated c-Myc activity, enhancing proliferation of cells and resistance [46]. Different strategies of breaking drug resistance might be necessary depending on mutation of VHL and HIF encoding genes. Table 1 indicates genes mutated in ccRCC cell lines resistant mTOR inhibitors as potential targets for combination therapy
Table 1. Impact of the mutated genes on mTOR network.
| Gene | Gene Function | Abnormality in Resistance | Cell Type | Impact on mTOR Network | Ref. |
|---|---|---|---|---|---|
| VHL | Tumor supressor gene | Inactivating mutation | ccRCC | Defective proteolysis of HIF1 α | [40] |
| MAPK upregulation (possible) | [40] | ||||
| IGFR1 upregulation | [55] | ||||
| Akt upregulation | [55] | ||||
| MTOR | Proliferation, angiogenesis, cell metabolism | Activating mutation | ccRCC | MTORC1 highly activated | [33] |
| TSC1 | Tumor supressor gene (possible) | Inactivating mutation | ccRCC | Uncoupling mTORC1 from REDD1 inhibtion | [33] |
| mouse embryonic fibroblasts | PDGFR upregulation | [63] | |||
| TSC2 | Tumor supressor gene (possible) | Inactivating mutation | ccRCC | Uncoupling mTORC1 from REDD1 inhibition. | [33] |
| mouse embryonic fibroblasts | PDGFR upregulation | [63] | |||
| REDD1 | Tumor supressor gene (possible) | Inactivating mutation | ccRCC | Uncoupling mTORC1 from REDD1 inhibition. | [34] |
|
BIRC5
(Survivin gene) |
Inhibitor of apoptosis | Upregulation | RCC | Upregulation of PI3K (might be possible) | [51] |
Novel dimensions in comprehensive approach the mechanisms of resistance
The First Dimension – the Analysis of Cell Metabolism
The unique biology of ccRCC gives rationale to consider this cancer as a metabolic disease [53]. In this section we would like to emphasize the importance of interactions of the mTOR network with other metabolic pathways.
The importance of cellular metabolism of ccRCC in overcoming resistance is demonstrated by a studies showing that targeting both IGF-1R and mTOR synergistically inhibits growth of RCC in vitro [54]. This might be related to a new recognized role for VHL protein in suppressing IGF1-R transcription and mRNA stability in hypoxia-independent manner [55]. The VHL gene inactivation leads to IGF1R up-regulation and Akt activation which potentially drives resistance in ccRCC [55].
More mechanisms of resistance in the metabolic network are possible to elucidate with dual inhibitors of PI3K/mTOR. In an ovarian cancer, dual inhibition of mTOR signaling leads to death of inner matrix-deprived cells in sferoids, whereas matrix-attached cells are resistant [56]. The matrix-associated resistance is mediated by inhibitor-induced up-regulation of cellular survival programs which involve the forkhead box (Foxo)-regulated transcription and cap-independent translation. Targeting of any one of up-regulated proteins, including B-cell lymphoma 2 (Bcl-2) proteins, epidermal growth factor receptor (EGFR), or IGF1R may enable circumvention of resistance to PI3K/mTOR inhibition. These results together demonstrate that acute adaptive responses to PI3K/mTOR inhibition in matrix-attached cells are similar to fundamental stress responses to nutrient and growth factor deprivation [56]. If the same metabolic networks of signaling pathways function in ccRCC matrix-attached cells remains to be investigated. However it has been shown so far that inhibition of AKT following mTORC2 depletion reduces levels of the forkhead box protein O1 (FoxO1) and the forkhead box protein O3a (FoxO3a) transcription factors, which regulate the expression of genes responsible for stress resistance, metabolism, cell-cycle arrest and apoptosis [57].
The energy status of the cell is mediated by mTORC1 through AMP-activated protein kinase (AMPK) which is a crucial sensor of intracellular energy status [58]. When cellular energy is low, AMPK is activated and stimulates TSC2, which reduces mTORC1 activation [59]. AMPK might represent a node of cellular energy and nutrient sensing network that interacts via AMPK-TSC1/2-mTOR and PI3K-Akt-mTOR pathways. Mutations in each of these genes result in dysregulation of metabolic pathways involved in oxygen, iron, energy or nutrient sensing in ccRCC [53].
The Second Dimension – the Analysis of Interactions with Microenvironment
Recent studies emphasize a role for the tumor micro- environment in cancer development [60]. Understanding the complex functional interrelation between the cellular and noncellular compartments of the tumor microenvironment is necessary to fully understand the mechanisms of resistance. The level of HIF-1α expression is usually low in normoxia, but this does not occur in tumor cells that express the membrane-type 1 matrix metalloproteinase (MT1-MMP). The cytoplasmic tail of MT1-MMP, which binds an HIF-1 suppressor protein called factor inhibiting HIF-1 (FIH-1) ensures inhibition of FIH-1 by the adaptor protein Mint3 in normoxia condition. To elucidate links between HIF-1, MT1-MMP/Mint3 and tumor growth signaling pathways, a panel of 252 signaling inhibitors was screened. The mTOR inhibitor was identified as a kinase with potential to inhibit phosphorylation of Mint3 that is required for FIH-1 inhibition. These data suggest a novel molecular link between the matrix associated MT1-MMP and mTOR signaling pathway [61].
Another study has revealed that dual inhibitors are more effective than temsirolimus in decreasing the viability and proliferation potential of RCC cell lines (Caki-1 and 786-O) in vitro by inducing cell cycle arrest and autophagy [46]. However experiments with xenografts has shown no difference in the inhibition of ccRCC development by a dual inhibitor versus temsirolimus. Simultaneously the levels of VEGF and platelet-derived growth factor (PDGF) were lower after temsirolimus administration. The authors concluded that temsirolimus may exert additional effects on the tumor microenvironment [62]. Possible mechanism of resistance might be related to the loss of TSC1/2 which suppresses platelet-derived growth factor receptor (PDGFR) expression in a rapamycin-sensitive manner [55, 63]. The mechanism in which mTOR signaling controls PDGFR expression remains to be elucidated. There is evidence for a physiological role of TSC1-mTOR signaling in endothelial cells for vascular development [56], therefore mTOR signaling in epithelial cells may also drive drug resistance. Acquisition of resistance reflected by vessel formation could be mediated by induction of VEGF-independent angiogenic pathways in which angiopoietin 1 and 2 bind to the kinase Tie2 and activates angiogenesis [54].
The Third Dimension: The Analysis of Cancer Stem Cells Resistance
In the classical approach to evolution of cancer drug resistance, cancer cells are selected and acquire drug resistance during drug exposure. These cells later give rise to a population of drug-resistant cells. However, according to the cancer stem cells (CSCs) concept, drug resistance is mostly result of the intrinsic or acquired resistance mechanisms accumulated in the population of CSCs. Stem cells proliferate long enough to accumulate all necessary mutations for cancer progression [64]. Regulation of growth and proliferation of stem cells is modulated via nutrient-sensing signaling pathways including mTOR and AMPK [65]. As mTOR signaling seems to be a key pathway in the homeostasis of CSC, we speculate that potential mechanisms of drug resistance to mTOR inhibitors arise in cancer stem cells. It has been shown that acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer [66]. Findings suggest that mTOR signaling is activated in CSC-like cells in nasopharyngeal carcinoma [67]. Moreover mTOR expression was associated with outcomes of colorectal cancer patients and predictive to poor prognosis in stage II patients [68]. mTOR inhibitors were demonstrated to suppress the stem-like cells [68].
In the case of ccRCC, the existence of stem cells is not well documented [69], because characteristics of the panel of unique surface markers for renal CSC is under investigation. However cancer stem -like cells in five human RCC cell lines were identified [70]. These cells displayed high potential for proliferation and self-renewal, as well as strong resistance related to the ABCB1 transporter. In vivo experiments also showed that they formed tumors in a NOD/SCID mice model [70]. The current understanding of the renal stem cell system is not complete, but it remains likely that the kidney harbors different stem cell pools with versatile characteristics [69]. Exact mechanisms of drug resistance to mTOR inhibitors are to be elucidated in further CSC research.
Overcoming MTOR inhibitors resistance- closing remarks
To elucidate mechanisms of resistance to mTOR inhibitors it seems to be useful to treat RCC as a metabolic disease and analyze mechanisms of resistance connected with the matrix microenvironment. Cancer stem cell research may give new profound reasons accounting for resistance. Full analysis of cancer, epithelial, and cancer stem cells will enable descriptions of the network of signaling pathways with contributions to mechanisms of resistance to mTOR inhibitors in ccRCC. A better understanding of the complex signaling network upstream and downstream mTOR has contributed so far to the development of novel agents targeting PI3K, mTORC1/2 and PI3K/mTORC1/2, which have demonstrated anti-tumor activity in preclinical models of RCC [71, 72]. However in vivo these agents may prove to be less effective than traditional mTOR inhibitors, because they do not display additional effects on the tumor microenvironment compared to those conferred by classical compounds [62]. Yet another emerging approach to overcome resistance is a use of dual inhibitors targeting autophagy [73]. Dual inhibitors of mTOR in combination with autophagy inhibitors has proven to enhance the suppression of growth and induction of apoptosis in ccRCC cell lines. In conclusion, we would like to underscore that targeting autophagy might be crucial to reverse resistance, as it seems to be intertwine in several mechanisms of resistance reviewed here.
Next we would like to highlight the clinical importance of mTORC2 targeting, as many pathways potentially engaged in resistance mechanisms lead to this complex. As prolonged rapamycin treatment reduces the levels of mTORC2, this unforeseen mechanism of action for mTOR inhibitors might target crucial mechanisms of resistance in certain cell types.
ACKNOWLEDGMENTS
Grant Support
This research was supported by the Military Institute of Medicine statutory founding no. 1/1744 (101). CS and AMC have been supported by the National Science Centre projects no. UMO-2011/01/B/NZ5/02822 and 2011/01/B/NZ4/01602. CS, AMC, ZFB, WS, AK have been supported by the Foundation’s for Polish Science TEAM project no. TEAM/ 2010-6/8. AMC has been supported by the Ministry of Science and Higher Education “Juventus” grant no. CRU/ WIM/275/2012. AK has been supported by MS&HE “Diamond grant” no. DI2012 007842.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors indicate no potential conflict of interest. There are no financial or non-financial competing interests.
REFERENCES
- 1.Gamelin E., Mertins S.D., Regis J.T., Mickley L., Abati A., Worrell R.A., et al. Intrinsic drug resistance in primary and metastatic renal cell carcinoma. J. Urol. 1999;162(1):217–224. doi: 10.1097/00005392-199907000-00071. [DOI] [PubMed] [Google Scholar]
- 2.Jonasch E., Futreal A., Davis I., Bailey S., Kim W.Y., Brugarolas J., et al. State-of-the-science: An update on renal cell carcinoma. Mol. Cancer Res. 2012;10(7):859–880. doi: 10.1158/1541-7786.MCR-12-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorević G., Matusan-Ilijas K., Babarović E., Hadzisejdić I., Grahovac M., Grahovac B., et al. Hypoxia inducible factor-1alpha correlates with vascular endothelial growth factor A and C indicating worse prognosis in clear cell renal cell carcinoma. J Exp Clin Cancer Res CR. 2009;28:40. doi: 10.1186/1756-9966-28-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicol D., Hii S.I., Walsh M., Teh B., Thompson L., Kennett C., et al. Vascular endothelial growth factor expression is increased in renal cell carcinoma. J. Urol. 1997;157(4):1482–1486. [PubMed] [Google Scholar]
- 5.Melichar B., Koralewski P., Ravaud A., Pluzanska A., Bracarda S., Szczylik C., et al. First-line bevacizumab combined with reduced dose interferon-alpha2a is active in patients with metastatic renal cell carcinoma. Ann Oncol Off J Eur Soc Med Oncol ESMO. 2008;19(8):1470–1476. doi: 10.1093/annonc/mdn161. [DOI] [PubMed] [Google Scholar]
- 6.Hutson TE, Bellmunt J, Porta C, Szczylik C, Staehler M, Nadel A, et al. Long-term safety of sorafenib in advanced renal cell carcinoma: follow-up of patients from phase III TARGET. 1990 doi: 10.1016/j.ejca.2010.06.121. [DOI] [PubMed] [Google Scholar]
- 7.Molina A.M., Feldman D.R., Voss M.H., Ginsberg M.S., Baum M.S., Brocks D.R., et al. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2012;118(7):1868–1876. doi: 10.1002/cncr.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sternberg CN, Hawkins RE, Wagstaff J, Salman P, Mardiak J, Barrios CH, et al. Sternberg CN, Hawkins RE, Wagstaff J, Salman P, Mardiak J, Barrios CH, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer Oxf Engl. 1990;49(6):1287–1296. doi: 10.1016/j.ejca.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Taneja S.S. Re: Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. J. Urol. 2012;188(2):412–413. doi: 10.1016/j.juro.2012.04.069. [DOI] [PubMed] [Google Scholar]
- 10.Bukowski R.M. Temsirolimus: a safety and efficacy review. Expert Opin. Drug Saf. 2012;11(5):861–879. doi: 10.1517/14740338.2012.713344. [DOI] [PubMed] [Google Scholar]
- 11.Motzer R.J., Escudier B., Oudard S., Hutson T.E., Porta C., Bracarda S., et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma : final results and analysis of prognostic factors. Cancer. 2010;116(18):4256–4265. doi: 10.1002/cncr.25219. [DOI] [PubMed] [Google Scholar]
- 12.Posadas E.M., Limvorasak S., Sharma S., Figlin R.A. Targeting angiogenesis in renal cell carcinoma. Expert Opin. Pharmacother. 2013;14(16):2221–2236. doi: 10.1517/14656566.2013.832202. [DOI] [PubMed] [Google Scholar]
- 13.Salvadori M. Antineoplastic effects of mammalian target of rapamycine inhibitors. World J. Transplant. 2012;2(5):74–83. doi: 10.5500/wjt.v2.i5.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venugopal B., Ansari J., Aitchison M., Tho L.M., Campbell R., Jones R.J. Efficacy of temsirolimus in metastatic chromophobe renal cell carcinoma. BMC Urol. 2013;13:26. doi: 10.1186/1471-2490-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porta C., Szczylik C., Escudier B. Combination or sequencing strategies to improve the outcome of metastatic renal cell carcinoma patients: a critical review. Crit. Rev. Oncol. Hematol. 2012;82(3):323–337. doi: 10.1016/j.critrevonc.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Hudes G., Carducci M., Tomczak P., Dutcher J., Figlin R., Kapoor A., et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007;356(22):2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y. Everolimus in renal cell carcinoma.Drugs Today Barc Spain. 1998;46(8):557–567. doi: 10.1358/dot.2010.46.8.1516824. [DOI] [PubMed] [Google Scholar]
- 19.Battelli C., Cho D.C. mTOR inhibitors in renal cell carcinoma. Therapy. 2011;8(4):359–367. doi: 10.2217/thy.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang B-H., Liu L-Z. Role of mTOR in anticancer drug resistance. Drug Resist Updat Rev Comment Antimicrob Anticancer Chemother. 2008;11(3):63–76. doi: 10.1016/j.drup.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laplante M., Sabatini D.M. mTOR signaling at a glance. J. Cell Sci. 2009;122(20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 23.Wang L., Rhodes C.J., Lawrence J.C. Activation of Mammalian Target of Rapamycin (mTOR) by Insulin Is Associated with Stimulation of 4EBP1 Binding to Dimeric mTOR Complex 1. J. Biol. Chem. 2006;281(34):24293–24303. doi: 10.1074/jbc.M603566200. [DOI] [PubMed] [Google Scholar]
- 24.Carrière A., Cargnello M., Julien L-A., Gao H., Bonneil E., Thibault P., et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol CB. 2008;18(17):1269–1277. doi: 10.1016/j.cub.2008.07.078. [DOI] [PubMed] [Google Scholar]
- 25.Chang F., Steelman L.S., Lee J.T., Shelton J.G., Navolanic P.M., Blalock W.L., et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 26.Inoki K., Li Y., Zhu T., Wu J., Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 27.Ma L., Chen Z., Erdjument-Bromage H., Tempst P., Pandolfi P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 28.Huang J., Manning B.D. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 2008;412(2):179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol CB. 2005;15(8):702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Harris T. E, Roth R. A, Lawrence J. C. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007;282(27):20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 31.Khaleghpour K., Pyronnet S., Gingras A-C., Sonenberg N. Translational Homeostasis: Eukaryotic Translation Initiation Factor 4E Control of 4E-Binding Protein 1 and p70 S6 Kinase Activities. Mol. Cell. Biol. 1999;19(6):4302–4310. doi: 10.1128/mcb.19.6.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choo A.Y., Yoon S-O., Kim S.G., Roux P.P., Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA. 2008;105(45):17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rini B.I., Atkins M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10(10):992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 34.Kucejova B., Pena-Llopis S., Yamasaki T., Sivanand S., Tran T.A., Alexander S., et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res MCR. 2011;9(9):1255–1265. doi: 10.1158/1541-7786.MCR-11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 36.Shanmugasundaram K., Block K., Nayak B.K., Livi C.B., Venkatachalam M.A., Sudarshan S. PI3K regulation of the SKP-2/p27 axis through mTORC2. Oncogene. 2013;32(16):2027–2036. doi: 10.1038/onc.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carracedo A., Ma L., Teruya-Feldstein J., Rojo F., Salmena L., Alimonti A., et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veilleux A., Houde V.P., Bellmann K., Marette A. Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes. Mol. Endocrinol. 2010;24(4):766–778. doi: 10.1210/me.2009-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y., Ehara H., Akao Y., Shamoto M., Nakagawa Y., Banno Y., et al. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem. Biophys. Res. Commun. 2000;278(1):140–143. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 40.Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D.A. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 2009;29(6):1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong X., Lin Z., Liang D., Fath D., Sang N., Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 2006;26(6):2019–2028. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang K., Li Z., Chen Y., Su C. The pharmacokinetics of a novel anti-tumor agent, beta-elemene, in Sprague-Dawley rats. Biopharm. Drug Dispos. 2005;26(7):301–307. doi: 10.1002/bdd.463. [DOI] [PubMed] [Google Scholar]
- 43.Zhan Y-H., Liu J., Qu X-J., Hou K-Z., Wang K-F., Liu Y-P., et al. β-Elemene induces apoptosis in human renal-cell carcinoma 786-0 cells through inhibition of MAPK/ERK and PI3K/Akt/ mTOR signalling pathways. Asian Pac J Cancer Prev APJCP. 2012;13(6):2739–2744. doi: 10.7314/apjcp.2012.13.6.2739. [DOI] [PubMed] [Google Scholar]
- 44.Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thoreen C.C., Kang S.A., Chang J.W., Liu Q., Zhang J., Gao Y., et al. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009;284(12):8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toschi A., Lee E., Gadir N., Ohh M., Foster D.A. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J. Biol. Chem. 2008;283(50):34495–34499. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordan J.D., Lal P., Dondeti V.R., Letrero R., Parekh K.N., Oquendo C.E., et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14(6):435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harada K., Miyake H., Kumano M., Fujisawa M. Acquired resistance to temsirolimus in human renal cell carcinoma cells is mediated by the constitutive activation of signal transduction pathways through mTORC2. Br. J. Cancer. 2013;109(9):2389–2395. doi: 10.1038/bjc.2013.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabatabaian F., Dougherty K., Di Fulvio M., Gomez-Cambronero J. Mammalian target of rapamycin (mTOR) and S6 kinase down-regulate phospholipase D2 basal expression and function. J. Biol. Chem. 2010;285(25):18991–19001. doi: 10.1074/jbc.M110.111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahalingam D., Medina E.C., Esquivel J.A., II, Espitia C.M., Smith S., Oberheu K., et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16(1):141–153. doi: 10.1158/1078-0432.CCR-09-1385. [DOI] [PubMed] [Google Scholar]
- 51.Yamada T., Horinaka M., Shinnoh M., Yoshioka T., Miki T., Sakai T. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int. J. Oncol. 2013;43(4):1080–1086. doi: 10.3892/ijo.2013.2042. [DOI] [PubMed] [Google Scholar]
- 52.Sato Y., Yoshizato T., Shiraishi Y., Maekawa S., Okuno Y., Kamura T., et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013;45(8):860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Donas J., Rodriguez-Antona C., Jonasch E. Molecular markers to predict response to therapy. Semin. Oncol. 2013;40(4):444–458. doi: 10.1053/j.seminoncol.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Linehan W.M., Srinivasan R., Schmidt L.S. The genetic basis of kidney cancer: a metabolic disease. Nat. Rev. Urol. 2010;7(5):277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardillo T.M., Trisal P., Arrojo R., Goldenberg D.M., Chang C-H. Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro. BMC Cancer. 2013;13:170. doi: 10.1186/1471-2407-13-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuen J.S., Cockman M.E., Sullivan M., Protheroe A., Turner G.D., Roberts I.S., et al. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene. 2007;26(45):6499–6508. doi: 10.1038/sj.onc.1210474. [DOI] [PubMed] [Google Scholar]
- 57.Muranen T., Selfors L.M., Worster D.T., Iwanicki M.P., Song L., Morales F.C., et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calnan D.R., Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 59.Hardie D.G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007;8(10):774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 60.Inoki K., Zhu T., Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 61.Fan F, Schimming A, Jaeger D, Podar K. Targeting the tumor microenvironment: focus on angiogenesis. J Oncol. 2012;2012:281261. doi: 10.1155/2012/281261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakamoto T., Weng J.S., Hara T., Yoshino S., Kozuka-Hata H., Oyama M., et al. HIF-1 regulation through crosstalk between mTOR and MT1-MMP. Mol. Cell. Biol. 2013 doi: 10.1128/MCB.01169-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H., Berel D., Wang Y., Li P., Bhowmick N.A., Figlin R.A., et al. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS One. 2013;8(1):e54918. doi: 10.1371/journal.pone.0054918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gil J., Stembalska A., Pesz K.A., Sasiadek M.M. Cancer stem cells: the theory and perspectives in cancer therapy. J. Appl. Genet. 2008;49(2):193–199. doi: 10.1007/BF03195612. [DOI] [PubMed] [Google Scholar]
- 65.Ochocki J.D., Simon M.C. Nutrient-sensing pathways and metabolic regulation in stem cells. J. Cell Biol. 2013;203(1):23–33. doi: 10.1083/jcb.201303110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang L., Graham P.H., Hao J., Ni J., Bucci J., Cozzi P.J., et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang C., Peng J., Jiang W., Zhang Y., Chen X., Wu X., et al. mTOR activation in immature cells of primary nasopharyngeal carcinoma and anti-tumor effect of rapamycin in vitro and in vivo. Cancer Lett. 2013 doi: 10.1016/j.canlet.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Cai Z., Ke J., He X., Yuan R., Chen Y., Wu X., et al. Significance of mTOR Signaling and Its Inhibitor Against Cancer Stem-Like Cells in Colorectal Cancer. Ann. Surg. Oncol. 2013 doi: 10.1245/s10434-013-3146-8. [DOI] [PubMed] [Google Scholar]
- 69.Axelson H., Johansson M.E. Renal stem cells and their implications for kidney cancer. Semin. Cancer Biol. 2013;23(1):56–61. doi: 10.1016/j.semcancer.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Huang B., Huang Y.J., Yao Z.J., Chen X., Guo S.J., Mao X.P., et al. Cancer stem cell-like side population cells in clear cell renal cell carcinoma cell line 769P. PLoS One. 2013;8(7):e68293. doi: 10.1371/journal.pone.0068293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Figlin R.A., Kaufmann I., Brechbiel J. Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: new strategies for overcoming resistance to VEGFR and mTORC1 inhibitors. Int J Cancer J Int Cancer. 2013;133(4):788–796. doi: 10.1002/ijc.28023. [DOI] [PubMed] [Google Scholar]
- 72.Cho D.C., Mier J.W. Dual inhibition of PI3-kinase and mTOR in renal cell carcinoma. Curr. Cancer Drug Targets. 2013;13(2):126–142. doi: 10.2174/1568009611313020003. [DOI] [PubMed] [Google Scholar]
- 73.Li H., Jin X., Zhang Z., Xing Y., Kong X. Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTor inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem. Funct. 2013;31(5):427–433. doi: 10.1002/cbf.2917. [DOI] [PubMed] [Google Scholar]