

Abstract
Clear - cell renal cell carcinoma (ccRCC) is a histological subtype of renal cell carcinoma - the most prevalent adult kidney cancer. Causes of ccRCC are not completely understood and therefore number of available therapies is limited. As a consequence of tumor chemo- and radioresistance as well as restrictions in offered targeted therapies, overall response rate is still unsatisfactory. Moreover, a significant group of patients (circa 1/4) does not respond to the targeted first-line treatment, while in other cases, after an initial period of stable improvement, disease progression occurs. Owing to this, more data on resistance mechanisms are needed, especially those concerning widely used, relatively lately approved and more successful than previous therapies - tyrosine kinase inhibitors (TKIs). Up to date, five TKIs have been licensed for ccRCC treatment: sunitinib (SUTENT®, Pfizer Inc.), sorafenib (Nexavar®, Bayer HealthCare/Onyx Pharmaceuticals), pazopanib (Votrient®, GlaxoSmithKline), axitinib (Inlyta®, Pfitzer Inc.) and tivozanib (AV-951®, AVEO Pharmaceuticals).
Researchers have specified different subsets of tyrosine kinase inhibitors potential resistance mechanisms in clear-cell renal cell carcinoma. In most papers published until now, drug resistance is divided into intrinsic and acquired, and typically multi-drug resistance (MDR) protein is described. Herein, the authors focus on molecular analysis concerning acquired, non-genetic resistance to TKIs, with insight into specific biological processes.
Keywords: Acquired drug resistance, tyrosine kinase inhibitors, sunitinib, sorafenib, pazopanib, axitinib, tivozanib, epithelial-mesenchymal transition, angiogenic switch, anti-angiogenic therapy, clear-cell renal cell carcinoma, non-genetic resistance mechanisms
Introduction
Clear-cell renal cell carcinoma (ccRCC) is the most common type of kidney cancer in adults and the 10th malignancy worldwide, with approximately 88 400 newly diagnosed patients each year in Europe. Its worldwide incidence and mortality rates rise by 2-3% per decade [1,2]. The etiology of ccRCC is unknown, however obesity, hyper- tension, smoking, unhealthy diet and diabetes are known risk factors [3]. Patients presented with localized ccRCC can be cured with partial or radical nephrectomy. Still, up to 30% of newly diagnosed patients develop metastases and among 20-30% post-surgery treatment cases recurrence is eventually noted. Regardless of the fact that many publications provide insight into the knowledge on probable resistance mechanisms in ccRCC, its actual origin still remains elusive. Lately, therapy for metastatic clear-cell renal cell carcinoma has significantly improved due to the introduction of several novel agents after the failure of initially successful treatment with interleukin 2 (IL-2) or interferon alpha (IFN-α), which were both causing severe toxic side effects in many cases [4]. Until a few years ago, immunotherapy was still the only option for metastatic ccRCC treatment. The novel therapies, including tyrosine kinase inhibitors, have been subsequently introduced [1,5,6].
Tyrosine kinases are signaling molecules and prototypic oncogenes, which play an important role in cancer development. They target various kinases including c-KIT, VEGF 1, 2 and 3, PDGFR-α and β, FLT3, RET, BRAF and CRAF (go to: Abbreviations) [5,6]. TKIs became the most successful class of drugs in the treatment of ccRCC, including sunitinib (SUTENT®, Pfizer Inc.), sorafenib (Nexavar®, Bayer HealthCare/Onyx Pharmaceuticals), pazopanib (Votrient®, GlaxoSmithKline), axitinib (Inlyta®, Pfitzer Inc.) and tivozanib (AV-951®, AVEO Pharmaceuticals) [5,7-12]. They all primarily function as anti-angiogenic agents, through the inhibition of tumor endothelium growth and tumor cell survival signaling impairment. As multi-targeted agents, they inhibit a number of receptors with varying potency [13]. Tyrosine kinases can be subdivided into two major groups – the first one consists of receptor tyrosine kinases (i.e. epithelial growth factor receptor: EGFR, ErbB/HER family members, vascular endothelial growth factor receptor: VEGFR and platelet-derived growth factor receptor: PDGFR) and the second one includes non-receptor cytoplasmic tyrosine kinases (i.e. SRC and FAK) [14]. Receptor tyrosine kinases perform a crucial role in the transduction of extracellular signals into the cell, while non-receptor tyrosine kinases take part mostly in intracellular communication [15].
Vascular endothelial growth factor (VEGF) is an essential molecule in the process of angiogenesis. VEGFR family comprises VEGFR-1, -2, and -3, which all mediate the angiogenic effect of VEGF ligands [16]. There are six different ligands for VEGFR: VEGF-A to VEGFR-E and placental-derived growth factor (PDGF). Those ligands bind to specific receptors on endothelial cells, mostly to VEGFR-2 (FLK-1/KDR), but also to VEGFR-1 (Flt-1) and -3. The binding of VEGF-A to VEGFR-1 is responsible for endothelial cell migration. VEGFR-2 induces endothelial cell proliferation, permeability, and survival and VEGFR-3 is related to lymphangiogenesis [17]. The role of endothelium in the process of resistance to TKIs in ccRCC will be described later on.
The impact of VEGF on the process of angiogenesis and also on cancer pathogenesis has given the rationale for design and development [16]. TKIs designed mainly for ccRCC therapy operate by four different mechanisms: 1) they compete either with adenosine triphosphate (ATP), or 2) with the substrate, or 3) with both, or 4) they act in an allosteric way [18]. In fact, most small-molecule kinase inhibitors discovered to date compete with ATP. On the basis of chemical and conformational changes, tyrosine kinase inhibitors can be classified into 3 categories. Type I kinase inhibitors recognize the active conformation of a kinase. In contrast, type II kinase inhibitors affect the inactive conformation of a kinase by an indirect competition with ATP. In this way, they occupy the hydrophobic pocket which is directly adjacent to the ATP-binding site. Type III kinase inhibitors are known as ‘covalent’ inhibitors [16,19].
Many anti-angiogenic TKIs are multi-targeted inhibitors. These agents simultaneously target kinases which are involved in several signaling pathways. The effect is mostly of a broader efficacy than in case of a single-targeted inhibitor. For example, VEGF and PDGF pathways both play important roles in angiogenesis. It has been suggested that a multi-targeted kinase inhibitor blocking VEGFR, as well as PDGFR signaling to inhibit vessel formation, will be more effective than a single-function inhibitor that targets only one of these pathways [15]. Unfortunately, resistance to such therapy will eventually develop regardless of TKIs therapy in ccRCC – which basically is the most vascularized solid tumor [20].
This article reviews the current state of the art concerning mechanisms of resistance to tyrosine kinase inhibitors, focusing mainly on the presence of a few very specific cell biology processes. It is essential that no ccRCC symptoms are in most cases found in early stages, as a result of which up to 30% of newly diagnosed patients present with metastases at diagnosis [21]. It is therefore needed to focus on understanding the role of many factors in this process, both those yet unknown and those neglected.
Possible mechanisms responsible for changes in tumor microenvironment in response to anti-angiogenic targeted therapy using tyrosine kinase inhibitors are: 1) the possibility to form populations of clonal cells which allow for the upregulation of alternative angiogenic signaling pathways, 2) elevated evasiveness of cells, and 3) intrinsic resistance to the hypoxia conditions [22]. A table providing chosen abnormalities and their connection with drug resistance is presented herein (Table 1).
Table 1. Abnormalities and alterations probably contributing to the mechanism of drug resistance to tyrosine kinase inhibitors in ccRCC.
|
Process
Name |
Cell Type
Involved |
Abnormality | Result |
Resistanceto TKIs
Developed |
|---|---|---|---|---|
| epithelial to mesenchymal transition | healthy epithelial cells | polarized epithelial cells convert into motile epithelial cells or to cells with stem cell-like properties | escape of cells from their biological structure; tumorigenesis; acquired resistance | all approved: sorafenib, sunitinib, axitinib, pazopanib, tivozanib |
| lysosomal sequestration | ccRCC cells | sunitinib is captured and stored in intracellular compartments (other than in ccRCC cells) instead of reaching cancer cells | low concentrations of sunitinib in plasma and serum, and finally in ccRCC cells; therapeutic concentrations not achieved | only sunitinib (proved until now; resistance to other TKIs possible) |
| increased pericyte coverage of tumor vessels | perivascular cells / vascular smooth muscle cells |
stabilized process of abnormally complicated vascular system formation; tumorigenesis | excessive angiogenesis of ccRCC, more aggressive tumor type | all approved: sorafenib, sunitinib, axitinib, pazopanib, tivozanib |
| angiogenic switch | vascular cells | multifactorial, excessive growth of tumor vascular system | ccRCC progression | all approved: sorafenib, sunitinib, axitinib, pazopanib, tivozanib |
| accumula-tion of bone marrow derived cells | vascular progenitor cells; pro-angiogenic monocytes; VEGFR-1+ hemiangio-cytes; CD11b+
myeloid cells |
bone marrow derived cells accumulation inside and around the tumor; new blood vessels supplying the arising tumor | ccRCC adaptation to hypoxia conditions; tumorigenesis | all approved: sorafenib, sunitinib, axitinib, pazopanib, tivozanib |
Drug resistance in CCRCC as a reversible phenomenon
Definition of Drug Resistance
Resistance to tyrosine kinase inhibitors, or drug resistance in general, is defined as a progression of a disease according to RECIST criteria, despite treatment application. This abbreviation stands for Response Evaluation Criteria in Solid Tumors (patient improves =”responds”, patient’s health state stays the same = “stabilizes”, or worsens = “progresses”) during treatment. The progression itself means a 20% or more increase in the number of lesions and their lateral size [22,23].
In general, sensitivity to targeted agents occurs when the survival of particular tumor is dependent on the constitutive activity of signaling pathways. Therefore, resistance to such agent develops, when a tumor becomes independent from the activity of drug targeted pathway. It may happen due to some genetic alterations, which typically cause the impossibility of proper drug binding as a consequence of impaired or lack of crucial proteins. Also, alternative signaling pathways may be activated, or specific molecule expression may increase as an answer to the inhibition, which naturally is its compensation [24].
In various publications, acquired and intrinsic (inherent) resistance is described [15,25,26]. Acquired resistance is defined as a progression of a disease after patients obtain an initial benefit from targeted therapies. Pre-existence of the excessively activated signaling pathways is believed to be mainly responsible for intrinsic resistance to TKIs, which means that patients do not respond to the treatment at all [25].
Intrinsic Resistance is Uncommon in ccRCC
Inherited TKI resistance is rather uncommon in ccRCC, on the contrary to other tumors with hypoxia-driven angio- genesis. For example, in lung cancer or in CML (chronic myeloid leukemia) mutations of genes encoding TKIs contribute to VEGFR inhibitors resistance development. However, this is a highly improbable scenario in kidney cancers in general, as such mutations would have to take place in endothelium, which is the main target of VEGFR inhibitors. Therefore, it is almost impossible that identical mutations would coexist within each metastasis [22]. Apart from these findings published in 2009 in Lancet, Huang et al. have published an original paper in Cancer Research a year later, in which they claim that sunitinib targets mainly ccRCC endothelium and is rather not connected with cancer cells. In this way, SUTENT inhibits tumor growth. Taking into account that sunitinib treatment is only somewhat successful, it may be a direct confirmation of described theory [27].
Epithelial-Mesenchymal Transition (EMT) Mechanism and Tumor Associated Macrophages (TAMs) as Elements of Resistance Reversibility
Studies have shown that in vitro resistance to TKIs is transient. Gotink et al. have proved in 2011 that sensitivity to sunitinib is gradually rebuilt after a 12-week period of drug-free culture. For example, prolonged exposure of tumor cells to sunitinib always results in drug resistance development in vitro [15,28,29].
One of the mechanisms which allow to interpret such phenomenon is epithelial to mesenchymal transition (EMT), where polarized epithelial cells convert into motile mesenchymal cells. This is a newly recognized phenomenon in the contemporary oncology era. Protein accumulation in their most modified state (with the highest number of transcriptional events) takes place in response to long-lasting extracellular stimuli, leading to cellular changes – in fact, often reversible [29,30,31]. This allows epithelial cells to ‘escape’ from their typical biological structure. β-catenin, supportive for cadherins, translocates from the cell membrane to the nucleus and further participates in EMT process [32]. Matrix metalloproteinases (MMPs) are up-regulated, allowing for the reduction of cell-cell adherence and cells’ penetration of the basement membrane. As a result, epithelial cells lose their polarity and change shape [33].
Reverse epithelial-mesenchymal transition and acquired resistance to sunitinib were investigated in ccRCC in xenograft studies. A histological examination from 2011 showed that EMT may be associated with acquired resistance to TKIs. Hammers et al. have obtained a biopsy from ccRCC skin metastasis. After transplanting sunitinib-resistant cells into nude mice, density of microvessels was reduced and pericytes coverage was impaired. Moreover, at the very beginning of the experiment, histology of the patient’s skin showed sarcomatoid skin lesions (which is typical for carcinomas) consisting of widely vascularized nodules. The latter had atypical spindle shape, facilitating cell movement. After injection all these features were lost. Mesenchymal marker, vimentin, as well as HIF-1α (which is involved in cell adaptation to the state of hypoxia; this, in return, induces VEGF and PDGF expression) exhibited elevated expression, which suggests that the reason for such state is EMT. These transcription factors also indirectly stimulate tumor growth [34,35].
Additionally, EMT was shown to be linked with a Sonic hedgehog signaling (Shh), which regulates embryonic cell growth, but when its ligands become activated, certain processes become also reliant on Shh – cell proliferation, differentiation, motility etc. Behnsawy et al. proved that ccRCC is dependent on Shh signaling and that Shh downstream transcriptional factor (Gli-1) positively regulates epithelial differentiation. If Gli-1 levels are low, it contributes to the metastatic cancer phenotype. Behnsawy’s team’s in vitro research showed that Shh induces EMT markers, such as E-cadherin [36].
To sum up, histological data showed that EMT cell phenotype significantly contributes to the occurrence of acquired resistance to tyrosine kinase inhibitors. EMT has been under research previously in other cancers: ovarian cancer, non-small cell lung cancer, pancreatic cancer, breast cancer etc. [37-39]. Authors of these studies state that epithelial to mesenchymal transition may be with confidence added to the list of potential TKIs resistance mechanisms. However, as it was previously mentioned, this resistance is reversible, so if patients receive clinical benefit from the treatment with tyrosine kinase inhibitor(s), they should be given a time-off treatment, mostly with anti-VEGF drugs. After such break it is highly possible that patients will respond to TKI treatment again [22, 37-39]. Resistance to TKIs is therefore a reversible and transient phenomenon, and such knowledge argues with the theory of primary genetic alterations’ significance, as these are by definition permanent [22].
It is significant as well that EMT and drug resistance are strictly connected with the presence of a so-called cancer stem cells (or: cancer stem-like cells, tumor-initiating cells). Cancer stem cells (CSCs) are defined as cells found within tumors or hematological cancers that possess features of normal stem cells, mainly the ability to give rise to all cell types. CSCs have therefore a tumor-forming potential, perhaps in contrast to other non-tumorigenic cancer cells. It is highly probable that CSCs will generate tumors or cancers through typical stem cell self-renewal and differentiation processes. Such cells probably exist as a separate subpopulation. It is believed that the development of specific therapies targeted at CSCs will result in the improvement of survival and quality of life of cancer patients, especially for those who suffer from metastases [40].
Huang et al. have lately defined CSCs presence in clear-cell renal cell carcinoma with side population (SP) phenotype in five human ccRCC cell lines. Authors indicate that their results reveal the properties of cancer stem cells in ccRCC, which may play important role in tumorigenesis, drug resistance, and excessive tumor angiogenesis [41]. Recently, EMT was shown to transform mammary epithelial cells to cells with stem cell-like properties, including the ability to self-renew and efficient tumor initiation [42]. Also, other evidence in the context of EMT connection with CSCs have been lately published, implying that epithelial-mesenchymal transition not only contributes to increased metastases occurrence, but also causes drug resistance [43].
Tumor-associated macrophages (TAMs) are also involved in the process of epithelial-mesenchymal transition. However, they primarily possess other functions. They were considered to have both negative and positive effect on tumorigenesis. In the latest review from 2012, the prognostic value of TAMs for survival in patients with solid tumors was described on the basis of extensive literature research, and still remains controversial [44].
Macrophages are innate myeloid cells released from bone marrow as immature monocyte precursors. They have ability to migrate into different tissues, where they undergo specific differentiation processes [45]. In response to wide variety of stimuli, macrophages can ‘transform’ into two characteristic phenotypes [44]. The most often activated M1 phenotype is physiological, and M1 macrophages are induced by interferon-c, possibly with lipopolysaccharide, and TNF-α. Tumor associated macrophages type 1 are a major component of leukocyte infiltrate and produce signal molecules [46]. Differentiation of M1 macrophages into alternative M2 type is induced by IL-4 or IL-13. M2 phenotype is mainly associated with tumor promotion and progression, regulating processes such as angiogenesis, tumor cell growth and downregulation of the antitumor response [47,48].
Inflammation-induced EMT has been previously shown, so it was hypothesized that M2-type TAMs are able to induce EMT [49,50]. According to Santoni et al. in their newest publication from 2013, because TAMs are connected with vascular density and they contribute to angiogenesis, they mostly benefit from PDGF, TGF-β, PIGHF and GRP overexpression. In case of tumors so enriched with vessels as ccRCC, these processes are crucial. M2 macrophages also have the ability to adapt to hypoxia conditions [39,51]. What is more, TAMs represent the vast majority of leukocyte population which infiltrate ccRCC as well as other tumors. When they undergo an alternative mechanism of activation, they contribute to poor prognosis in ccRCC. Instead of classical activation, interleukin-4, -10 and -13 induce macrophages to acquire M2-like state which, on the contrary to physiological M1 state, results in tumor necrosis factor alpha (TNF-α) overexpression [52]. Moreover, as a component of ccRCC microenvironment, TAMs contribute to higher vessel density, PDGF, VEGF or even placental growth factor (PIGF) overproduction (PIGF is a VEGF homolog also stimulating tumor angiogenesis) [53]. It was also shown that if TAMs are detected in serum at high levels, this represents poor clinical outcome. Dannenmann et al. state that ccRCC may even attract these macrophages when they are still undifferentiated and ‘change’ them into M2 phenotype. Their analysis confirmed that only M2, but not M1 macrophage phenotype is associated with more advanced tumor stage [54]. All in all, TAMs are key components in EMT process and represent a promising target for anti-ccRCC therapy.
Proven and probable processes causing resistance to TKIS in CCRCC
Angiogenic Escape/Angiogenic Switch
Both current and past researches have shown that anti-angiogenic therapy is only to some extent successful in the treatment of ccRCC, which is a highly vascularized tumor. Still, this approach should be definitely continued and improved to finally overcome resistance to TKIs. Concerning sunitinib, progression develops usually about 1 year after the beginning of the treatment. Huang et al. presented some crucial mechanisms of angiogenic switch in sunitinib-resistant ccRCC cells. They were mostly connected with elevated expression of interleukin-8, which was secreted in excessive amounts from tumor cells into the plasma. IL-8 is a member of CXC chemokines family and a potent angiogenic factor. However, IL-8 neutralizing antibody, when co-administered with sunitinib, has resensitised ccRCC to TKIs treatment [55].
Huang et al. have also studied the mechanisms of resistance to TKIs by excluding the possibility of primary mutations. To do this, they sequenced receptors of tyrosine kinases genes - no mutations were found in FLT1 (VEGFR-1), KDR (VEGFR-2), FLT4 (VEGFR-3), PDGFR-α, PDGFR-β, c-KIT, and RET genes. This means that probably resistance to TKIs is mediated via VEGF/VEGFR mutation-independent mechanism and such data supports the earlier hypotheses that drug resistance to TKIs occurs as a result of action of angiogenic factors other than VEGF [55-57]. Regardless of the fact that VEGF is a crucial mediator of angiogenesis in physiological processes as a endothelial cells mitogen, it can be also present on cancer cells [15].
The hypothesis that the angiogenesis over-induction may be a hallmark of ccRCC progression is widely accepted nowadays [58,59]. The ‘angiogenic switch’ was documented not only in the in vitro, but in the in vivo studies as well (ccRCC animal models). Evidence for acquired resistance as a result of neo-angiogenesis phenomenon have been also described [57,60-63].
To sum up, both resistance and escape from anti-angiogenic therapy are multifactorial; drug resistance in clear-cell renal cell carcinoma, as well as in many other tumors where TKIs are of primary use as a targeted therapy, significantly lowers treatment efficacy.
Increased Pericyte Coverage of ccRCC Vessels
Pericytes have been discovered over 100 years ago as perivascular cells that are wrapped around blood vessels (peri means - around; cyte means – the cell). They are also called Rouget cells, mural cells or, since they have contractile fibers, vascular smooth muscle cells (VSMCs) [64]. Pericytes consist of a prominent nucleus and a small amount of cytoplasm. They are known for its supportive function to the microvasculature [65]. On the other hand, endothelial cells form the vessel layer, and mural cells are braiding the surface of the vascular tube, playing a crucial role in endothelial cells’ proliferation, migration and stabilization. Those, in turn, stimulate pericytes’ expansion and activation of their precursor cells population. Undoubtedly, a kind of physiological balance between the number of endothelial cells and pericytes exists and is thoroughly controlled by many, not necessarily all known, signaling pathways [66]. Lately, pericytes have been highlighted as critical for pathological over-angiogenesis in tumors and therefore are now perceived as potentially new therapeutic targets, especially for such well-vascularized tumors as clear-cell renal cell carcinoma [67,68]. Perivascular cells mainly stabilize blood vessels formation, however, they also possess many other functions; their contraction regulates blood flow. Interestingly, in several organs, such as brain, liver and kidneys, pericytes perform specific functions and owing to this, they have additional names in these organs. Pericytes belonging to the subgroup of the glomerular capillaries in the kidney are called mesangial cells and account for circa 30% of the glomeruli population. In kidneys, they form a special surface for blood ultrafiltration [66,67]. Moreover, pericytes are necessary for the proper wound healing process, during the menstrual cycle and pregnancy. Under pathological conditions, such as diabetic retinopathy and tumor growth, pericytes are also of high importance. The difference is that in pathological states the newly formed vessels do not stop growing and are under constant reconstruction, which leads to the formation of abnormally complicated vascular system [68].
In 2013, Cao et al. have published data on pericyte coverage of blood vessels, stating that highly differentiated vessels belonging to the general tumor vasculature is an independent unfavorable prognostic factor for patients with clear-cell renal cell carcinoma. The authors observed that higher number of pericyte-generated microvessels in serum is correlated with much more aggressive type of ccRCC and with its higher resistance to treatment as well [69]. Earlier research from 2010 revealed the failure of additional pericytes targeting while cancer treatment did not increase the antitumor effect already generated by tyrosine kinase inhibitors [70]. Studies confirmed that signaling pathways other than those related to VEGF may be essential in excessive angiogenic process in ccRCC. Yet unpublished observations of Casanovas and Kerbel, about which Bergers et al. have mentioned in their known review 2010 showed that TKI treatment as well as dual targeting of monocytes and endothelial cells, lead to higher incidence of metastasis in various tumors [71].
Interestingly, pericyte depletion (‘exhaustion’) in cancer was shown to cause epithelial to mesenchymal transition associated with increased hypoxia conditions. Moreover, impaired pericyte coverage of blood vessels paradoxically leads to defective tumor vasculature formation and increased metastasis, and at the same time inhibits tumor growth [72].
Reduced Drug Levels in Plasma and Serum
During patients treatment and molecular laboratory research as well, it is essential to keep in mind that apart from targeting inadequate molecules, unknown signaling pathways etc., the other possible factors contributing to the resistance mechanism in ccRCC are reduced drug levels in plasma. It was proved that in the case of sunitinib and axitinib, the higher drug concentrations in plasma, the higher clinical benefit [73]. Moreover, it was reported that sunitinib tends to inhibit ccRCC proliferation only at clinically relevant concentrations [74]. Basically, a typical resistance model considering reduced drug levels is the increased drug efflux which results in a decreased intracellular TKIs concentrations in cells which subsequently develop resistance. The ultimate idea to overcome, at least partially, such difficulties, seems to optimize the dose of therapy administered to ccRCC patients. A few years ago, authors of a prominent meta-analysis showed that the higher dose of sunitinib, the longer time with no progression. What is more, after significantly increased doses of this tyrosine kinase inhibitor, a probability of achieving at least a partial or even a complete response was much higher in metastatic renal cell carcinomas. Unfortunately, this was observed only in case of sunitinib administration, as patients receiving higher doses of sorafenib mostly do not tolerate its high concentrations [75]. It has been further proposed that serum drug levels rather than whole blood drug levels should be taken into consideration while adjusting TKIs dose in order to obtain maximum efficiency and minimum resistance. The only available data in this matter were published by Khalil et al. in 2011 and were suggesting that such strategy should be successful in the improvements of drug tolerance [76,77].
Drug-level mechanism of resistance is therefore related to the impaired influx and excessive efflux of the drug components. As a result, effective concentration is not achieved, which leads to the lack of proper response to TKIs treatment. Irregular blood flow or even the anatomical/ functional defects in tumor vasculature may of course affect drug response, but still, drug levels and drug plasma sequestration (described in the sub-chapter below) are mainly influencing the level of resistance to TKIs [78].
Lysosomal Sunitinib Sequestration
Most of currently approved, mostly anti-cancer drugs, used as an alternative to standard chemotherapy, are chemically weak bases. This makes them substrates in the process of sequestration (substances capture and their long-term storage) into intracellular compartments with acidic features. Such compartments may be lysosomes, which capture drugs via ion trapping mechanism [79].
Recent data revealed that sunitinib is sequestered in lysosomes while treatment of clear-cell renal cell carcinoma. Gotink et al. have investigated last year intracellular sunitinib concentrations in renal cell carcinoma to establish the rule that resistance to sunitinib develops, since in resistant cells this tyrosine kinase inhibitor is sequestered in some specific cell compartments. To do this, they analyzed the subcellular sunitinib distribution and showed that this drug, which is biochemically a hydrophobic, weak base, is sequestered in acidic lysosomes. This mechanism was assessed as reversible and mainly connected with sunitinib because of its chemical features [74].
Potential mechanisms of drug-induced changes in lysosomal volumes are currently being investigated. Such results may help to rationalize how sequestration may work when other tyrosine kinase inhibitors are taken into consideration [74,79].
Accumulation of Bone Marrow Derived Cells Around and Inside the Tumor
Bone marrow derived cells (BMDCs) comprise: vascular progenitor cells, pro-angiogenic monocytes, VEGFR-1+ hemiangiocytes and CD11b+ myeloid cells [80]. These cells modulate expression of a wide variety of cytokines, growth factors, enzymes (mainly proteases) etc. They are recruited as a result of hypoxia occurrence, which in turn is caused by vascular regression [71]. The latter occurs due to anti-angiogenic therapy, mainly using tyrosine kinase inhibitors. The problem connected with BMDCs is that they possess the ability to create new blood vessels which supply the arising tumor. This means that hypoxia, triggered in response to excessive angiogenesis, contributes to tumor adaptation to its conditions by means of BDMCs [81].
Conclusion and future directions
Anti-angiogenic agents are more efficient rather than previous therapeutic approaches, using, for instance, interferon [82]. Iacovelli et al. meta-analyzed the incidences of positive responses for the first-line treatment in metastatic renal cell carcinoma and showed that this is an extremely rare event. In fact, the number of patients reaching complete response (CR) when treated with tyrosine kinase inhibitors (sunitinib and sorafenib) accounts for less than 3%, which is nonetheless a better result than achieved by using standard chemo-, radio- or hormone therapy [4].
Drug resistance in clear-cell renal cell carcinoma occurs to enable cancer cells’ survival in hypoxia conditions, but this is not the main point. It seems that ccRCC cells are putting all their efforts to gain angiogenic potential which would be as independent of VEGF, as possible [22]. Clinical studies performed until now have demonstrated obvious linkages between blocking angiogenic pathways and clinical treatment efficacy [25,77]. The adoption of alternative signaling pathways to compensate for the inhibition of ‘traditional’ VEGF/VEGFR signaling is probably a common mechanism for the resistance development [55]. Still, actual resistance mechanisms in ccRCC remain elusive. Hypothetically, chemoresistance may result from inherent or acquired genetic alterations, composition changes in the microenvironmental vasculature of compensatory character, as well as from over-production of additional pro-angiogenic growth factors, increased pericyte coverage of blood vessels and accumulation of bone marrow derived cells near ccRCC cells [34] (Fig. 1).
Fig. (1).
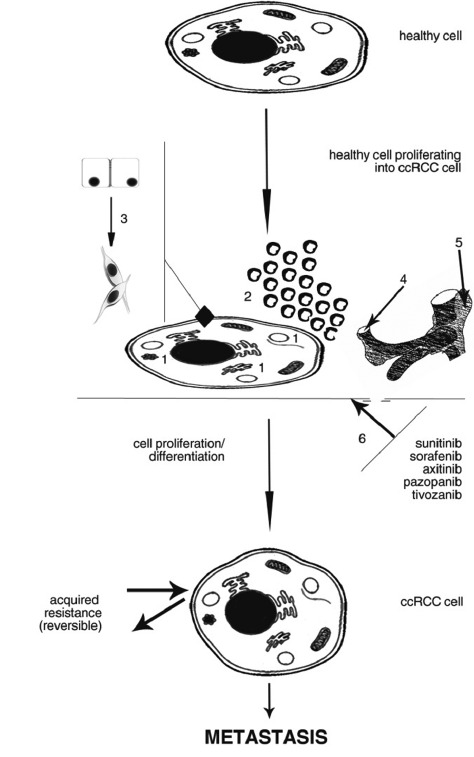
Mechanism of healthy cell differentiation into ccRCC cell. Sample processes have been marked by: 1: lysosomal sequestration; 2: accumulation of BDMCs; 3: EMT; 4: angiogenic switch; 5: increased pericyte coverage; 6: reduced drug levels in serum/plasma. When these processes are activated - no matter if only one of them, or more parallely/subsequently, drug resistance and therefore metastasis will probably occur.
In conclusion, many novel therapeutic approaches are currently under intense investigation, which in turn may provide some additional treatment options for patients who eventually tend to progress after VEGF- and/or PDGR- targeted treatment. Predictive markers are needed for proper therapeutic approach selection. Resistance itself is a really broad term, which by defining it only considering RECIST criteria brings some limitations. This is a drawback mainly for molecular biologists/biotechnologists or other participants of molecular research teams working on resistance mechanisms in oncology. Contemporarily, therapeutic strategies such as dose escalation, drug combinations, parallel inhibition of signaling pathways and extensive laboratory research are implemented to overcome ccRCC metastasis. Resistance to TKIs treatment in ccRCC may be delayed by the restoration of angiostatic signaling, but nonetheless it eventually develops [59]. Revealing the truth about TKIs resistance mechanisms in clear-cell renal cell carcinoma treatment may shed light upon both therapeutic as well as scientific approaches and bring hope for terminally ill patients.
ACKNOWLEDGMENTS
Grant Support
This research was supported by the Military Institute of Medicine statutory founding no. 1/1744 (101). CS and AMC have been supported by the National Science Centre projects no. UMO-2011/01/B/NZ5/02822 and 2011/01/B/NZ4/01602. CS, AMC, ZFB, WS, AK have been supported by the Foundation’s for Polish Science TEAM project no. TEAM/ 2010-6/8. AMC has been supported by the Ministry of Science and Higher Education “Juventus” grant no. CRU/ WIM/275/2012. AK has been supported by MS&HE “Diamond grant” no. DI2012 007842.
LIST OF ABBREVIATIONS
- ATP
adenosine triphosphate
- BMDCs
bone marrow derived cells
- BRAF
human gene that makes a protein called B-Raf; also referred to as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B1
- ccRCC
clear-cell renal cell carcinoma
- CD11b
cluster of differentiation 11b molecule
- c-KIT
proto-oncogene; encodes a transmembrane kinase which is related to the receptors for colony-stimulating factor type 1 and platelet-derived growth factor, as well as to the immunoglobulin superfamily
- CML
chronic myeloid leukemia
- CR
complete response
- CRAF
RAF proto-oncogene serine/threonine-protein kinase also known as proto-oncogene c-RAF or simply c-Raf or even Raf-1
- CSCs
cancer stem cells
- CXC
CXC chemokine receptor
- EGFR
epidermal growth factor receptor
- EMT
epithelial to mesenchymal transition
- ErbB
HER2, also known as Neu, ErbB-2, CD340 or p185, is a protein that in humans is encoded by the ERBB2 gene; a member of the epidermal growth factor receptor family
- FLK1
KDR, see: PDGF
- FLT1
FMS-related tyrosine kinase 1, see: VEGF-1
- FLT3
FMS-related tyrosine kinase 3
- FLT4
FMS-related tyrosine kinase 4, see: VEGF-2, VEGF-3
- Gli-1
sonic hedgehog downstream transcriptional factor
- GRP
gastrin-releasing peptide
- HER
human epithelial growth factor receptor
- HIF-1α
hypoxia-inducible factor 1-α
- IL-α
interleukin-α
- KDR
kinase insert domain receptor
- MMPS
metalloproteinases
- PDGF-α
platelet-derived growth factor α
- PDGF-β
platelet-derived growth factor β
- PIGHF
placental induced growth factor
- RET
protooncogene encoding a receptor tyrosine kinase for members of the glial cell line-derived neurotrophic factor family of extracellular signaling molecules
- Shh
sonic hedgehog
- SP
side population
- TAMs
tumor associated macrophages
- TGF-β
tumor growth factor beta
- TKIs
tyrosine kinase inhibitors
- TNF-α
tumor necrosis factor alpha
- VEGF-1,-2,-3
vascular endothelial growth factor 1, 2, 3
- VSMCs
vascular smooth muscle cells
AUTHORS’ DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST
The authors indicate no potential conflict of interest.
REFERENCES
- 1.Gupta K., Miller J.D., Li J.Z., et al. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): A literature review. Cancer Treat. Rev. 2008;34:193–205. doi: 10.1016/j.ctrv.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Ljungberg B., et al. The epidemiology of renal cell carcinoma. Eur. Urol. 2011;60:615–621. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 3.Murai M., Oya M. Renal cell carcinoma: etiology, incidence and epidemiology. Curr. Opin. Urol. 2004;14:229–233. doi: 10.1097/01.mou.0000135078.04721.f5. [DOI] [PubMed] [Google Scholar]
- 4.Iacovelli R., Alesini D., Palazzo A., et al. Targeted therapies and complete responses in first line treatment of metastatic renal cell carcinoma. A meta-analysis of published trials. Cancer Treat. Rev. 2013 doi: 10.1016/j.ctrv.2013.09.003. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 5.Escudier B., Eisen T., Stadler W.M., et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007;356(2):125–134. doi: 10.1056/NEJMoa060655. [Erratum in: N Engl J Med. 2007; 357]. [2]. [DOI] [PubMed] [Google Scholar]
- 6.Motzer R.J., Hutson T.E. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007;356(2):115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 7.Coppin C. Sunitinib for advanced renal cell cancer. Biologics. 2008;2(1):97–105. doi: 10.2147/btt.s1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tafreshi A., Thientosapol E., Liew M.S., et al. Efficacy of sorafenib in advanced renal cell carcinoma independent of prior treatment, histology or prognostic group. Asia Pac. J. Clin. Oncol. 2013;••• doi: 10.1111/ajco.12122. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 9.Cella D., Escudier B., Rini B., et al. Patient-reported outcomes for axitinib vs sorafenib in metastatic renal cell carcinoma: phase III (AXIS) trial. Br. J. Cancer. 2013;108(8):1571–1578. doi: 10.1038/bjc.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rini B.I., Escudier B., Tomczak P., et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378(9807):1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 11.Sternberg C.N., Davis I.D., Mardiak J., et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J. Clin. Oncol. 2010;28(6):1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 12.Motzer R.J., Nosov D., Eisen T., et al. Tivozanib Versus Sorafenib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma: Results From a Phase III Trial. J. Clin. Oncol. 2013;31(30):3791–3799. doi: 10.1200/JCO.2012.47.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhargava P., Robinson M.O. Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr. Oncol. Rep. 2011;13:103–111. doi: 10.1007/s11912-011-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arora A., Scholar E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005;315:71–79. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 15.Gotink K.J., Verheul H.M. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicklin D.J., Ellis L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol Of J Am Soc Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 17.Parikh A.A., Ellis L.M. The vascular endothelial growth factor family and its receptors. Hematol. Oncol. Clin. North Am. 2004;18:951–971. doi: 10.1016/j.hoc.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Posner I., Engel M., Gazit A., Levitzki A. Kinetics of inhibition by tyrphostins of the tyrosine kinase activity of the epidermal growth factor receptor and analysis by a new computer program. Mol. Pharmacol. 1994;45:673–683. [PubMed] [Google Scholar]
- 19.Johnson L.N. Protein kinase inhibitors: contributions from structure to clinical compounds. Q. Rev. Biophys. 2009;42:1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- 20.Karashima T., Fukuhara H., Tamura K., et al. Expression of angiogenesis-related gene profiles and development of resistance to tyrosine-kinase inhibitor in advanced renal cell carcinoma: characterization of sorafenib-resistant cells derived from a cutaneous metastasis. Int. J. Urol. 2013;20(9):923–930. doi: 10.1111/iju.12084. [DOI] [PubMed] [Google Scholar]
- 21.Figlin R.A., Kaufmann I., Brechbiel J. Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: new strategies for overcoming resistance to VEGFR and mTORC1 inhibitors. Int. J. Cancer. 2013;133(4):788–796. doi: 10.1002/ijc.28023. [DOI] [PubMed] [Google Scholar]
- 22.Rini B.I., Atkins M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10(10):992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 23.Eisenhauer E.A., Therasse P., Bogaerts J., et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Rosa R., Damiano V., Nappi L. Angiogenic and signalling proteins correlate with sensitivity to sequential treatment in renal cell cancer. Br. J. Cancer. 2013;109(3):686–693. doi: 10.1038/bjc.2013.360. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Khattak M., Larkin J. Sequential therapy with targeted agents in metastatic renal cell carcinoma: beyond second-line and overcoming drug resistance. World J. Urol. 2013 doi: 10.1007/s00345-012-103-z. [DOI] [PubMed] [Google Scholar]
- 26.Buczek M., Escudier B., Bartnik E., et al. Resistance to tyrosine kinase inhibitors in clear-cell renal cell carcinoma: From the patient's bed to molecular mechanisms. Biochim. Biophys. Acta. 2013 doi: 10.1016/j.bbcan.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Huang D., Ding Y., Li Y., et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70(3):1053–1062. doi: 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- 28.Ravaud A., Digue L., Trufflandier N., et al. VEGFR TKI 'resistance' or transient clinical insensitivity to VEGFR TKI in metastatic renal cell carcinoma. Ann. Oncol. 2010;21(2):431–432. doi: 10.1093/annonc/mdp548. [DOI] [PubMed] [Google Scholar]
- 29.Grünwald V., Weikert S., Seidel C., et al. Efficacy of sunitinib re-exposure after failure of an mTOR inhibitor in patients with metastatic RCC. Onkologie. 2011;34(6):310–314. doi: 10.1159/000328575. [DOI] [PubMed] [Google Scholar]
- 30.Harada K., Miyake H., Kusuda Y., Fujisawa M. Expression of epithelial-mesenchymal transition markers in renal cell carcinoma: impact on prognostic outcomes in patients undergoing radical nephrectomy. BJU Int. 2012;110(1):E1131–E1137. doi: 10.1111/j.1464-410X.2012.11297.x. [DOI] [PubMed] [Google Scholar]
- 31.Hugo H., Ackland M.L., Blick T., et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007;213(2):374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 32.Klymkowsky M.W., Savagner P. Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am. J. Pathol. 2009;174(5):1588–1593. doi: 10.2353/ajpath.2009.080545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 34.Hammers H.J., Verheul H.M., Salumbides B., et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol. Cancer Ther. 2010;9(6):1525–1535. doi: 10.1158/1535-7163.MCT-09-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ackland M.L., Newgreen D.F., Fridman M., et al. Epidermal growth factor-induced epithelial-mesenchymal transition in human breast carcinoma cells. Lab. Invest. 2003;83(3):435–448. doi: 10.1097/01.lab.0000059927.97515.fd. [DOI] [PubMed] [Google Scholar]
- 36.Behnsawy H.M., Shigemura K., Meligy F.Y., et al. Possible role of sonic hedgehog and epithelial-mesenchymal transition in renal cell cancer progression. Korean J. Urol. 2013;54(8):547–554. doi: 10.4111/kju.2013.54.8.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghersi G. Roles of molecules involved in epithelial/mesenchymal transition during angiogenesis. Front. Biosci. 2008;1(13):2335–2355. doi: 10.2741/2848. [DOI] [PubMed] [Google Scholar]
- 38.Frederick B.A., Helfrich B.A., Coldren C.D., et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007;6(6):1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 39.Yu M., Bardia A., Wittner B.S., et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Insan M.B., Jaitak V. New Approaches to Target Cancer Stem Cells: Current Scenario. Mini Rev. Med. Chem. 2013 doi: 10.2174/13895575113136660107. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41.Huang B., Huang Y.J., Yao Z.J., et al. Cancer stem cell-like side population cells in clear cell renal cell carcinoma cell line 769P. PLoS One. 2013;8(7):e68293. doi: 10.1371/journal.pone.0068293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollier B.G., Evans K., Mani S.A. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J. Mammary Gland Biol. Neoplasia. 2009;14(1):29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 43.Singh A., Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Q-w, Liu L, Gong C-y, et al. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heusinkveld M., van der Burg S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2009;9:216. doi: 10.1186/1479-5876-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capece D, Fischietti M, Verzella M. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages Volume. 2013 doi: 10.1155/2013/187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang X., et al. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013;332(1):3–10. doi: 10.1016/j.canlet.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 48.de Vries M., Briaire-de Bruijn I., Malessy M.J., et al. Tumor-associated macrophages are related to volumetric growth of vestibular schwannomas. Otol. Neurotol. 2013;34(2):347–352. doi: 10.1097/MAO.0b013e31827c9fbf. [DOI] [PubMed] [Google Scholar]
- 49.Liu C.Y., Xu J.Y., Shi X.Y., et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 2013;93(7):844–854. doi: 10.1038/labinvest.2013.69. [DOI] [PubMed] [Google Scholar]
- 50.Ikemoto S., Yoshida N., Narita K., et al. Role of tumor-associated macrophages in renal cell carcinoma. Oncol. Rep. 2003;10(6):1843–1849. [PubMed] [Google Scholar]
- 51.Santoni M., Massari F., Amantini C., et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2013 doi: 10.1007/s00262-013-1487-6. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006;25(3):409–416. doi: 10.1007/s10555-006-9005-3. [DOI] [PubMed] [Google Scholar]
- 53.Mantovani A., Sozzani S., Locati M., et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 54.Dannenmann S.R., Thielicke J., Stöckli M., et al. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. OncoImmunology. 2013;2(3):e23562. doi: 10.4161/onci.23562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang D., Ding Y., Zhou M., et al. Interleukin-8 mediates resistance to antiangiogenic agent. Cancer Res. 2010;70(3):1063–1071. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mizukami Y., Jo W.S., Duerr E.M., et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 2005;11(9):992–997. doi: 10.1038/nm1294. [DOI] [PubMed] [Google Scholar]
- 57.Porta C., Paglino C., Imariso I., et al. Changes in circulating pro-angiogenic cytokines, other than VEGF, before progression to sunitinib therapy in advanced renal cell carcinoma patients. Oncology. 2013;84(2):115–122. doi: 10.1159/000342099. [DOI] [PubMed] [Google Scholar]
- 58.Saylor P., Escudier B., Michaelson M.D. Importance of fibroblast growth factor receptor in neovascularization and tumor escape from antiangiogenic therapy. Clin. Genitourin. Cancer. 2012;10(2):77–83. doi: 10.1016/j.clgc.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Bhatt R.S., Wang X., Zhang L., et al. Renal cancer resistance to antiangiogenic therapy is delayed by restoration of angiostatic signaling. Mol. Cancer Ther. 2010;9(10):2793–2802. doi: 10.1158/1535-7163.MCT-10-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bergers D., Hanahan D. Modes of resistance to antiangiogenic therapy. Nat. Rev. Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergers G., Benjamin L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 62.Cao Y., Zhong W., Sun Y. Improvement of antiangiogenic cancer therapy by understanding the mechanisms of angiogenic factor interplay and drug resistance. Semin. Cancer Biol. 2009;19(5):338–343. doi: 10.1016/j.semcancer.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Tamaskar I., Dhillon J., Pili R. Resistance to angiogenesis inhibitors in renal cell carcinoma. Clin. Adv. Hematol. Oncol. 2011;9(2):101–110. [PubMed] [Google Scholar]
- 64.Hirschi K.K., d'Amore P.A. Control of angiogenesis by the pericyte: molecular mechanisms and significance. EXS. 1997;79(49) doi: 10.1007/978-3-0348-9006-9_18. [DOI] [PubMed] [Google Scholar]
- 65.Allt G., Lawrenson J.G. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169(1):1–11. doi: 10.1159/000047855. [DOI] [PubMed] [Google Scholar]
- 66.Ribatti D., Nico B., Crivellato E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011;55(3):261–268. doi: 10.1387/ijdb.103167dr. [DOI] [PubMed] [Google Scholar]
- 67.Armulik A., Genové G., Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 68.Hall A.P. Review of the pericyte during angiogenesis and its role in cancer and diabetic retinopathy. Toxicol. Pathol. 2006;34(6):763–775. doi: 10.1080/01926230600936290. [DOI] [PubMed] [Google Scholar]
- 69.Cao Z., Shang B., Zhang G., et al. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim. Biophys. Acta. 2013;1836(2):273–286. doi: 10.1016/j.bbcan.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 70.Nisancioglu M.H., Betsholtz C., Genové G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res. 2010;70(12):5109–5115. doi: 10.1158/0008-5472.CAN-09-4245. [DOI] [PubMed] [Google Scholar]
- 71.Pàez-Ribes M., Allen E., Hudock J., et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooke V.G., LeBleu V.S., Keskin D. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. 2012;21(1):66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Escudier B., Eisen T., Stadler W.M., et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007;356(2):125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 74.Gotink K.J., Broxterman H.J., Labots M., et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin. Cancer Res. 2011;17(23):7337–7346. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Houk B.E., Bello C.L., Poland B., et al. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother. Pharmacol. 2010;66(2):357–371. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- 76.Khalil B., Hudson J., Williams R., et al. An individualised dose/schedule strategy for sunitinib in metastatic renal cell cancer (mRCC) on progression-free survival (PFS): Correlation with dynamic microbubble ultrasound (DCE-US) data. J. Clin. Oncol. 2011:29. [abstr.e15149]. [Google Scholar]
- 77.Ravaud A., Gross-Goupil M. Overcoming resistance to tyrosine kinase inhibitors in renal cell carcinoma. Cancer Treat. Rev. 2012 doi: 10.1016/j.ctrv.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 78.Sierra J.R., Cepero V., Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase therapy. Mol. Cancer. 2010;9(75) doi: 10.1186/1476-4598-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Logan R., Kong A., Krise J.P., et al. Evaluating the Roles of Autophagy and Lysosomal Trafficking Defects in Intracellular Distribution-Based Drug-Drug Interactions Involving Lysosomes. J. Pharm. Sci. 2013;102(11):4173–4180. doi: 10.1002/jps.23706. [DOI] [PubMed] [Google Scholar]
- 80.Azam F., Mehta S., Harris A.L. Mechanisms of resistance to antiangiogenesis therapy. Eur. J. Cancer. 2010;46(8):1323–1332. doi: 10.1016/j.ejca.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 81.Dewhirst M.W., Cao Y., Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and therapy response. Nat. Rev. Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cristofanilli M., Charnsangavej C., Hortobagyi G.N. Angiogenesis modulation in cancer research: novel clinical approaches. Nat. Rev. Drug Discov. 2002;1(6):415–426. doi: 10.1038/nrd819. [DOI] [PubMed] [Google Scholar]