

Abstract
Objective
Lupus develops when genetically predisposed people encounter environmental agents, such as ultraviolet light, silica, infections, and cigarette smoke, that cause oxidative stress, but how oxidative damage modifies the immune system to cause lupus flares is unknown. We previously showed that inhibiting DNA methylation in CD4+ T cells by blocking ERK pathway signaling is sufficient to alter gene expression, and that the modified cells cause lupus-like autoimmunity in mice. We also reported that T cells from patients with active lupus have decreased ERK pathway signaling, have decreased DNA methylation, and overexpress genes normally suppressed by DNA methylation. This study was undertaken to test whether oxidizing agents decrease ERK pathway signaling in T cells, decrease DNA methyltransferase levels, and cause demethylation and overexpression of T cell genes similar to that found in T cells from patients with active lupus.
Methods
CD4+ T cells were treated with the oxidizers H2O2 or ONOO−. Effects on ERK pathway signaling were measured by immunoblotting, DNA methyltransferase 1 (DNMT-1) levels were measured by reverse transcriptase–polymerase chain reaction (RT-PCR), and the methylation and expression of T cell genes were measured using flow cytometry, RT-PCR, and bisulfite sequencing.
Results
H2O2 and ONOO− inhibited ERK pathway signaling in T cells by inhibiting the upstream regulator protein kinase Cδ, decreased DNMT-1 levels, and caused demethylation and overexpression of genes previously shown to be suppressed by DNA methylation in T cells from patients with active lupus.
Conclusion
Our findings indicate that oxidative stress may contribute to human lupus flares by inhibiting ERK pathway signaling in T cells to decrease DNMT-1 and cause DNA demethylation.
Lupus remains a poorly understood disease causing disability and early mortality in those affected. Familial clustering and the identification of genetic susceptibility loci indicate that genes contribute to lupus (1), but incomplete concordance in identical twins (2) and reports that drugs like procainamide and hydralazine cause a lupus-like disease in genetically predisposed people (3) indicate that an environmental component is also required. Commonly encountered environmental agents, such as ultraviolet (UV) light, cigarette smoke, and silica, and physiologic stressors like infections are also associated with lupus flares (4–6), but how these agents might trigger lupus flares is unclear. However, UV light, smoking, silica, infections, and other stressors stimulate reactive oxygen species production (7), suggesting that oxidative damage to proteins or other molecules may contribute to lupus flares.
Studies from several groups demonstrate that human lupus flares are characterized by oxidative damage to serum proteins and other molecules. The protein modifications include nitration, caused by superoxide (O2−) combining with nitric oxide (NO) to form peroxynitrite (ONOO−) (8,9), as well as increased levels of protein carbonyls, decreased protein thiol levels, and others (10,11). Lupus T cells in particular are subjected to oxidative stress. T cells from patients with active lupus have increased mitochondrial oxidative phosphorylation, resulting in O2− generation that modifies proteins, either directly by oxidation or indirectly by combining with intracellular NO (10). How T cell protein oxidation might contribute to lupus flares, though, is unclear.
Work from our group demonstrates that exogenous agents that inhibit DNA methylation can contribute to the development of lupus-like autoimmunity by modifying CD4+ T cell chromatin structure and gene expression. We reported that procainamide and hydralazine are DNA methylation inhibitors (12), and that treating normal CD4+ T cells with the DNA methyltransferase inhibitor 5-azacytidine, or procainamide or hydralazine, inhibits methylation of genes including ITGAL (CD11a) and TNFSF7 (CD70), increasing their expression and changing the T cells from antigen-specific “helper” cells into autoreactive cells (3). We also reported that T cells modified with these and other DNA methylation inhibitors are sufficient to cause lupus in animal models (3). Importantly, the same changes in T cell DNA methylation, gene expression, and cellular function are found in CD4+ T cells from patients with active lupus (3,7).
Impaired ERK pathway signaling in T cells contributes to the epigenetic changes. The signaling impairment causes a failure to up-regulate DNA methyltransferase 1 (DNMT-1) during mitosis, so DNA methylation patterns are not completely copied from the parent to the daughter cells, causing demethylation and overexpression of genes that contribute to lupus flares (3). Evidence that CD4+ T cells treated with the ERK pathway inhibitors hydralazine or U0126 have hypomethylated DNA and are sufficient to cause lupus in murine models (13), and that transgenic mice with an inducible T cell–specific ERK pathway defect similarly develop demethylated T cell DNA and lupus-like autoimmunity (14,15) demonstrates that impaired ERK pathway signaling in T cells is sufficient to cause lupus-like autoimmunity. More recent studies demonstrate that the ERK pathway defect in lupus T cells is due to protein kinase Cδ (PKCδ) nitration and that hydralazine also inhibits PKCδ (16,17).
Since UV light, infections, silica, and smoking stimulate oxidative stress (7), and increased mitochondrial oxidative phosphorylation generates reactive oxygen species like O2− and ONOO− in T cells from patients with active lupus (8–11), we hypothesized that reactive oxygen species generated from environmental exposures and/or increased mitochondrial activity may inhibit ERK pathway signaling in T cells, thereby decreasing DNMT-1 expression to cause demethylation and overexpression of genes that contribute to lupus flares. We therefore treated normal human CD4+ T cells with H2O2 or ONOO− and tested whether these oxidizers inhibit ERK pathway signaling and cause demethylation and overexpression of methylation-sensitive genes, similar to the signaling and epigenetic changes found in lupus T cells.
MATERIALS AND METHODS
Subjects
Healthy men and women without chronic inflammatory diseases or other significant illnesses who were younger than 60 years of age (mean ± SD age 38 ± 16 years; n = 15) were recruited by advertising. These studies were approved by the University of Michigan Institutional Review Board.
Chemicals and reagents
H2O2, hydralazine, and N-acetylcysteine (NAC) were purchased from Sigma-Aldrich, and ONOO− was purchased from Calbiochem.
T cell isolation and culture
Peripheral blood mononuclear cells (PBMCs) were isolated by density-gradient centrifugation and then cultured in complete tissue culture medium (RPMI 1640/10% fetal bovine serum) and stimulated for 18–24 hours with 1 μg/ml of phytohemagglutinin (PHA; Murex) as previously described (18). The cells were then washed 3 times with phosphate buffered saline (PBS) and cultured alone or with 50 μM H2O2, 50 μM ONOO−, 10 μM hydralazine, and/or 1 mM NAC for another 72 hours. CD4+ cells were then isolated by negative selection using magnetic beads (CD4+ T cell isolation kit; Miltenyi Biotec), according to the manufacturer's instructions, and used immediately.
Flow cytometry
Cy5-conjugated anti-CD3, fluorescein isothiocyanate (FITC)–conjugated anti-CD8, Cy5-conjugated anti-CD8, Cy5-conjugated anti-CD4, phycoerythrin (PE)–conjugated anti-CD158b (killer cell immunoglobulin-like receptor 2DS2 [KIR-2DS2]), PE-conjugated anti-NKB1 (KIR-3DL1), and FITC-conjugated anti-CD28 were obtained from BD PharMingen. PE-conjugated anti-CD158d (KIR-2DL4) was obtained from R&D Systems and PE-conjugated anti-CD158a/h (KIR-2DL1/2DS2) and PE-conjugated anti-CD158b1/b2 (KIR-2DL2/2DL3/2DS2) were obtained from Beckman Coulter Immunotech. Five microliters of each PE-conjugated anti-KIR antibody was mixed to form a cocktail and used to stain the cells (19). All labeling procedures were performed on ice in PBS containing 10% horse serum and normal human AB serum (Invitrogen) and sodium azide. Cell staining was performed as previously described (20), and the stained cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Reverse transcriptase–polymerase chain reaction (RT-PCR)
Total RNA was isolated using the RNeasy Mini kit (Qiagen). KIR-2DL4, CD70, DNMT-1, and β-actin transcripts were measured in bead-purified T cells using a Rotor-Gene 3000 (Corbett Robotics) and the QuantiTect SYBR Green RT-PCR kit (Qiagen) according to the manufacturer's instructions. CD70, DNMT-1, and β-actin primers and amplification protocols have been described previously (20–22). Primers designed to match polymorphic positions unique to KIR-2DL4 genes have been described previously (20,23).
ERK pathway immunoblot assays
Purified CD4+ T cells were suspended in RPMI 1640 supplemented with 10% fetal calf serum, 2 mM glutamine and penicillin/streptomycin and then left unstimulated or stimulated with 50 ng/ml phorbol myristate acetate (PMA) for 15 minutes at 37°C. Where indicated, cells were incubated in the presence or absence of hydralazine (10 μM), H2O2 (50 μM), or ONOO− (50 μM) for 60 minutes before PMA stimulation. Following stimulation, the cells were lysed, then total and phosphorylated ERK, MEK, RAF, and PKCδ were measured by immunoblotting as described (16). The following primary antibodies were used at a 1:1,000 dilution: anti–phospho-Raf (Ser338), anti–phospho–MEK-1/2 (Ser217/221), anti–MEK-1/2, and anti–phospho-PKCδ (T505) (Cell Signaling Technology). Polyclonal rabbit anti-active MAPK (1:5,000) was purchased from Promega. Secondary antibodies included anti-rabbit IgG horseradish peroxidase (HRP; 1:2,000) (Cell Signaling Technology) and anti-mouse IgG HRP (1:4,000; Sigma-Aldrich).
Pyrosequencing
Genomic DNA was isolated from T cells using the DNeasy Blood and Tissue kit (Qiagen) and then bisulfite treated using the EZ DNA Methylation-Gold kit (Zymo Research). CD70 and KIR-2DL4 promoter methylation were determined by Pyrosequencing as described previously (20).
Statistical analysis
Statistical analyses were performed using Student's t-test or analysis of variance (ANOVA) (24) in Systat software.
RESULTS
Increased methylation-sensitive T cell gene expression after treatment with H2O2 or ONOO−
CD4+ T cell TNFSF7 (CD70) and KIR genes are silenced primarily by DNA methylation, and inhibiting the replication of DNA methylation patterns during mitosis is sufficient to demethylate their respective regulatory regions and induce gene expression (20,22). These genes are also aberrantly demethylated and expressed on CD4+ T cells from patients with active lupus (25). Initial experiments were conducted to test whether oxidizing agents induce the expression of these genes. We selected H2O2 because this oxidizer is generated by mitochondrial oxidative phosphorylation and superoxide dismutase in metabolically active T cells such as those described in lupus flares (8), and ONOO− because this oxidizer contributes to protein nitration in lupus (10).
PBMCs from healthy individuals were stimulated for 18 hours with PHA and treated with the indicated concentrations of ONOO− or H2O2 for 72 hours. CD4+ T cell CD70 and KIR expression were then measured by flow cytometry and RT-PCR using protocols similar to those previously used to activate these and other epigenetically silenced T cell genes with DNMT inhibitors such as 5-azacytidine (18,20,22). As shown in Figure 1A, normal CD4+ T cells did not express KIR proteins, and ONOO− caused a dose-dependent increase in the number of KIR-expressing CD4+ T cells. Figure 1B similarly shows that relatively few CD4+ T cells normally express CD70, and that ONOO− increased the percentage of CD4+CD70+ T cells, similar to its effects on KIR. H2O2 also increased the percentages of CD4+KIR+ T cells (Figure 1C) and CD4+CD70+ T cells (Figure 1D).
Figure 1.
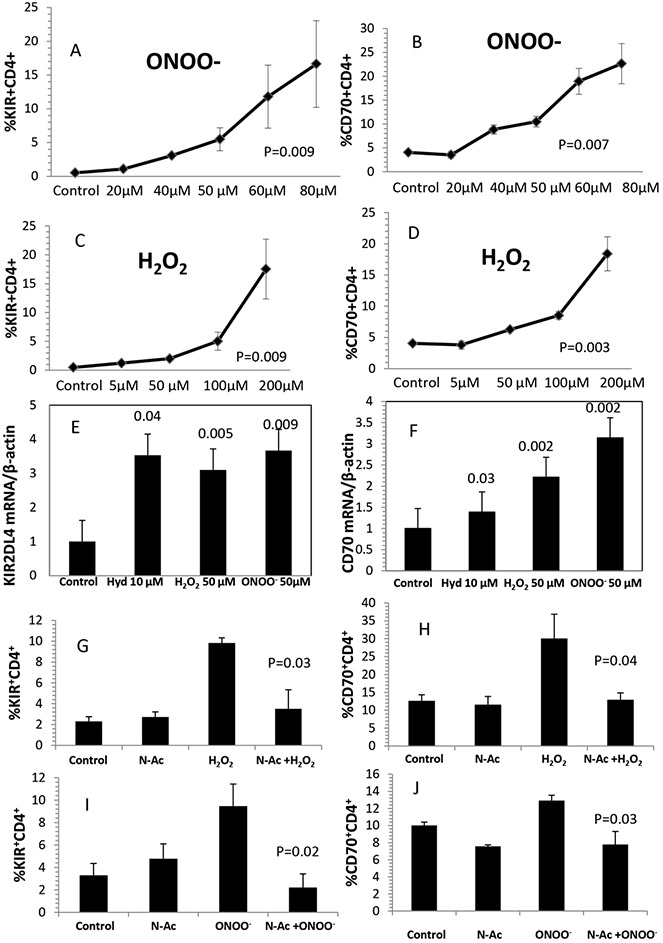
Effects of oxidative stress on killer cell immunoglobulin-like receptor (KIR) and CD70 gene expression in T cells. A–D, Peripheral blood mononuclear cells (PBMCs) from 3 healthy young donors were stimulated with phytohemagglutinin (PHA), treated with ONOO− (A and B) or H2O2 (C and D), and then stained with fluorochrome-conjugated antibodies to CD4, CD8, and KIR proteins (A and C) or fluorochrome-conjugated anti-CD70 (B and D) and analyzed by flow cytometry. Values are the mean ± SEM of 3 determinations. P values were determined by analysis of variance. E and F, PBMCs from 4 healthy young donors were stimulated with PHA and then treated with the indicated concentrations of hydralazine (Hyd), H2O2, or ONOO− as in A–D. CD4+ cells were then purified and KIR-2DL4 (E) or CD70 (F) transcripts were measured by reverse transcriptase–polymerase chain reaction. Results are presented relative to untreated T cells. Bars show the mean ± SEM. Values above the bars are P versus control, by t-test. G–J, PBMCs from healthy young donors were stimulated and then cultured alone (control) or with 1 mM N-acetylcysteine (NAC), 50 μM H2O2, 50 μM ONOO−, 1 mM NAC plus 50 μM H2O2, or 1 mM NAC plus 50 μM ONOO−. The cells were then stained for CD4 and KIR (G and I) or CD4 and CD70 (H and J) and analyzed as above. Bars show the mean ± SEM from 3 independent experiments in G and I and from 5 independent experiments in H and J. P values are versus H2O2 in G and H and versus ONOO− in I and J.
The effects of H2O2 and ONOO− on CD4+ T cell KIR and CD70 gene expression were confirmed at the messenger RNA (mRNA) level. The KIR gene locus contains ∼14 closely related genes, and most people inherit a different repertoire of KIR genes. Everyone, however, has KIR2DL4 (26). CD4+ T cells from healthy volunteers were stimulated with PHA and treated with 50 μM H2O2 or 50 μM ONOO−, and 72 hours later KIR2DL4 and CD70 mRNA were measured by RT-PCR, and the results were expressed relative to β-actin. Hydralazine (10 μM), which inhibits DNA methylation by blocking ERK pathway signaling through its effects on PKCδ (16), was included as a control. All 3 agents increased KIR mRNA levels (Figure 1E) and CD70 mRNA levels (Figure 1F), consistent with our previous findings with other DNA methylation inhibitors (19,22).
We next tested whether the antioxidant NAC inhibited the effects of H2O2 and ONOO− on CD4+ T cell gene expression. NAC is a thiol-containing antioxidant that combines rapidly with reactive oxygen species (27), does not inhibit T cell proliferation (28), and has been reported to decrease oxidative stress and flare severity in lupus patients (29). As shown in Figure 1G, 1 mM NAC had no effect on T cell KIR expression, while H2O2 increased KIR expression, and NAC prevented the effect of H2O2 on KIR expression (P = 0.03, H2O2 alone versus H2O2 and NAC). The combination of H2O2 and NAC also inhibited the effect of H2O2 on T cell CD70 expression (P = 0.04) (Figure 1H). Similarly, Figure 1I shows that NAC inhibited the effect of ONOO− on CD4+ KIR expression (P = 0.02), and Figure 1J shows the same inhibitory effect on CD70 expression induced by ONOO− (P = 0.03). These results demonstrate that the effects of these oxidizing agents on CD4+ T cell gene expression can be prevented with a clinically tested and safe antioxidant.
Controls included comparing the effects of ONOO−, H2O2, and hydralazine on CD4+CD28+ and CD4+CD28− T cells. CD28− T cells are a “senescent” subset with shortened telomeres and decreased replicative potential. This subset is present in small numbers in younger (<60 years of age) healthy people, but increases with further aging. The subset develops earlier in patients with chronic inflammatory diseases like rheumatoid arthritis and also with chronic stimulation in vitro, likely due to replicative stress (30). Importantly, this subset overexpresses a repertoire of methylation-sensitive genes similar to those overexpressed by experimentally demethylated T cells (21,31). We used flow cytometry to compare the effects of ONOO−, H2O2, and hydralazine on KIR and CD70 expression by this subset in the same subjects. Hydralazine, H2O2, and ONOO− all increased KIR expression on the CD4+CD28+ and CD4+CD28− subsets (P < 0.05 for all 3 treatments versus untreated controls) (data available from the author upon request). However, while hydralazine, H2O2, and ONOO− increased CD70 expression on the CD4+CD28+ subset, the effect of this oxidizer on CD70 expression by the CD4+CD28− subset was much smaller, and was of borderline significance only for H2O2 (P = 0.048). In all cases except hydralazine, the increase was somewhat greater on the CD28+ subset than on the CD28− subset (P < 0.05). However, since the CD4+CD28− subset represents a small subset within the total CD4+ population (mean ± SD 6.3 ± 2.2% in this cohort), we used unfractionated CD4+ cells in further experiments.
Inhibition of T cell DNA methylation by H2O2 and ONOO−
Since the CD70 and KIR genes are suppressed by DNA methylation in CD4+ T cells, we hypothesized that H2O2 and ONOO− may inhibit methylation of the same regulatory regions in or near the TNFSF7 (CD70) and KIR2DL4 genes previously identified by our group in CD4+ lupus T cells, as well as in CD4+ T cells treated with other DNA methylation inhibitors (20,22). PBMCs from healthy subjects were stimulated with PHA and treated with 50 μM H2O2 or 50 μM ONOO−, and 72 hours later CD4+ T cells were isolated and methylation of their regulatory regions was measured by Pyrosequencing.
Figure 2A shows the effects of H2O2 and ONOO− on KIR2DL4 methylation relative to the methylation of the same CG pair in untreated T cells (P < 0.001; n = 4 experiments), and Figure 2B shows their effects on the methylation of TNFSF7, also relative to untreated T cells (P < 0.001; n = 3 experiments). Both oxidizing agents caused a significant decrease in the methylation of the same sequences previously shown to suppress transcription when methylated in vitro (20,22). Importantly, the same sequences demethylate in CD4+ T cells from patients with active lupus but not those with inactive lupus (19,20,22).
Figure 2.
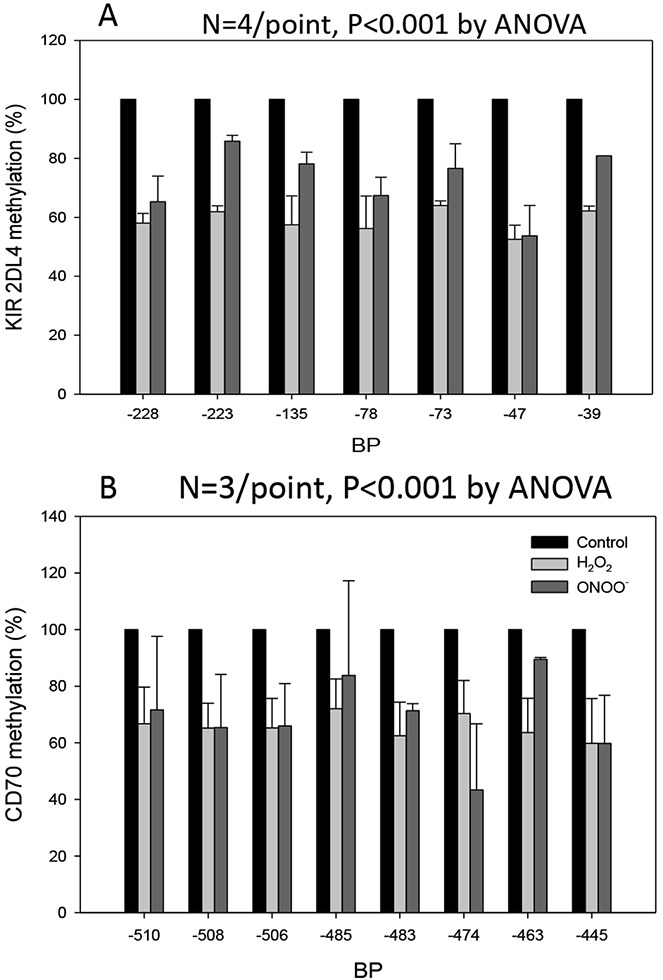
H2O2 and ONOO− inhibit T cell DNA methylation. Peripheral blood mononuclear cells from healthy young donors were stimulated with phytohemagglutinin and then washed and cultured alone or with 50 μM H2O2 or 50 μM ONOO− added on days 1, 2, and 3. CD4+ cells were then isolated and DNA purified, and killer cell immunoglobulin-like receptor 2DL4 (KIR2DL4) (A) and TNFSF7 (CD70) (B) promoter methylation was measured by bisulfite sequencing. Results are presented as the average methylation of dC bases in CG pairs numbered relative to the transcription start site. Bars show the mean ± SEM of 4 experiments for KIR2DL4 and 3 experiments for TNFSF7. P values are versus control for each sequence. ANOVA = analysis of variance.
Controls included comparing the methylation status of 3 CG pairs located −440, −428, and −389 bp 5′ of the TNFSF7 transcription start site. CD4+ T cells from 4 healthy donors were left untreated or treated with H2O2 or ONOO−, and methylation of the same sequences was compared using bisulfite sequencing. We previously demonstrated that these 3 CG pairs remain methylated in CD4+ T cells treated with the DNMT inhibitors 5-azacytidine and procainamide, with the MEK inhibitors PD98059 and U0126, and with the PKCδ inhibitor hydralazine (22). H2O2 and ONOO− had no significant effect on the methylation of these CG pairs (data are available online at http://sitemaker.umich.edu/brlab/home).
T cell DNA methylation patterns are replicated during mitosis primarily by DNMT-1. We tested whether H2O2 and ONOO− decrease CD4+ T cell DNMT-1 levels. PBMCs from 4 healthy subjects were stimulated with PHA for 18 hours and then cultured alone or with 50 μM H2O2 or 50 μM ONOO− as described in the Figure 1 legend. Twenty-four hours later, CD4+ cells were purified, RNA was isolated, DNMT-1 transcripts were measured by RT-PCR, and the results were expressed relative to β-actin. Hydralazine (10 μM), which inhibits T cell DNA methylation by decreasing DNMT-1 levels through inhibitory effects on PKCδ activation and ERK pathway signaling (16), was included as a positive control. Figure 3 shows that all 3 agents caused a similar decrease in DNMT-1 levels.
Figure 3.
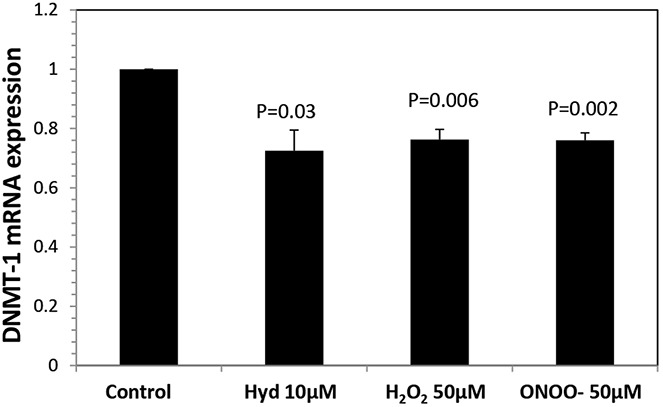
DNMT-1 mRNA levels in CD4+ T cells treated with H2O2 or ONOO−. Peripheral blood mononuclear cells from 4 young donors were stimulated and cultured alone or with 10 μM hydralazine (Hyd), 50 μM H2O2, or 50 μM ONOO− as described in the Figure 1 legend. CD4+ cells were isolated 24 hours later, and RNA was isolated and DNMT-1 transcripts were measured relative to β-actin. Bars show the mean ± SEM. P values are versus control.
Since both H2O2 and ONOO− decreased DNMT-1 levels, we next determined if H2O2 and ONOO− also inhibited ERK pathway signaling, similar to hydralazine and the defect in lupus T cells (16). PBMCs from healthy young controls were stimulated with PHA for 18 hours, and then CD4+ T cells were isolated, treated with 50 μM H2O2, 50 μM ONOO−, or 10 μM hydralazine for 60 minutes, and then restimulated with PMA (32). Fifteen minutes later the cells were lysed, and cytoplasmic proteins were then fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes, and serially probed with antibodies to the active dually phosphorylated forms of ERK-1/2, phospho–MEK-1/2 Ser217/221, and Raf-1 p-Ser338. β-actin was used as a loading control. Figure 4A shows representative immunoblots of phospho-Raf, phospho-MEK, phospho-ERK, and actin in PMA-stimulated T cells that were left untreated or treated with hydralazine, H2O2, or ONOO−. Figure 4B shows the mean ± SEM of the densitometric analyses from 6 independent experiments comparing the effects of the test reagents on PMA-stimulated Raf phosphorylation, while Figure 4C shows the effects on MEK, and Figure 4D shows the effects on ERK. All 3 reagents decreased ERK pathway signaling, indicating a common pathway for the effects of hydralazine, H2O2, and ONOO− on CD4+ T cell gene expression.
Figure 4.
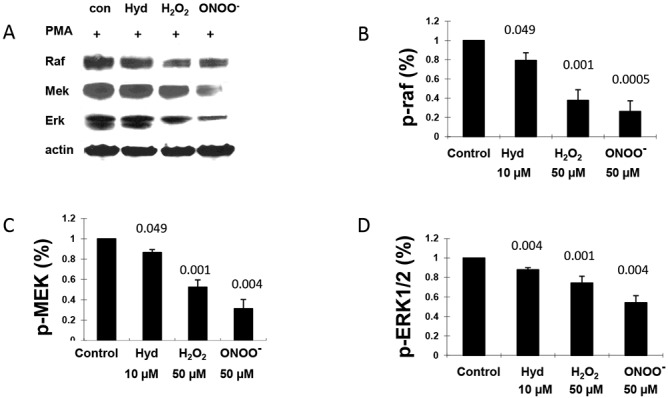
Decreased ERK pathway signaling in CD4+ T cells treated with H2O2 or ONOO−. CD4+ T cells from 6 healthy young donors were treated with 10 μM hydralazine (Hyd), 50 μM H2O2, or 50 μM ONOO− for 60 minutes and then stimulated with phorbol myristate acetate (PMA) for 15 minutes. The cells were then lysed and Raf, MEK, and ERK phosphorylation was measured by immunoblotting. A, Representative immunoblots of Raf, MEK, and ERK phosphorylation in untreated cells and cells treated with hydralazine, H2O2, or ONOO−. B–D, Phosphorylation of Raf (B), MEK (C), and ERK (D) in cells treated with hydralazine, H2O2, or ONOO−. Bars show the mean ± SEM. Values above the bars are P versus control.
PKCδ inactivation
We previously showed that oxidative damage to PKCδ contributes to the ERK pathway signaling defect in lupus T cells (16), and that hydralazine also inhibits PKCδ (33). We therefore investigated if H2O2 and ONOO− also inactivate T cell PKCδ. CD4+ T cells were isolated from healthy young controls, treated with 10 μM hydralazine, 50 μM H2O2, or 50 μM ONOO− for 60 minutes, and then stimulated with PMA as in Figure 4. Hydralazine, H2O2, and ONOO− all inhibited PKCδ phosphorylation (Figure 5).
Figure 5.
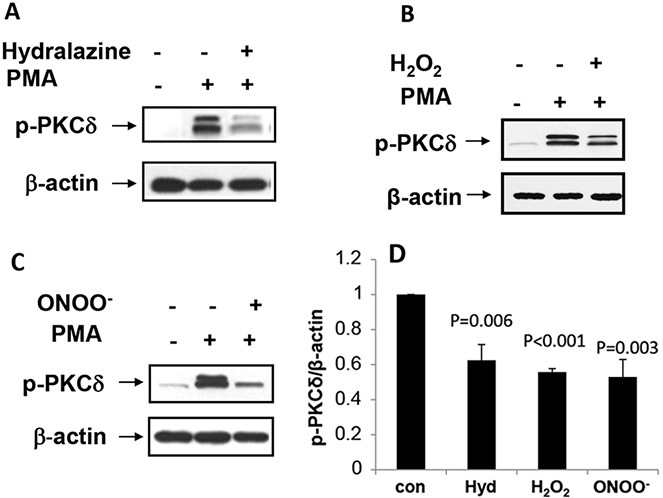
Protein kinase Cδ (PKCδ) inactivation in CD4+ T cells treated with hydralazine (Hyd), H2O2, or ONOO−. A, CD4+ T cells from a healthy young donor were cultured with 10 μM hydralazine for 60 minutes, stimulated with phorbol myristate acetate (PMA) for 15 minutes, and then lysed, and PKCδ phosphorylation was measured by immunoblotting. β-actin was used as a loading control. B, CD4+ T cells from a healthy young donor were cultured with 50 μM H2O2 and stimulated with PMA, and PKCδ phosphorylation was measured by immunoblotting. C, CD4+ T cells from a healthy young donor were cultured with 50 μM ONOO− for 60 minutes and stimulated with PMA for 15 minutes, and PKCδ phosphorylation was measured by immunoblotting. D, PKCδ phosphorylation was quantified in CD4+ T cells treated with hydrazaline, H2O2, or ONOO−. Bars show the mean ± SEM from 6 similar experiments for each treatment. P values are versus control.
DISCUSSION
T cells are typically activated during the course of an immune response, and the increased metabolic demands stimulate mitochondrial oxidative phosphorylation, resulting in ATP generation to provide energy for the synthetic reactions. However, oxidative phosphorylation also generates reactive oxygen intermediates (ROIs) such as O2− (8). T cell stimulation also generates NO which acts as an intracellular signaling molecule (8). Importantly, NO can combine with O2− to form ONOO−, which nitrates intracellular molecules, including proteins, DNA, and lipids (10). The consequences of these covalent modifications on CD4+ T cell function in normal immune responses are incompletely understood, but chronic stimulation and consequent oxidative stress may result in the death of some T cells through apoptosis (34) or necrosis (8) as well as the epigenetic modifications described above.
The effects of oxidative stress on T cell function and gene expression appear to be clinically silent in most people who have ongoing inflammatory immune responses, but who lack a sufficient number of genes predisposing to lupus and perhaps other forms of autoimmunity. In contrast, lupus tends to flare when people with a sufficiently large total genetic risk for lupus encounter environmental agents, such as UV light, infections, silica, cigarette smoke, and others, that stimulate inflammatory responses (4–6). Evidence for the oxidative damage comes from reports that serum proteins from patients with active lupus have elevated levels of 3-nitrotyrosine, caused by ONOO− reacting with Tyr, as well as elevated levels of protein-bound carbonyls relative to healthy controls (9). ONOO− can also oxidize lipids including low-density lipoproteins (LDLs), and circulating complexes of antiphospholipid antibodies and oxidized LDLs are found in lupus patients with antiphospholipid antibody syndrome (35). The inflammatory response caused by the autoimmune flare may also further increase the amount of ROIs generated.
The experiments presented above demonstrate that treating dividing T cells with H2O2 or ONOO− demethylates T cell DNA by decreasing ERK pathway signaling, decreasing DNMT-1 expression, and preventing the replication of DNA methylation patterns during mitosis, similar to previous results with the PKCδ inhibitor hydralazine (16) as well as with the DNMT-1 inhibitors 5-azacytidine and procainamide, and the MEK inhibitors PD98059 and U0126. Further, the same KIR2DL4 sequences demethylate in CD4+ T cells from patients with active but not inactive lupus (19), consistent with our previous reports that the methylation status of some T cell DNA sequences is less stringently maintained than others (36–38). Demethylation of regulatory elements then increases the expression of T cell genes normally suppressed at least in part by DNA methylation, including CD70 and KIR (20,22). We also found that 1 mM (163 mg/liter) NAC, an antioxidant, prevented these epigenetic effects. This observation further supports a role for ROIs in gene demethylation and activation, and suggests that environmental agents that cause inflammation may stimulate generation of reactive oxygen species that have similar epigenetic effects on T lymphocyte gene expression in vivo. This is clinically relevant because 600 mg of NAC can be safely administered twice a day for 2 days to prevent contrast-induced kidney damage (39), and one 600 mg dose gives a peak blood level of 465 mg/liter (40). This suggests that NAC may also prevent or treat lupus flares, as proposed by Lai et al (29).
Importantly, inhibiting T cell PKCδ activation and downstream ERK pathway signaling is sufficient to cause lupus-like autoimmunity in animal models. We reported that inhibiting CD4+ T cell DNA methylation, either by treatment with direct DNMT-1 inhibitors such as procainamide and 5-azacytidine, or with ERK pathway inhibitors such as U0126 or hydralazine, causes demethylation and overexpression of genes including CD11a, perforin, CD70, and the KIR gene family (3,20), and that injecting CD4+ T cells treated with either DNMT-1 inhibitors or ERK pathway inhibitors is sufficient to cause lupus-like autoimmunity in syngeneic mice (13). Importantly, procainamide and hydralazine cause drug-induced lupus (3). Further, MEK is a key signaling molecule in the ERK pathway, and inducing expression of a dominant negative MEK selectively in T cells also causes lupus-like autoimmunity in transgenic mice by decreasing DNMT-1 levels (14,15), indicating the importance of ERK pathway defects in lupus-like diseases.
Abnormal ERK pathway signaling in T cells may similarly contribute to flares in patients with lupus. Lupus T cells are characterized by multiple intracellular signaling abnormalities, including decreased ERK pathway signaling, increased mammalian target of rapamycin signaling, enhanced Ca2+ fluxes, TCRζ dysregulation, decreased Elf-1 levels, increased PP2A expression, and others (41,42). The mechanism(s) causing these abnormalities are unclear, and whether the abnormalities are secondary to the disease process or contribute to disease flares is unknown. However, since decreased ERK pathway signaling in T cells causes lupus-like autoimmunity in animal models (14,15), the ERK pathway signaling defect found in T cells from patients with active lupus may be causative rather than secondary to the consequences of a lupus flare. The role of oxidative stress in the other lupus T cell signaling abnormalities is unknown.
Linking oxidative stress to epigenetic changes may have direct therapeutic relevance to lupus. First, avoiding environmental agents that cause oxidative stress may delay disease onset and prevent flares. Second, antioxidants like NAC or others may have therapeutic benefit by scavenging the reactive oxygen species generated during inflammatory responses, possibly preventing flares or decreasing flare severity (29).
In summary, agents causing oxidative stress are implicated in triggering lupus flares (4–6). The findings of the present study indicate that the ROIs mediating oxidative stress can contribute to epigenetic changes in T cells that resemble those found in patients with active lupus (3,25,43) and that cause lupus-like autoimmunity in animal models (14). These studies thus suggest that environmental agents that cause inflammatory responses may trigger lupus flares in genetically predisposed people through effects on T cell signaling pathways, and that antioxidants may help prevent lupus onset and flares.
Acknowledgments
The authors thank Ms Sushma Yarlagadda for expert technical assistance and Ms Patricia Bergeron for expert administrative assistance.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Richardson had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Li, Gorelik, Strickland, Richardson.
Acquisition of data. Li, Gorelik, Strickland.
Analysis and interpretation of data. Li, Gorelik, Strickland, Richardson.
REFERENCES
- 1.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72(Suppl 2):ii56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdanos DP, Smyk DS, Rigopoulou EI, Mytilinaiou MG, Heneghan MA, Selmi C, et al. Twin studies in autoimmune disease: genetics, gender and environment. J Autoimmun. 2012;38:J156–69. doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol. 2007;3:521–7. doi: 10.1038/ncprheum0573. [DOI] [PubMed] [Google Scholar]
- 4.Cooper GS, Gilbert KM, Greidinger EL, James JA, Pfau JC, Reinlib L, et al. Recent advances and opportunities in research on lupus: environmental influences and mechanisms of disease. Environ Health Perspect. 2008;116:695–702. doi: 10.1289/ehp.11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zandman-Goddard G, Solomon M, Rosman Z, Peeva E, Shoenfeld Y. Environment and lupus-related diseases. Lupus. 2012;21:241–50. doi: 10.1177/0961203311426568. [DOI] [PubMed] [Google Scholar]
- 6.Rogers MA, Levine DA, Blumberg N, Fisher GG, Kabeto M, Langa KM. Antigenic challenge in the etiology of autoimmune disease in women. J Autoimmun. 2012;38:J97–102. doi: 10.1016/j.jaut.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson B, Somers E. Environmental exposures, epigenetic changes and the risk of lupus. Lupus. doi: 10.1177/0961203313499419. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy G, Koncz A, Fernandez D, Perl A. Nitric oxide, mitochondrial hyperpolarization, and T cell activation. Free Radic Biol Med. 2007;42:1625–31. doi: 10.1016/j.freeradbiomed.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oates JC, Shaftman SR, Self SE, Gilkeson GS. Association of serum nitrate and nitrite levels with longitudinal assessments of disease activity and damage in systemic lupus erythematosus and lupus nephritis. Arthritis Rheum. 2008;58:263–72. doi: 10.1002/art.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oates JC, Gilkeson GS. The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clin Immunol. 2006;121:243–50. doi: 10.1016/j.clim.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan PE, Sturgess AD, Davies MJ. Increased levels of serum protein oxidation and correlation with disease activity in systemic lupus erythematosus. Arthritis Rheum. 2005;52:2069–79. doi: 10.1002/art.21130. [DOI] [PubMed] [Google Scholar]
- 12.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140:2197–200. [PubMed] [Google Scholar]
- 13.Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal–regulated kinase pathway signaling. Arthritis Rheum. 2003;48:746–56. doi: 10.1002/art.10833. [DOI] [PubMed] [Google Scholar]
- 14.Strickland FM, Hewagama A, Lu Q, Wu A, Hinderer R, Webb R, et al. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J Autoimmun. 2012;38:J135–43. doi: 10.1016/j.jaut.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–78. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T cell protein kinase Cδ activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J Immunol. 2007;179:5553–63. doi: 10.4049/jimmunol.179.8.5553. [DOI] [PubMed] [Google Scholar]
- 17.Gorelik GJ, Yarlagadda S, Patel D, Richardson BC. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum. 2012;64:2964–74. doi: 10.1002/art.34503. [published erratum appears in Arthritis Rheum 2014;66:769]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Q, Richardson B. Methods for analyzing the role of DNA methylation and chromatin structure in regulating T lymphocyte gene expression. Biol Proced Online. 2004;6:189–203. doi: 10.1251/bpo89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basu D, Liu Y, Wu A, Yarlagadda S, Gorelik GJ, Kaplan MJ, et al. Stimulatory and inhibitory killer Ig-like receptor molecules are expressed and functional on lupus T cells. J Immunol. 2009;183:3481–7. doi: 10.4049/jimmunol.0900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Kuick R, Hanash S, Richardson B. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin Immunol. 2009;130:213–24. doi: 10.1016/j.clim.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Chen Y, Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4+CD28− T cells. Clin Immunol. 2009;132:257–65. doi: 10.1016/j.clim.2009.03.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005;174:6212–9. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 23.Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–63. doi: 10.1016/s1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- 24.Herzog E, Galvez J, Roks A, Stolk L, Verbiest M, Eilers P, et al. Tissue-specific DNA methylation profiles in newborns. Clin Epigenetics. 2013;5:8. doi: 10.1186/1868-7083-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel DR, Richardson BC. Dissecting complex epigenetic alterations in human lupus. Arthritis Res Ther. 2013;15:201. doi: 10.1186/ar4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Middleton D, Williams F, Halfpenny IA. KIR genes. Transpl Immunol. 2005;14:135–42. doi: 10.1016/j.trim.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Gillissen A, Nowak D. Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy. Respir Med. 1998;92:609–23. doi: 10.1016/s0954-6111(98)90506-6. [DOI] [PubMed] [Google Scholar]
- 28.Novaes R, Freire-de-Lima CG, de Albuquerque RC, Affonso-Mitidieri OR, Espindola O, Lima MA, et al. Modulation of glutathione intracellular levels alters the spontaneous proliferation of lymphocyte from HTLV-1 infected patients. Immunobiology. 2013;218:1166–74. doi: 10.1016/j.imbio.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64:2937–46. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broux B, Markovic-Plese S, Stinissen P, Hellings N. Pathogenic features of CD4+CD28− T cells in immune disorders. Trends Mol Med. 2012;18:446–53. doi: 10.1016/j.molmed.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Gorelik GJ, Strickland FM, Richardson BC. Decreased ERK and JNK signaling contribute to gene overexpression in “senescent” CD4+CD28− T cells through epigenetic mechanisms. J Leukoc Biol. 2010;87:137–45. doi: 10.1189/jlb.0809562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J. 2013;452:195–209. doi: 10.1042/BJ20130220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorelik G, Barreiro Arcos ML, Klecha AJ, Cremaschi GA. Differential expression of protein kinase C isoenzymes related to high nitric oxide synthase activity in a T lymphoma cell line. Biochim Biophys Acta. 2002;1588:179–88. doi: 10.1016/s0925-4439(02)00163-1. [DOI] [PubMed] [Google Scholar]
- 34.Tirosh O, Aronis A, Melendez JA. Mitochondrial state 3 to 4 respiration transition during Fas-mediated apoptosis controls cellular redox balance and rate of cell death. Biochem Pharmacol. 2003;66:1331–4. doi: 10.1016/s0006-2952(03)00481-7. [DOI] [PubMed] [Google Scholar]
- 35.Passam FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular pathophysiology of the antiphospholipid syndrome: the role of oxidative post-translational modification of β2 glycoprotein I. J Thromb Haemost. 2011;9(Suppl 1):275–82. doi: 10.1111/j.1538-7836.2011.04301.x. [DOI] [PubMed] [Google Scholar]
- 36.Lu Q, Kaplan M, Ray D, Ray D, Zacharek S, Gutsch D, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002;46:1282–91. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 37.Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, et al. DNA methylation and chromatin structure regulate T cell perforin gene expression. J Immunol. 2003;170:5124–32. doi: 10.4049/jimmunol.170.10.5124. [DOI] [PubMed] [Google Scholar]
- 38.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007;179:6352–8. doi: 10.4049/jimmunol.179.9.6352. [DOI] [PubMed] [Google Scholar]
- 39.Rehman T, Fought J, Solomon R. N-acetylcysteine effect on serum creatinine and cystatin C levels in CKD patients. Clin J Am Soc Nephrol. 2008;3:1610–4. doi: 10.2215/CJN.01560408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izzedine H, Guerin V, Launay-Vacher V, Bernard M, Deray G. Effect of N-acetylcysteine on serum creatinine level. Nephrol Dial Transplant. 2001;16:1514–15. doi: 10.1093/ndt/16.7.1514. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. 2010;9:173–8. [PMC free article] [PubMed] [Google Scholar]
- 42.Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:207. doi: 10.1186/ar3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel DR, Richardson BC. Epigenetic mechanisms in lupus. Curr Opin Rheumatol. 2010;22:478–82. doi: 10.1097/BOR.0b013e32833ae915. [DOI] [PMC free article] [PubMed] [Google Scholar]