

Abstract
Advanced prostate cancer is characterized by incurable castration-resistant progression and osteoblastic bone metastasis. While androgen deprivation therapy remains the primary treatment for advanced prostate cancer, resistance inevitably develops. Importantly, mounting evidence indicates that androgen receptor (AR) signaling continues to play a critical role in the growth of advanced prostate cancer despite androgen deprivation. While the mechanisms of aberrant AR activation in advanced prostate cancer have been extensively studied, the downstream AR target genes involved in the progression of castration resistance are largely unknown. Here, we identify WNT7B as a direct AR target gene highly expressed in castration-resistant prostate cancer (CRPC) cells. Our results show that expression of WNT7B is necessary for the growth of prostate cancer cells and that this effect is enhanced under androgen-deprived conditions. Further analyses reveal that WNT7B promotes androgen-independent growth of CRPC cells likely through the activation of protein kinase C isozymes. Our results also show that prostate cancer-produced WNT7B induces osteoblast differentiation in vitro through a direct cell–cell interaction, and that WNT7B is upregulated in human prostate cancer xenografts that cause an osteoblastic reaction when grown in bone. Taken together, these results suggest that AR-regulated WNT7B signaling is critical for the growth of CRPC and development of the osteoblastic bone response characteristic of advanced prostate cancer.
Introduction
Prostate cancer is dependent on androgens for growth. While androgen deprivation therapy for advanced prostate cancer effectively reduces tumor burden, the disease typically recurs as incurable castration-resistant prostate cancer (CRPC). The progression of CRPC is often associated with metastases that occur primarily in bone (1). While both osteolytic (bone lysing) and osteoblastic (bone forming) processes occur during prostate cancer bone metastasis, prostate cancer predominantly yields osteoblastic lesions, in contrast to the mostly osteolytic lesions observed in other cancers (lung, kidney, and breast). Studies suggest that tumor-induced osteoblastic and osteolytic activity may play different roles in promoting tumor growth (2). While anti-osteolytic therapies have been developed, they show limited effectiveness against osteoblastic bone disease in prostate cancer. Antiosteoblastic approaches still lack therapeutic targets and remain restricted to palliative radiotherapy and systemic chemotherapy (3). Increased understanding of the mechanisms underlying castration resistance and osteoblastic bone metastases will provide opportunities to develop more effective therapies for advanced prostate cancer.
WNT signaling plays a central role in both developmental and oncogenic processes. There are 19 closely related WNT genes identified in humans. Secreted WNT proteins bind to the Frizzled receptors and other coreceptors at the plasma membrane and initiate canonical or noncanonical intracellular signaling cascades (4). Canonical WNT signaling stabilizes intracellular β-catenin by preventing its phosphorylation-dependent degradation, resulting in transcriptional activation of WNT target genes. Noncanonical WNT signaling activates intracellular kinases such as c-Jun-NH2 kinase (JNK), Ca2+/calmodulin-dependent protein kinase II (CaMKII), and protein kinase C (PKC). Aberrant WNT signaling has been linked to a number of cancers. In prostate cancer, nuclear β-catenin was frequently detected in advanced prostate cancer despite the infrequency of β-catenin mutations (∼5%) in tumors (5, 6). Gene expression profiles of prostate cancer often show overexpression of WNT ligands or underexpression of secreted WNT inhibitors (7–10). These changes are likely to be involved in enhancement of nuclear β-catenin and prostate cancer development and progression.
It is well known that AR signaling is essential during all phases of prostate cancer development and progression. While β-catenin is a coactivator for TCF/LEF transcription factors in the canonical WNT pathway, it also interacts with AR and enhances AR-mediated transcription in the presence of androgen (11–13). Although cooperative gene regulation by AR and TCF/LEF transcription factors has been reported, these 2 transcription factors often compete for β-catenin, with both signaling pathways promoting cell growth (14, 15). The relative importance of AR and TCF/LEF factors in prostate cancer growth depends on the expression levels of each component at any given time, as well as the local androgen levels. In the presence of androgen, activation of WNT/β-catenin signaling in AR-positive prostate cancer cells often acts through AR-dependent mechanisms rather than classical TCF/LEF-dependent mechanisms (16).
Emerging evidence suggests that WNT signaling plays an important role during progression to CRPC. WNT signaling supports the survival and growth of prostate epithelial cells after androgen deprivation (17, 18). Enhancement of the WNT/β-catenin pathway modulates AR signaling and augments AR transcriptional activity under androgen-deprived conditions (19). Interaction between AR and WNT signaling pathways in CRPC is also significantly increased in vivo (20). Recent exome sequencing further revealed a significant enrichment of mutations in WNT signaling components in castration-resistant compared with paired castration-sensitive tumors (21). While these results support the importance of WNT signaling in prostate cancer progression, the WNT members critical for CRPC growth and the mechanisms of WNT regulation and signaling in CRPC remain unclear.
Given the critical role of WNT signaling in the differentiation and activity of osteoblasts (22), WNT signaling is likely to contribute to the development of prostate cancer osteoblastic bone metastasis. This is supported by evidence that the WNT antagonist DKK1 strongly inhibits bone formation in osteoblastic lesions (9). It is unclear, however, which WNT ligands induce osteoblastic activity in prostate cancer bone metastasis and why osteoblastic lesions are often observed in prostate cancer given that WNT signaling is activated in other cancers as well.
In the present study, we have established a new connection between AR and WNT signaling with important implications for prostate cancer progression and bone metastasis. Our data show that WNT7B expression is regulated by AR, WNT7B remains at a high level in CRPC cells after androgen deprivation, and that WNT7B activates a noncanonical WNT signaling pathway promoting prostate cancer growth and survival after androgen deprivation. Furthermore, we show that prostate cancer-produced WNT7B is associated with osteoblastic responses in the bone. Given that AR is expressed in almost all prostate cancer cells and remains active even under castrated conditions, AR-regulated WNT7B signaling is likely one of the critical mechanisms underlying osteoblastic bone metastasis in advanced prostate cancer.
Materials and Methods
Cell lines and materials
LNCaP, C4-2B, 22RV1, HCT116, ST2, and C3H10T1/2 cells were maintained in cell culture medium with 10% FBS as previously described (23–25). LAPC4 cells (from Dr. Charles Sawyers, University of California, Los Angeles; ref. 26) were maintained in RPMI-1640 with 20% FBS. All cell lines were frozen within 2 passages after receipt from the cell bank and used within less than 6 months after resuscitation. All prostate cancer cell lines were authenticated on the basis of growth, morphology, and RNA-sequencing results. Recombinant human WNT3A (R&D Systems) and 5α-dihydrotestosterone (DHT, Sigma-Aldrich) were purchased.
Stable prostate cancer cell lines with WNT7B knockdown or overexpression
For stable WNT7B knockdown, C4-2B cells were infected with lentiviruses encoding short hairpin RNA (shRNA) against human WNT7B or LacZ control for 24 hours, and selected with puromycin (1.5 μg/mL). Pooled populations were maintained in RPMI-1640 containing 10% FBS and puromycin. WNT7B knockdown efficiency was examined by quantitative reverse transcription PCR (qRT-PCR). The shRNA lentiviral vectors were obtained from the RNAi Core at Washington University (St. Louis, MO). The shRNA sequences are listed in Supplementary Table S1. The pLKO.1 vector system was used to produce lentiviral particles. For stable WNT7B over-expression, viruses expressing mouse WNT7B and coexpressing GFP via an internal ribosome entry site were generated as previously described (25, 27). LNCaP, C4-2B, and LAPC4 cells were infected with viruses expressing WNT7B or empty vector, and more than 90% infection efficiency was achieved as determined by GFP detection.
Western blot analysis
Whole-cell lysates were prepared from prostate cancer cells as indicated, proteins were separated on 4%–15% SDS-PAGE gels (Bio-Rad), and transferred onto polyvinylidene difluoride membranes. Blotted proteins were detected by the following antibodies: anti-WNT7B (AF3460; 1:1,000, R&D Systems), anti-AR (ab74272; 1:1,000, Abcam), anti-β-tubulin (sc-80011; 1:4,000, Santa Cruz Technology), anti-phospho-β-catenin (T41/S45; 9565, 1:1,000, Cell Signaling), anti-β-catenin (9582; 1:1,000, Cell Signaling), anti-phospho-MARCKS (2741; 1:250, Cell Signaling), anti-MARCKS (5607; 1:1,000, Cell Signaling), anti-lamin B1 (ab16048; 1:1,000, Abcam), anti-PKCα (sc-208; 1:2,000, Santa Cruz Technology), anti-PKCδ (sc-937; 1:1,000, Santa Cruz Technology). Nuclear proteins were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagent Kit (Thermo Scientific). Membrane proteins were extracted using Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Scientific).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) analyses were conducted as described previously (23). The primers for quantitative real-time PCR (qPCR) are listed in Supplementary Table S1.
qRT-PCR
qRT-PCR was conducted as described previously (23). Values are means ± SDs of triplicate wells. The primers are listed in Supplementary Table S1.
RNA interference
A predesigned pool of 4 siRNA duplexes (Thermo Scientific) or single siRNA duplexes (Qiagen or IDT) against WNT7B, AR, β-catenin, PKCα, PKCδ, and MARCKS were purchased. A nonspecific pool of 4 siRNA duplexes (siNS1) and 2 different nonspecific single siRNA duplexes (siNS2 and siNS3) were used as controls. All siRNA sequences are listed in Supplementary Table S1. Cells were transfected with siRNA as indicated at a final concentration of 15 nmol/L using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies) according to the manufacturer's instruction.
Cell proliferation
LNCaP (1 × 104 cells/well), C4-2B (6 × 103 cells/well), or 22RV1 (1 × 104 cells/well) cells were plated in 96-well plates and transfected with gene-specific siRNA. Cells were maintained in phenol red-free RPMI-1640 containing 5% charcoal-stripped serum (CSS) with ethanol or 10 nmol/L DHT for 3 or 5 days. The number of viable cells was analyzed using the CCK-8 kit (Dojindo Molecular Technologies).
Soft agar colony formation assay
C4-2B cells with stable WNT7B knockdown or over-expression were plated in 24-well plate (2,000 cells/well). Cells were suspended in RPMI-1640 containing 10% FBS and 0.5% agar, and overlaid onto a solid layer of the same medium containing 1.0% agar. After 2-week incubation, colonies were counted and photographed at × 40 magnification.
Apoptosis assay
LNCaP, C4-2B, or 22RV1 cells were grown in phenol red-free RPMI-1640 containing 5% CSS with ethanol or 10 nmol/L DHT for 3 days after WNT7B or nonspecific siRNA transfection. Cell apoptosis was determined by caspase-3 and -7 activities using Caspase-Glo 3/7 assay kit (Promega) or by DNA fragmentation using terminal deoxynucleotidyl transferase—mediated dUTP nick end labeling (TUNEL) assay kit (Promega) according to the manufacturer's instruction.
Luciferase assay
C4-2B (3 × 104 cells/well) and HCT116 cells (1 × 104 cells/well) were plated in 48-well plates and grown in RPMI-1640 containing 5% FBS for 24 hours. Cells were transfected with TOPFlash or FOPFlash luciferase reporter plasmids (200 ng/well, Addgene) using Lipofectamine LTX Reagent (Life Technologies). pRL-TK Renilla luciferase reporter (10 ng/well, Promega) was cotransfected as an internal control. After plasmid transfection, cells were treated with or without recombinant WNT3A (50 ng/mL, R&D systems) for 24 hours. Firefly and Renilla luciferase activities were measured using Dual-Glo Luciferase Assay System (Promega). The results are represented as Firefly/Renilla ratio.
Osteoblast differentiation assay
ST2 and C3H10T1/2 cells (1 × 105 cells/well) transduced to overexpress WNT7B or vector control were seeded in a 12-well plate in α-minimum essential medium (α-MEM) and basal medium eagle (BME), respectively, with 10% FBS in the presence of 50 μg/mL ascorbic acid and 50 mmol/L β-glycerophosphate. After 1 week, the expression levels of osteoblast differentiation markers, alkaline phosphatase (ALP), and bone sialoprotein (BSP) were measured by qRT-PCR. ALP enzymatic activity was also measured as previously described (25, 27). After 3 weeks of incubation, mineralization was examined using von Kossa staining as previously described (25, 27).
In coculture experiments, ST2 or C3H10T1/2 cells (1 × 105 cells/well) were seeded in a 12-well plate together with prostate cancer cells at 1:1 ratio. Prostate cancer cells were stably overexpressing WNT7B or transfected with WNT7B siRNA 1 day before coculture. The cells were grown in α-MEM or BME medium with 10% FBS in the presence of 50 μg/mL ascorbic acid and 50 mmol/L β-glycerophosphate. ST2 or C3H10T1/2 osteoblast differentiation was examined as described above.
Microarray data analysis
For gene expression from prostate cancer xenograft tumors, RNA was extracted from 24 LuCaP prostate cancer xenografts as described previously (28) and amplified and hybridized to Agilent 44K whole human genome expression oligonucleotide microarrays (Agilent Technologies). Probe labeling and hybridization were conducted following the Agilent suggested protocols, and fluorescent array images were collected using the Agilent DNA microarray scanner G2565BA. Agilent Feature Extraction Software was used to grid, extract, and normalize data. Differences in gene expression were analyzed via Welch test with the Benjamini– Hochberg correction for multiple testing. WNT7B expression data from clinical prostate cancer tumors were obtained from the NCBI GEO database GSE2443 (29) and GSE3325 (30), and analyzed using limma (31).
Results
WNT7B expression is regulated by the AR in CRPC cells
Using RNA-seq, we examined WNT gene expression in androgen-dependent LNCaP and LNCaP-derived castration-resistant C4-2B cells in the presence and absence of 10 nmol/L DHT (23). Of 19 WNT genes, WNT7B was the only WNT member found to be upregulated after DHT stimulation in both LNCaP and C4-2B cells. RNA-seq results for all WNT genes are presented in Supplementary Table S2. WNT7B expression results were confirmed by qRT-PCR and Western blot analysis (Fig. 1A and B). Notably, the high level of WNT7B expression in C4-2B cells in the absence of androgen is comparable with the DHT-induced WNT7B expression levels in LNCaP cells, implying that WNT7B may play a role in androgen-independent growth of C4-2B cells.
Figure 1.

WNT7B is an AR target gene in prostate cancer cells. A, qRT-PCR results showing expression of WNT7B in LNCaP and C4-2B cells. Cells were grown in RPMI-1640 medium with 5% CSS for 3 days followed by the treatment with ethanol or 10 nmol/L DHT for 16 hours. B, Western blot analysis showing WNT7B protein levels under the same conditions as in (A). C, ChIP-seq results showing 2 AR-binding sites at the WNT7B locus in LNCaP and C4-2B cells. D, ChIP-qPCR analyses were conducted to confirm the ChIP-seq results. The prostate specific antigen (PSA) enhancer site was used as a positive control. AR occupancy at the negative control region was defined as 1. E, C4-2B cells were grown in RPMI-1640 medium with 5% CSS for 2 days followed by AR siRNA transfection. qRT-PCR analyses of WNT7B mRNA levels were conducted 3 days after AR siRNA transfection. AR protein levels were examined by Western blot analysis in parallel (inset). *, P < 0.05; **,P < 0.01, determined by a 2-tailed Student t test. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
ChIP-seq analyses of LNCaP and C4-2B cells revealed one strong AR binding site approximately 1.7 kb downstream of the transcription start site (within the first intron) and one weak AR binding site approximately 700 bp downstream of the 3′ end of the WNT7B gene (Fig. 1C). Both binding sites were validated by an independent site-specific ChIP-qPCR analysis (Fig. 1D). Interestingly, we observed AR occupancy (5-fold enrichment over the control)at the weak AR-binding site (Site 1) in the absence of androgen in C4-2B cells but not in LNCaP. This is consistent with the fact that AR protein is more active, stable, and constitutively nuclear in CRPC cells (32) and may explain the higher basal expression ofWNT7B in C4-2B cells under androgen-deprived conditions. Furthermore, the basal expression level of WNT7B in C4-2B cells was decreased more than 70% after knockdown of AR using 2 different AR siRNA (Fig. 1E), indicating that WNT7B expression depends on AR even in androgen-deprived conditions. We further examined AR-mediated WNT7B expression in an additional CRPC 22RV1 cell line. Notably, the AR was constitutively bound to the WNT7B locus (Site 2) in 22RV1 cells (5-fold enrichment over the control, Supplementary Fig. S1A), which may be due to the predominant expression of an AR splice variant lacking the ligand-binding domain in this cell line (33). WNT7B expression levels in the absence of androgen were similar to those in the presence of androgen (Supplementary Fig. S1B) but decreased about 40% after RNA interference against AR (Supplementary Fig. S1C and S1D). Taken together, these results show that WNT7B is an AR target gene in both androgen-dependent and CRPC cells, and that WNT7B remains at a high level in CRPC cells in the absence of androgen due to distinct AR-binding events.
WNT7B is required for the growth of both androgen-dependent and castration-resistant prostate cancer cells
To investigate the potential oncogenic role of WNT7B in prostate cancer cells, we examined LNCaP and C4-2B cell proliferation after knockdown of WNT7B. A high knockdown efficiency of WNT7B after RNA interference was confirmed by Western blot analysis (Fig. 2A). Knock-down of WNT7B significantly inhibited LNCaP and C4-2B growth (Fig. 2B). Notably, WNT7B RNA interference abolished prostate cancer cell growth and resulted in cell death in the absence of androgen. Further analyses revealed that prostate cancer cell apoptosis substantially increased 3 days after WNT7B siRNA transfection as determined by caspase-3 and -7 activity assays (Fig. 2C) as well as TUNEL assays (Fig. 2D; Supplementary Fig. S2). In line with the cell proliferation results, the most prominent apoptosis was observed in androgen-deprived conditions. To test whether overexpression of WNT7B promotes C4-2B proliferation, we established WNT7B-overexpressing LNCaP and C4-2B stable cell lines (Fig. 2E). Overexpression of WNT7B had little effect on LNCaP and C4-2B cell proliferation (Fig. 2F). To further assess the role of WNT7B in prostate cancer progression, we conducted anchorage-independent colony formation assays on soft agar using C4-2B cells. Stable knockdown of WNT7B through lentiviral shRNA transduction (Supplementary Fig. S3) significantly inhibited C4-2B cell anchorage-independent growth as indicated by both colony number and size (Fig. 2G), whereas overexpression of WNT7B significantly increased C4-2B cell anchorage-independent growth (Fig. 2H), suggestive of a potential role of WNT7B in prostate cancer progression. In addition, we found that WNT7B is also required for CRPC 22RV1 cell growth in both the presence and absence of androgen (Supplementary Fig. S4). Taken together, these results support the notion that WNT7B is required for the growth of both androgen-dependent prostate cancer cells and CRPC cells under androgen-deprived conditions.
Figure 2.

WNT7B is necessary for androgen-dependent and CRPC cell growth. A, WNT7B protein levels were examined by Western blot analysis 3 days after WNT7B siRNA transfection. B, LNCaP and C4-2B cell proliferation was examined using CCK8 assays 3 and 5 days after WNT7B siRNA transfection in the presence or absence of 10 nmol/L DHT. C, Apoptosis assay with LNCaP and C4-2B cells showing caspase-3 and -7 activity 3 days after WNT7B siRNA transfection in the presence or absence of 10 nmol/L DHT. The results are presented as mean ± SD of 2 independent experiments. D, TUNEL assays were conducted under the same conditions as in (C). A total of 10 fields were photographed. Total number of cells and the number of TUNEL-positive cells in each field were counted and percentage of apoptotic cells was calculated. E, WNT7B-overexpressing stable prostate cancer cell lines were confirmed by Western blot analysis. F, LNCaP and C4-2B cells overexpressing WNT7B or vector control were grown in the presence or absence of 10 nmol/L DHT for 5 days before CCK8 assays. G, C4-2B cells with stable WNT7B knockdown (shWNT7B-1 and -2) or control knockdown (shLacZ) were examined in soft agar colony formation assays. The assays were conducted in RPMI-1640 medium containing 10% FBS. The colony number is presented as mean ± SD of 3 independent wells. The experiment was conducted for at least 3 independent times. H, WNT7B-overexpressing or vector control C4-2B cells were used in soft agar colony formation assays as in (G). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant, determined by a 2-tailed Student t test.
Previous studies have shown that WNT7B is expressed in a subset of high-grade prostate cancer and bone metastasis specimens but not in normal prostate tissues (34). To further investigate whether WNT7B is critical for prostate cancer progression, we analyzed 2 publicly available prostate cancer microarray datasets (29, 30). Comparison between 10 untreated androgen-dependent primary tumors versus 10 androgen-independent primary tumors revealed that WNT7B is significantly upregulated at the primary site of patients with metastatic CRPC (Supplementary Fig. S5A). WNT7B is also expressed at a higher level in hormone-refractory metastatic tumors compared with localized prostate tumors (Supplementary Fig. S5B). These results are consistent with a role for WNT7B in promoting CRPC tumor growth.
WNT7B promotes CRPC growth after androgen deprivation through a noncanonical PKC pathway
We next sought to investigate mechanisms of the WNT7B-induced intracellular cascades responsible for C4-2B cell growth under androgen-deprived conditions. β-catenin is an important cofactor in the regulation of canonical WNT signaling as well as AR signaling. Therefore, we first investigated whether WNT7B alters β-catenin levels, phosphorylation, and nuclear translocation. Our results show that knockdown of endogenous WNT7B in C4-2B cells in the absence of androgen resulted in increased Thr41/Ser45 phosphorylation of β-catenin and decreased total protein levels of β-catenin (Fig. 3A). In contrast, overexpression of WNT7B slightly decreased Thr41/Ser45 phosphorylation of β-catenin, yet had little effect on total β-catenin protein levels. Importantly, while WNT7B had a modest effect on total β-catenin levels, no differences in nuclear β-catenin protein levels were detected. Confocal microscopy confirmed these results; β-catenin staining was present predominately at the cell membrane and diffused in the cytoplasm, and weak nuclear β-catenin staining was unaltered after knockdown or overexpression of WNT7B (Supplementary Fig. S6). We were unable to detect significant TCF/LEF activity in C4-2B cells using TOPFlash reporter assays (Fig. 3B), and knockdown of β-catenin did not inhibit C4-2B cell growth in the absence of androgen (Fig. 3C). These results indicate that β-catenin/TCF transcriptional signaling is unlikely to be involved in WNT7B-induced androgen-independent growth of C4-2B cells.
Figure 3.

WNT7B promotes androgen-independent C4-2B cell growth in a β-catenin–independent manner. A, Western blot analysis results showing β-catenin, phosphorylated β-catenin (p-β-catenin), MARCKS and phosphorylated MARCKS (p-MARCKS) protein levels in C4-2B cells after knockdown (siNS1 vs. siWNT7B) or overexpression (Vector vs. WNT7B) of WNT7B in the absence of androgen. B, luciferase assays in HCT116 and C4-2B (with or without WNT7B overexpression) cells after transient transfection of TOPFlash and FOPFalsh reporters. Cells were treated with or without recombinant WNT3A. C, C4-2B cell proliferation was examined using CCK8 assays 3 and 5 days after β-catenin siRNA (siβ-catenin) transfection in the absence of DHT. Two different siβ-catenin were used and β-catenin protein levels were examined by Western blot analysis 3 days after siβ-catenin transfection in C4-2B cells (inset).
We have previously reported that WNT7B induces osteoblast differentiation through the PKCδ-mediated noncanonical WNT signaling pathway (25). In this study, we tested whether WNT7B stimulates androgen-independent C4-2B growth through PKC signaling, as shown in osteoblasts. We observed that phosphorylation of MARCKS, a prototypic substrate of PKC, was significantly reduced after knockdown of WNT7B and enhanced after overexpression of WNT7B in C4-2B cells in the absence of androgen (Fig. 3A). The total protein levels of MARCKS were slightly decreased after WNT7B knockdown but not increased after WNT7B overexpression, indicating that alterations of phosphorylated MARCKS were unlikely to be attributable to changes in total protein levels. In fact, the change in total MARCKS levels may be secondary to altered phosphorylation, as phosphorylated MARCKS is more protected from proteolytic cleavage than the dephosphorylated form (35). Previous studies have shown that PKCα and PKCδ are abundantly expressed in prostate cancer cells and exhibit distinct functional properties (36). Phosphorylation of MARCKS was effectively reduced by knockdown of either PKCα or PKCδ (Fig. 4A and 4B), implicating both PKC isozymes in regulation of MARCKS phosphorylation. Furthermore, knockdown of PKCα or PKCδ inhibited C4-2B cell growth in the absence of androgen (Fig. 4C and 4D), suggesting that both PKC isozymes are required for androgen-independent C4-2B cell proliferation. Interestingly, our RNA-seq and ChIP-seq results suggest that PKCα and PKCδ are also direct AR target genes in C4-2B cells (Supplementary Fig. S7 and Supplementary Table S2). As a result, AR may bypass WNT7B and upregulate PKCα and PKCδ directly in the presence of androgen, leading to functional compensation. This mechanism may explain why knockdown of WNT7B has less inhibitory effects on androgen-dependent prostate cancer growth (Fig. 2B). Finally, knockdown of MARCKS also substantially affected C4-2B cell growth (Fig. 4E), suggesting an important function of this gene in cell proliferation. Taken together, our results have suggested a previously undescribed PKC-mediated noncanonical pathway in CRPC by which WNT7B promotes C4-2B cell growth after androgen deprivation.
Figure 4.

WNT7B promotes androgen-independent C4-2B cell growth through the PKC signaling pathway. A and B, PKCα, PKCδ, phosphorylated MARCKS (p-MARCKS), and MARCKS protein levels were examined by Western blot analysis 3 days after PKCα (A) or PKCδ (B) RNA interference in C4-2B cells in the absence of androgen. C–E, C4-2B cell proliferation was examined using CCK8 assays 3 and 5 days after transfection with siPKCα (C), siPKCδ (D), or siMARCKS (E) in the absence of androgen. MARCKS protein levels were examined by Western blot analysis 3 days after MARCKS RNA interference in C4-2B cells (inset). ***, P < 0.001, between nonspecific siRNA and gene-specific siRNA as determined by a 2-tailed Student t test.
WNT7B promotes the development of osteoblastic bone reactions
The importance of WNT signaling in bone development has been well established (27). Consistent with our previous results (25), we observed WNT7B-induced osteoblast differentiation in mouse bone marrow–derived stromal ST2 cells and mouse pluripotent mesenchymal C3H10T1/2 cells (Supplementary Fig. S8A and S8B). Increased osteoblastic activity after WNT7B overexpression was shown by increases in ALP enzymatic activity and mineralization. It should be noted that overexpression of WNT7B in C4-2B cells did not increase ALP activity and mineralization of C4-2B cells (Supplementary Fig. S8C).
We next asked whether prostate cancer-produced WNT7B promotes osteoblast differentiation. Studies have suggested that WNT7B is highly insoluble after posttranslational modification and that WNT7B activation is likely mediated through a direct cell–cell interaction (37). We observed enriched WNT7B protein on C4-2B cell membrane using immunofluorescence and Western blot analyses (Supplementary Fig. S9A and S9B). Conditioned medium from LNCaP and C4-2B cell culture had little effect on ST2 cell differentiation despite overexpression of WNT7B by these cells (Supplementary Fig. S9C), suggesting that the lipid-modified WNT7B is likely linked to plasma membrane. We then cocultured C4-2B with ST2 cells in osteogenic medium and observed moderate increases in ALP activity and mineralization of ST2 cells. Both effects were diminished after WNT7B knockdown in C4-2B cells (Fig. 5A). The mRNA levels of osteoblast differentiation markers, ALP and BSP, were decreased more than 50% as determined by qRT-PCR. Because we used primers specific for mouse genes, the mRNA levels of ALP and BSP reflected the expression of these 2 genes in ST2 cells. The expression of ALP and BSP were barely detectable in C4-2B cells using these primers (Supplementary Fig. S10). In addition, coculture of C4-2B cells overexpressing WNT7B with ST2 cells markedly enhanced ALP activity, mineralization, and ALP and BSP mRNA levels in ST2 cells (Fig. 5B). Similar results were obtained with 2 additional prostate cancer cell lines. Overexpression of WNT7B in LNCaP or LAPC4 cells markedly promoted ST2 osteoblast differentiation in the coculture system (Supplementary Fig. S11). To examine whether these effects are specific for ST2 cells, we conducted similar assays on C3H10T1/2 cells. Coculture of WNT7B-overexpressing C4-2B cells with C3H10T1/2 cells dramatically enhanced ALP activity, mineralization, and ALP and BSP mRNA levels in C3H10T1/2 cells (Fig. 5C). Similar effects were observed when C3H10T1/2 cells were cocultured with WNT7B-overexpressing LAPC4 cells (Supplementary Fig. S12). Thus, WNT7B produced from androgen-dependent and -independent prostate cancer cell lines can induce osteoblast differentiation.
Figure 5.

C4-2B-produced WNT7B promotes osteoblast differentiation. A, C4-2B cells were transfected with WNT7B siRNA (siWNT7B) or nonspecific siRNA (siNS1). After 24 hours, C4-2B cells were cocultured with ST2 cells in osteogenic medium. To determine osteoblastic activity of ST2 cells, alkaline phosphatase staining was conducted on day 7 and von Kossa staining for calcium deposits was conducted on day 21 (left). Mouse ALP and BSP mRNA levels were examined by qRT-PCR on day 7 (right). B, WNT7B-overexpressing C4-2B cells or vector control cells were cocultured with ST2 cells in osteogenic medium. The same assays were conducted as in (A). C, WNT7B-overexpressing C4-2B cells or vector control cells were cocultured with C3H10T1/2 cells in osteogenic medium. The same assays were conducted as in (A). *, P < 0.05; **, P < 0.01, determined by a 2-tailed Student t test.
WNT7B is upregulated in human prostate cancer tumors that cause osteoblastic lesions in bone metastasis
To further investigate the involvement of WNT7B in the osteoblastic reaction elicited by human prostate cancer tumor growth in the bone, we queried the levels of WNT7B in LuCaP prostate cancer xenografts (Supplementary Table S3). LuCaP xenografts were established from primary and metastatic human prostate tumors. Twenty-one androgen-dependent LuCaP xenografts were grown in intact mice, whereas 3 castration-resistant variants (LuCaP 23.1V, LuCaP 35V, and LuCaP 96V) were grown in castrated mice. Of 24 xenograft tumors, 4 were AR-negative neuroendocrine tumors and expressed very low levels of WNT7B, consistent with the fact that WNT7B is an AR target gene. These 4 samples were excluded from further analyses. The xenograft tumors were grouped on the basis of whether they did or did not elicit an osteoblastic reaction when growing in the tibia of intact mice. Xenografts that elicit a large volume of new bone formation were designated as osteoblastic tumors, whereas the remaining tumors were classified as nonosteoblastic. Comparison of the mRNA expression levels of 8 WNT ligands in osteoblastic (n = 5) and nonosteoblastic (osteolytic, mixed, and no reaction, n = 15) xenografts showed that only WNT7B was overexpressed in prostate cancer xenografts that caused an osteoblastic reaction (Fig. 6). No differences were detected for WNTs3, 4, 5A, 5B, 6, 10B, and 11. Notably, the tumor with the highest WNT7B expression level (LuCaP 23.1) induces a robust bone reaction in the bone [see Fig. 2 in ref. 38]. A LuCaP 23.1-derived CRPC xenograft tumor (LuCaP 23.1V) continued to express WNT7B at a very high level and elicit osteoblastic lesions (Supplementary Table S3). These results indicate that WNT7B may play an important role in the osteoblastic reaction resulting from growth of prostate cancer in the bone.
Figure 6.

WNT7B is overexpressed in prostate cancer xenograft tissues causing osteoblastic lesions. WNT gene expression levels from microarray studies were analyzed between prostate cancer LuCaP xenograft tissues with osteoblastic lesions (n = 5) versus nonosteoblastic lesions (n = 15).
Discussion
Despite androgen deprivation, AR signaling persists in CRPC through a variety of mechanisms. While it is believed that a subset of AR target genes remain transcriptionally active, major efforts have been made to understand which AR target genes are responsible for the growth of CRPC. Here, we report that WNT7B is an AR-regulated gene that is persistently upregulated in CRPC cells and promotes CRPC cell proliferation under androgen-deprived conditions. Our data also show that WNT7B produced by prostate cancer cells induces osteoblastic bone lesions, and that the WNT7B paracrine signal is mediated through a direct cell–cell interaction (Fig. 7).
Figure 7.
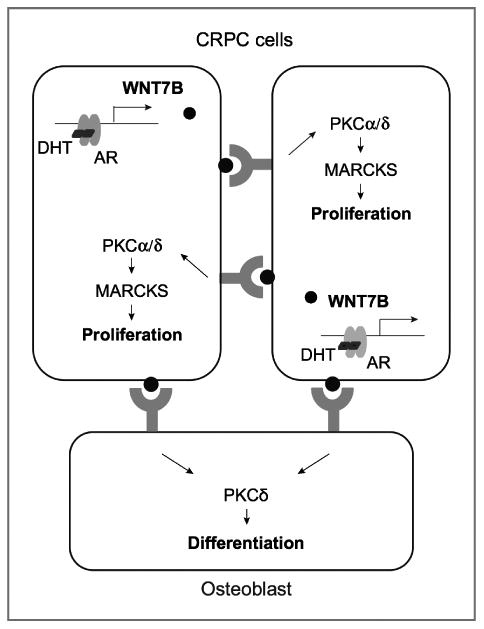
Schematic model of WNT7B signaling in CRPC cells and osteoblasts.
WNT-induced signaling pathways are cell- and context-dependent. We have shown that WNT7B promotes androgen-independent CRPC cell proliferation likely through a PKC-mediated noncanonical WNT pathway. A similar pathway was observed in WNT7B-induced osteoblast differentiation (25). PKC proteins are a family of serine/threonine kinases involved in the transduction of signals for cell proliferation, differentiation, and apoptosis. These isozymes may exhibit overlapping or opposing functions. While PKCα and PKCδ generally function as proapoptotic proteins, they can also act as antiapoptotic proteins (39, 40). Whether pro- or antiapoptotic functions are exhibited depends on cell type, stimulus, and stage of cancer development and progression. The specific role of each PKC isozyme in different types of cancers is not completely understood (36). It is well documented that PKCα and PKCδ mediate phorbol ester-induced apoptosis in prostate cancer cells (41). Our data, however, suggest a novel oncogenic function of these 2 PKC isozymes through their role in WNT7B-induced proliferation of CRPC cells. MARCKS is a ubiquitously expressed substrate of PKC involved in cell adhesion, spreading, motility, and mitogenesis through actin cytoskeletal regulation (42, 43). Upon phosphorylation by PKC, MARCKS translocates from the cell membrane to the cytosol, leading to actin rearrangement and enhanced cell migration and invasion. While our data show that MARCKS is required for the growth of CRPC cells, it is conceivable that WNT7B-induced phosphorylation of MARCKS may increase metastatic activity as observed in other cancer cells (44, 45).
Prostate cancer cells secrete a number of osteogenic factors, such as endothlin-1 (ET-1), bone morphogenetic proteins (BMP), and WNTs. These factors promote differentiation of osteoblasts that, in turn, release growth factors that facilitate prostate cancer growth in bone. As a result, prostate cancer cells, capable of producing factors that induce bone formation, tend to survive and proliferate in bone. Several lines of evidence indicate that WNT signaling is likely the primary determinant of tumor-induced bone remodeling. It is well known that increasing DKK1 expression may convert prostate cancer cells from an osteoblastic to an osteolytic type. ET-1 may also increase bone formation by suppression of DKK1 (46). WNTs promote BMP expression and modulate osteoblastic activity in prostate cancer bone metastasis (47). The present study further supports the notion that AR-mediated upregulation of WNT signaling might play a major role in osteoblastic bone metastasis in advanced prostate cancer. Interestingly, WNT7B has been identified as an AR target gene in ER-negative breast cancer cells (48), and approximately 10%–15% of breast cancer bone metastases display predominantly osteoblastic lesions (49). Whether these bone lesions are associated with high AR activity and WNT7B expression is unknown.
Current antiandrogen therapies for advanced prostate cancer have focused on blocking AR ligand binding and androgen synthesis. While these novel therapies have been a major breakthrough, increases in survival have been measured in months. Our findings suggest a unique opportunity for development of more effective therapies by targeting the AR/WNT7B/PKC signaling cascade, which would inhibit prostate cancer proliferation and prevent the formation of a metastatic reservoir in the bone.
Supplementary Material
Acknowledgments
The authors thank Drs. David R. Piwnica-Worms, John R. Edwards, Scot Matkovich (Washington University in St. Louis), and Robert L. Vessella (University of Washington, Seattle) for valuable discussions.
Grant Support: This work was supported by the Concern Foundation, the Siteman Cancer Center Developmental Research Award in Prostate Cancer Research, and the BRIGHT Institute at Washington University School of Medicine/P50 Molecular Imaging Center Pilot Research Fund.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Authors' Contributions: Conception and design: D. Zheng, T. Zhou, L. Jia
Development of methodology: D. Zheng, K.N. Weilbaecher, L. Jia
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): D. Zheng, J. Chen, Z. Qi, E. Corey, L. Jia
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): D. Zheng, K.F. Decker, T. Zhou, E. Corey, L. Jia
Writing, review, and/or revision of the manuscript: D. Zheng, K.F. Decker, T. Zhou, Z. Qi, K.N. Weilbaecher, E. Corey, F. Long, L. Jia
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): T. Zhou, J. Chen, L. Jia
Study supervision: L. Jia
References
- 1.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz A, Lin SH. Osteolytic and osteoblastic bone metastases: two extremes of the same spectrum? Recent Results Cancer Res. 2012;192:225–33. doi: 10.1007/978-3-642-21892-7_11. [DOI] [PubMed] [Google Scholar]
- 3.Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8:357–68. doi: 10.1038/nrclinonc.2011.67. [DOI] [PubMed] [Google Scholar]
- 4.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–77. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 5.Voeller HJ, Truica CI, Gelmann EP. Beta-catenin mutations in human prostate cancer. Cancer Res. 1998;58:2520–3. [PubMed] [Google Scholar]
- 6.Saha B, Arase A, Imam SS, Tsao-Wei D, Naritoku WY, Groshen S, et al. Overexpression of E-cadherin and beta-catenin proteins in metastatic prostate cancer cells in bone. Prostate. 2008;68:78–84. doi: 10.1002/pros.20670. [DOI] [PubMed] [Google Scholar]
- 7.Thiele S, Rauner M, Goettsch C, Rachner TD, Benad P, Fuessel S, et al. Expression profile of WNT molecules in prostate cancer and its regulation by aminobisphosphonates. J Cell Biochem. 2011;112:1593–600. doi: 10.1002/jcb.23070. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, et al. A novel role for Wnt/Ca2 +signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS One. 2010;5:e10456. doi: 10.1371/journal.pone.0010456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65:7554–60. doi: 10.1158/0008-5472.CAN-05-1317. [DOI] [PubMed] [Google Scholar]
- 10.Wissmann C, Wild PJ, Kaiser S, Roepcke S, Stoehr R, Woenckhaus M, et al. WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J Pathol. 2003;201:204–12. doi: 10.1002/path.1449. [DOI] [PubMed] [Google Scholar]
- 11.Masiello D, Chen SY, Xu Y, Verhoeven MC, Choi E, Hollenberg AN, et al. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol Endocrinol. 2004;18:2388–401. doi: 10.1210/me.2003-0436. [DOI] [PubMed] [Google Scholar]
- 12.Mulholland DJ, Cheng H, Reid K, Rennie PS, Nelson CC. The androgen receptor can promote beta-catenin nuclear translocation independently of adenomatous polyposis coli. J Biol Chem. 2002;277:17933–43. doi: 10.1074/jbc.M200135200. [DOI] [PubMed] [Google Scholar]
- 13.Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, et al. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002;277:11336–44. doi: 10.1074/jbc.M111962200. [DOI] [PubMed] [Google Scholar]
- 14.Mulholland DJ, Read JT, Rennie PS, Cox ME, Nelson CC. Functional localization and competition between the androgen receptor and T-cell factor for nuclear beta-catenin: a means for inhibition of the Tcf signaling axis. Oncogene. 2003;22:5602–13. doi: 10.1038/sj.onc.1206802. [DOI] [PubMed] [Google Scholar]
- 15.Amir AL, Barua M, McKnight NC, Cheng S, Yuan X, Balk SP. A direct beta-catenin-independent interaction between androgen receptor and T cell factor 4. J Biol Chem. 2003;278:30828–34. doi: 10.1074/jbc.M301208200. [DOI] [PubMed] [Google Scholar]
- 16.Cronauer MV, Schulz WA, Ackermann R, Burchardt M. Effects of WNT/beta-catenin pathway activation on signaling through T-cell factor and androgen receptor in prostate cancer cell lines. Int J Oncol. 2005;26:1033–40. doi: 10.3892/ijo.26.4.1033. [DOI] [PubMed] [Google Scholar]
- 17.Placencio VR, Sharif-Afshar AR, Li X, Huang H, Uwamariya C, Neilson EG, et al. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res. 2008;68:4709–18. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu X, Wang Y, Jiang M, Bierie B, Roy-Burman P, Shen MM, et al. Activation of beta-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate. 2009;69:249–62. doi: 10.1002/pros.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verras M, Brown J, Li X, Nusse R, Sun Z. Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res. 2004;64:8860–6. doi: 10.1158/0008-5472.CAN-04-2370. [DOI] [PubMed] [Google Scholar]
- 20.Wang G, Wang J, Sadar MD. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008;68:9918–27. doi: 10.1158/0008-5472.CAN-08-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–92. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2012;13:27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- 23.Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012;40:10765–79. doi: 10.1093/nar/gks888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D, et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009;5:e1000597. doi: 10.1371/journal.pgen.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, et al. Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev Cell. 2007;12:113–27. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3:402–8. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- 27.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 28.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Best CJ, Gillespie JW, Yi Y, Chandramouli GV, Perlmutter MA, Gath-right Y, et al. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin Cancer Res. 2005;11:6823–34. doi: 10.1158/1078-0432.CCR-05-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 32.Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–8. [PubMed] [Google Scholar]
- 33.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–49. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li ZG, Yang J, Vazquez ES, Rose D, Vakar-Lopez F, Mathew P, et al. Low-density lipoprotein receptor-related protein 5 (LRP5) mediates the prostate cancer-induced formation of new bone. Oncogene. 2008;27:596–603. doi: 10.1038/sj.onc.1210694. [DOI] [PubMed] [Google Scholar]
- 35.Spizz G, Blackshear PJ. Protein kinase C-mediated phosphorylation of the myristoylated alanine-rich C-kinase substrate protects it from specific proteolytic cleavage. J Biol Chem. 1996;271:553–62. doi: 10.1074/jbc.271.1.553. [DOI] [PubMed] [Google Scholar]
- 36.Caino MC, von Burstin VA, Lopez-Haber C, Kazanietz MG. Differential regulation of gene expression by protein kinase C isozymes as determined by genome-wide expression analysis. J Biol Chem. 2011;286:11254–64. doi: 10.1074/jbc.M110.194332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, et al. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–21. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corey E, Quinn JE, Bladou F, Brown LG, Roudier MP, Brown JM, et al. Establishment and characterization of osseous prostate cancer models: intra-tibial injection of human prostate cancer cells. Prostate. 2002;52:20–33. doi: 10.1002/pros.10091. [DOI] [PubMed] [Google Scholar]
- 39.Basu A, Pal D. Two faces of protein kinase Cdelta: the contrasting roles of PKCdelta in cell survival and cell death. Sci World J. 2010;10:2272–84. doi: 10.1100/tsw.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lahn M, Sundell K, Gleave M, Ladan F, Su C, Li S, et al. Protein kinase C-alpha in prostate cancer. BJU Int. 2004;93:1076–81. doi: 10.1111/j.1464-410X.2003.04784.x. [DOI] [PubMed] [Google Scholar]
- 41.Gavrielides MV, Gonzalez-Guerrico AM, Riobo NA, Kazanietz MG. Androgens regulate protein kinase Cdelta transcription and modulate its apoptotic function in prostate cancer cells. Cancer Res. 2006;66:11792–801. doi: 10.1158/0008-5472.CAN-06-1139. [DOI] [PubMed] [Google Scholar]
- 42.Disatnik MH, Boutet SC, Pacio W, Chan AY, Ross LB, Lee CH, et al. The bi-directional translocation of MARCKS between membrane and cytosol regulates integrin-mediated muscle cell spreading. J Cell Sci. 2004;117:4469–79. doi: 10.1242/jcs.01309. [DOI] [PubMed] [Google Scholar]
- 43.Rombouts K, Mello T, Liotta F, Galli A, Caligiuri A, Annunziato F, et al. MARCKS actin binding capacity mediates actin filament assembly during mitosis in human hepatic stellate cells. Am J Physiol Cell Physiol. 2012;303:C357–67. doi: 10.1152/ajpcell.00093.2012. [DOI] [PubMed] [Google Scholar]
- 44.Chen X, Rotenberg SA. PhosphoMARCKS drives motility of mouse melanoma cells. Cell Signal. 2010;22:1097–103. doi: 10.1016/j.cellsig.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Techasen A, Loilome W, Namwat N, Takahashi E, Sugihara E, Puapairoj A, et al. Myristoylated alanine-rich C kinase substrate phosphorylation promotes cholangiocarcinoma cell migration and metastasis via the protein kinase C-dependent pathway. Cancer Sci. 2010;101:658–65. doi: 10.1111/j.1349-7006.2009.01427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clines GA, Mohammad KS, Bao Y, Stephens OW, Suva LJ, Shaughnessy JD, Jr, et al. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol. 2007;21:486–98. doi: 10.1210/me.2006-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2008;68:5785–94. doi: 10.1158/0008-5472.CAN-07-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20:119–31. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–64. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.