

Abstract
DNA-based vaccines, while highly immunogenic in mice, generate significantly weaker responses in primates. Therefore, current efforts are aimed at increasing their immunogenicity, which include optimizing the plasmid/gene, the vaccine formulation and method of delivery. For example, co-immunization with molecular adjuvants encoding an immunomodulatory protein has been shown to improve the antigen (Ag)-specific immune response. Thus, the incorporation of enhancing elements, such as these, may be particularly important in the influenza model in which high titered antibody (Ab) responses are critical for protection. In this regard, we compared the ability of plasmid-encoded high-mobility group box 1 protein (HMGB1), a novel cytokine in which we have previously mutated in order to increase DNA vaccine immunogenicity, with boost Ag-specific immune responses during DNA vaccination with influenza A/PR/8/34 nucleoprotein or the hemagglutinin of A novel H1N1/09. We show that the HMGB1 adjuvant is capable of enhancing adaptive effector and memory immune responses. Although Ag-specific antibodies were detected in all vaccinated animals, a greater neutralizing Ab response was associated with the HMGB1 adjuvant. Furthermore, these responses improved CD8 T+-cell effector and memory responses and provided protection against a lethal mucosal influenza A/PR/8/34 challenge. Thus, co-immunization with HMGB1 has strong in vivo adjuvant activity during the development of immunity against plasmid-encoded Ag.
Keywords: HMGB1, DNA vaccine, adaptive immunity, Ag presentation, A/PR/8/34 viral challenge, H1N1/09 (swine flu)
INTRODUCTION
DNA vaccines have many conceptual advantages for vaccine development.1 They can be constructed with many safety features while retaining the specificity of a subunit vaccine. As DNA vaccine plasmids are non-live, non-replicating and non-spreading, there is no risk of either reversion to a disease causing form or secondary infection. The ability to express the vaccine alone or in conjunction with additional vaccine elements, such as adjuvants, also provides design flexibility; the inclusion of specialized molecular adjuvants expressing cytokines, costimulatory molecules and/or immunomodulatory proteins may help enhance or tailor the vaccine-elicited protective immune response against a particular pathogen. Furthermore, long-term expression of the DNA vaccine-encoded immunogen, cost-effectiveness, stability, antigen (Ag) presentation by both major histocompatibility complex class I and class II molecules, ease of development and production, and elicitation of cell-mediated as well as Ab-mediated immune responses are some of the other advantages of this vaccine platform.
However, despite these numerous advantages, the major limiting factor of ‘first generation’ DNA vaccines has been their poor immunogenicity in primates. Currently, many strategies are being developed to overcome this pitfall, including plasmid/gene optimization, vaccine formulation, methods of delivery and using adjuvants encoding an immunomodulatory proteins that can either be expressed in combination with or within the vaccine construct.2–7 Multiple laboratories have reported that co-injection of plasmids encoding cytokines (interleukin-15 or interleukin-12),6,8 chemokines,9 or co-stimulatory molecules such as CD80 and CD86 (refs 6, 8, 10), can produce an immunomodulatory effect on the resulting immune responses. Therefore, it is hoped that, by inclusion of multiple optimizing factors including adjuvanting by the expression of immunomodulatory proteins, ‘next generation’ DNA vaccines will yield better protective efficacy.
In this regard, we have previously reported that high-mobility group box 1 protein (HMGB1), a novel cytokine that can function as an inflammatory agent, when combined with human immunodeficiency virus-1 (HIV-1)-Ag-encoded DNA vaccine results in the enhancement of antibody (Ab) responses and the CD8+ T cell interferon-γ (IFN-γ response).11 Thus, the use of this adjuvant during DNA vaccination is likely to increase inflammation by stimulating the recruitment and activation of dendritic cells (DCs) to sites of vaccine-encoded Ag production. Here we expanded the scope of these initial studies to evaluate the capacity of the HMGB1 adjuvant to enhance immunity during DNA vaccination using an important flu Ag system as well as its ability to provide protective immunity against a viral challenge. We observed that the HMGB1 adjuvant was able to markedly enhance both effector and memory immune responses, which resulted in improved levels of cytotoxic T lymphocytes as well as enhanced Ab titers. Furthermore, the HMGB1 adjuvant improved survival in a lethal influenza challenge model as well as increasing immunity and neutralizing Ab responses against the hemagglutinin (HA) from pandemic influenza A subtype H1N1/09 virus.
RESULTS
Secretion of HMGB1 enhances Ag-presenting cell (APC) maturation and splenic DCs counts in vivo
HMGB1 is reported to activate macrophages and DCs.12 We have shown previously that HMGB1 protein promoted in vitro bone marrow-derived DCs activation/maturation as defined by increased CD83, CD86 and CCR7 expression, as well as by enhanced secretion of the chemokine macrophage inflammatory protein-2.11 To test the adjuvant effects of HMGB1 on Ag presentation and the induction of immunity, we immunized mice three times, 2 weeks apart, with plasmid encoded-Nucleoprotein (pNP),13 with or without pHMGB1, and collected the quadriceps muscles and draining lymph nodes at 1 week following the final immunization. As shown in Figure 1a, co-immunization of pNP with pHMGB1 drove higher numbers of APCs expressing CD83 (brown color) when compared with pNP injected with pVax1 in the infiltrative areas of the electroporated quadriceps muscle.
Figure 1.

HMGB1 enhances APC maturation. (a) Immunohistochemical staining of CD83+ cells (brown color) infiltrating the muscle after pNP vaccination with pHMGB1 adjuvant. Representative pictures of sections from pNP+pVax1 and pNP+pHMGB1 groups are shown. (b) Inguinal lymphonodal cells harvested at 7 days after the final i.m. injection of pNP alone or in combination with pHMGB1 were stained for the markers of activation CD80 and CD86, and flow cytometric analysis was performed. Empty vector pVax1 was used as a negative control and bar graphs. (c) At day 14 post immunization, splenic sections from pHMGB1-adjuvanted mice (side scatter channel (SSC) with pNP or pGag) were stained for CD11c+ (brown color) and negative cells were blue in color. Original magnification is ×100 and boxes (the insert picture at the right-bottom side) are ×400. (d) Quantification of CD11c+ cells. The results are representative of three independent experiments.
In inguinal lymph nodes of the intramuscular (i.m.) immunized mice, we observed that injection of pNP alone was not sufficient to upregulate CD80 and CD86 (Figure 1b) in a 7-day time frame. Indeed, the percentage of cells expressing these markers did not significantly vary among the three groups: pVax1, pNP and pHMGB1 alone. However, co-administration of pNP along with the HMGB1 construct dramatically increased the expression of the costimulatory molecules; the percentage of CD80+ cells is 4.0% for pNP+pHMGB1 versus 0.7% for pNP alone, and the percentage of CD86+ cells is 11.7% for pNP+pHMGB1 versus 2.9% for pNP alone (Figure 1c).
To investigate the basis for the markedly enhanced APCs biodistribution and to examine the specific cell types influenced by HMGB1 secretion, we performed immunohistochemistry in the marginal zones of the spleen. Strikingly, CD11c+ cells rapidly accumulated in the spleens from HMGB1 co-immunized animals when compared with those of splenic sections from pVax1-immunized mice (Figure 1d). Furthermore, similar biodistribution patterns of DCs were observed for another immunogenic plasmid, HIV-1 Gag-expressing pGag, when co-immunized with pHMGB1. Altogether, the CD11c+ cells showed a typical splenic DCs distribution, with strong staining in the marginal zones and within both the T-cell areas and the red pulp. Thus, co-immunization of molecular plasmid adjuvant pHMGB1 with several plasmid expressed Ag enhanced the activation and maturation of APCs in key immunological sites.
HMGB1 expression boosts DNA vaccination immunogenicity
We next analyzed the ability of adjuvant pHMGB1 to increase the cellular immune response to plasmid vaccination by a standard IFN-γ enzyme-linked immunosorbent spot (ELISPOT) assay. We have shown previously that a significant enhancement of cytotoxic T lymphocytes response after coinjection with chemokines indicate that the enhancement of cytolytic activity was Ag specific and CD8+ T cell dependent.14 As shown in Figure 2, mice were immunized three times, 2 weeks apart, with pNP alone or in combination with pHMGB1 (Figure 2a). Co-immunization with HMGB1-encoding plasmid induced a higher number of nucleoprotein (NP)-specific IFN-γ secreting T cells when compared with NP alone-vaccinated mice; ELISPOT counts were 1132±46 in the HMGB1 mice versus 753±29 for the pNP alone group (Figures 2b and c). Furthermore, when compared immunization with or without electroporation (EP), the levels of NP-specific immune responses were almost half of those in i.m. immunization group compared with EP group. Therefore, co-immunization of pNP with pHMGB1 plus EP resulted in an increased NP-specific cellular response in mice.
Figure 2.

Electroporation increase of NP-specific cellular responses by pHMGB1 co-immunization. Effects of adjuvant pHMGB1 on the induction of NP-specific cellular immune responses were measured in the spleens using the standard IFN-γ ELISPOT assay. (a) Immunization schedule for the murine study is shown. (b, c) Immunogenicity of NP. Splenocytes were harvested at 7 days following the third immunizations, 2 weeks apart, administered i.m. with EP (b) or with out EP (c). Negative control immunized animals receiving three injections of empty vector control plasmid (pVax1). Splenocytes were stimulated with multiple pools of overlapping peptides spanning the entire length of the NP and then IFN-γ spot-forming units (SFUs) per million splenocytes were enumerated. Experiments were performed independently at least three times with similar results.
Next, to determine whether co-immunization with HMGB1-encoding plasmid could increase the proliferative capacity of NP-specific CD8+ and CD4+ T lymphocytes, a standard carboxyfluorescein succinimidyl ester proliferation assay was performed at 1 month following the final immunization using peptide pools spanning the influenza NP. After 4 days of in vitro incubation of carboxyfluorescein succinimidyl ester-labeled splenocytes from immunized mice with NP peptides, cells were washed and stained as described above. On average, co-administration of pNP along with the HMGB1 construct increased the percentage of total proliferating cells; the addition of the pHMGB1 resulted in a 42% increase in the percentage of total proliferating CD8+ cells and 86% for CD4+ cells (Figures 3a and b). Therefore, the expression of HMGB1 during vaccination boosted the proliferative capacity of memory CD8+ T cells.
Figure 3.
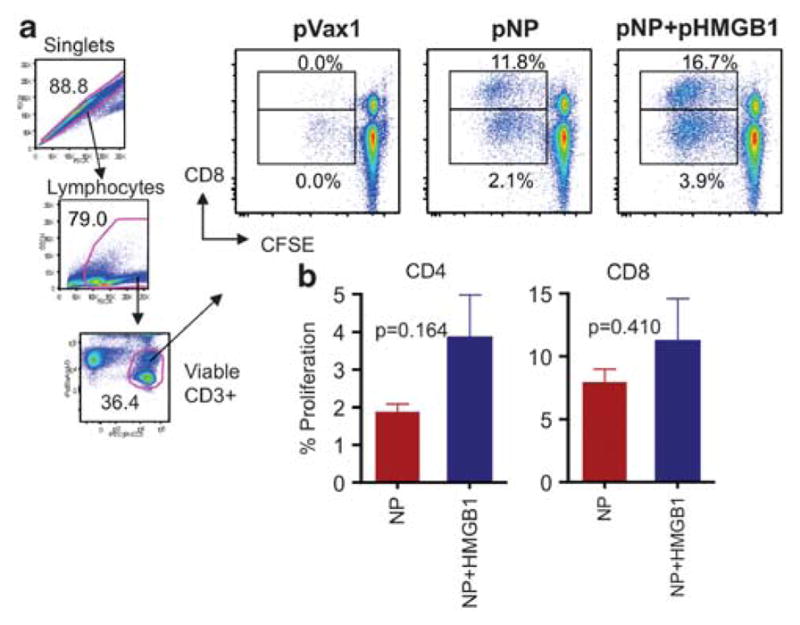
HMGB1-elicited T-cell proliferative responses to NP. After the third immunization, splenocytes were harvested and stimulated with NP to determine NP-specific CD8+ and CD4+ T-cell proliferation capacity. (a) Singlets, lymphocytes and viable CD3+ cells were gated and representative dot plots from each group of mice are shown for NP-specific CD8+ and CD4+ T cells. Average peptide-specific responses are displayed. (b) Proliferative responses for NP are shown as group mean responses ± s.d. with pVax1 values subtracted. Error bars represent s.d. Similar results were observed in two independent experiments. Statistical analysis was performed with Graphpad Prism 5 (Graphpad Software Inc.).
Plasmid HMGBI mediates protection against a lethal mucosal influenza challenge
On the basis of observation that pHMGB1 co-injection resulted in the enhancement of effector and memory CD8+ T-cell responses (Figure 3), we next addressed whether this adjuvant could enhance protection of BALB/c mice against a lethal influenza (influenza A/PR/8/34) challenge. Mice were infected intranasally at 30 days post infection (d.p.i.) with a normally lethal dose of A/PR/8/34 influenza. The survival curves (Figure 4a) and weight variation charts (Figure 4b) are shown for each group of mice. Naive animals became infected, showing rapid weight loss and labored breathing within 5 d.p.i., and −90% succumbed to lethal criteria by 9 d.p.i. However, only 60% of the animals that received immunization with pNP alone died by 9 d.p.i. Furthermore, these animals were followed for 5 days by which time the animals had lost −30% of their total body weight (Figure 4b). Although pNP alone showed only 40% protection from death, pNP in combination with pHMGB1 was able to protect 60% of the vaccinated animals from death, which also exhibited improved morbidity (decreased weight loss post challenge, with the average maximum weight loss in the pNP group at 23.1%±1.3%, and in the pNP+pHMGB1 group at 18.7%±2.1%; Figure 4b).
Figure 4.
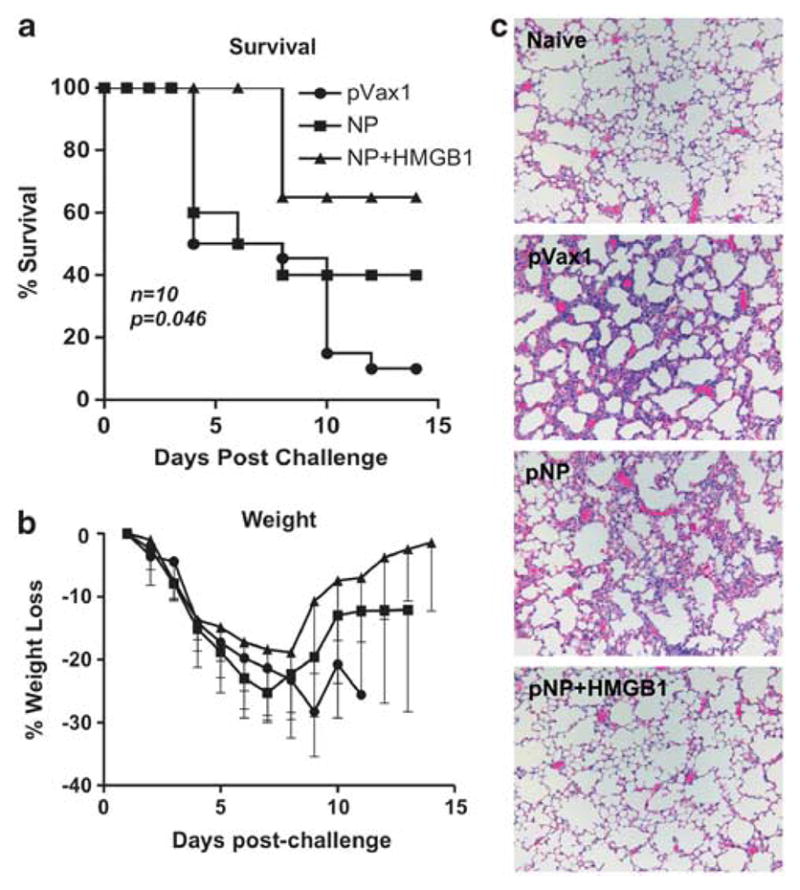
Protective efficacy of the HMGB1-adjuvanted pNP. At 4 weeks after the third immunization, mice were challenged intranasally with 10 lethal dose 50 of A/Puerto Rico/8/34 virus. (a) Kaplan–Meier curve showing survival percentage of each experimental group over the course of 14 days. (b) Average weight loss among survivors of each group tracked over 14 days. Similar results were observed in two independent experiments with at least n=10 per group for each experiment. (c) Histopathological analysis of lung tissue from vaccinated and challenged BALB/c mice. Mice were challenged with the influenza A/Puerto Rico/8/34 strain at 1 month after the final immunization. The mice were killed at 4 days post challenge, and lung tissue was collected for histological review. Representative micrographs from pVax1, pNP and pNP+pHMGB1 groups are shown along with a tissue section of a naive mouse lung for comparison.
Histopathological changes of the lung tissue from BALB/c mice following A/PR/8/34 influenza challenge were also evaluated. Paraffin-embedded sections of lungs collected four d.p.i. were stained with hematoxylin and eosin and micrographs were isolated for histopathological analysis. Mice inoculated with pVax1 vector alone showed severe hypertrophy of the alveolar lining cells and extensive infiltration of lymphocytes (Figure 4c). The vaccination with NP-encoding plasmid decreased the severity of the infiltration but foci of initial pneumonia could still be observed. In contrast, administration of pHMGB1 along with pNP dramatically reduced the histological changes of the lung tissue in response to A/PR/8/34 infection, and only few and negligible foci of inflammation could be observed in the interalveolar septa. Altogether, co-immunization of pHMGB1 with pNP enhanced the capacity for development of protective immunity in mice against a normally lethal challenge with the A/PR/8/34 influenza virus.
Construction and in vitro expression of pH109
We previously developed a synthetic consensus NP protein, which when expressed from a DNA construct generated protective immunity against an H5N1 influenza virus in a non-human primate infection model.15 However, it is unlikely that NP-specific Abs have a significant role in protection, as the virus-associated NP is an internal protein which is sequestered from the surface of the virus. Thus, to investigate the ability for the HMGB1 plasmid adjuvant to improve humoral immunity, we next focused on the viral surface HA protein for the induction of neutralizing Abs and developed a novel H1 consensus Swine 1909 vaccine, which will likely be useful in eliciting an anti-Swine 1909 humoral response. The current studies were designed to test this novel pH109 vaccine, and also to extend these findings in order to improve humoral immunity using the adjuvant pHMGB1.
As summarized in Figure 5a, several modifications were designed after generation of the consensus sequence and cloning, including the incorporation of a human immunoglobulin E (IgE) leader peptide to help increase protein expression.16 Gel electrophoresis was used to confirm the insertion and size of the pH109 (Figure 5b). To confirm in vitro expression of HA by pH109, 293T cells were transfected and both supernatants and cell lysates were collected at 48 h later. HA protein levels were then measured by western immunoblotting using HA-specific Abs (Figures 5c and d). Thus, pH109 effectively expressed the consensus sequence of the novel H1N1/09 HA protein in vitro.
Figure 5.
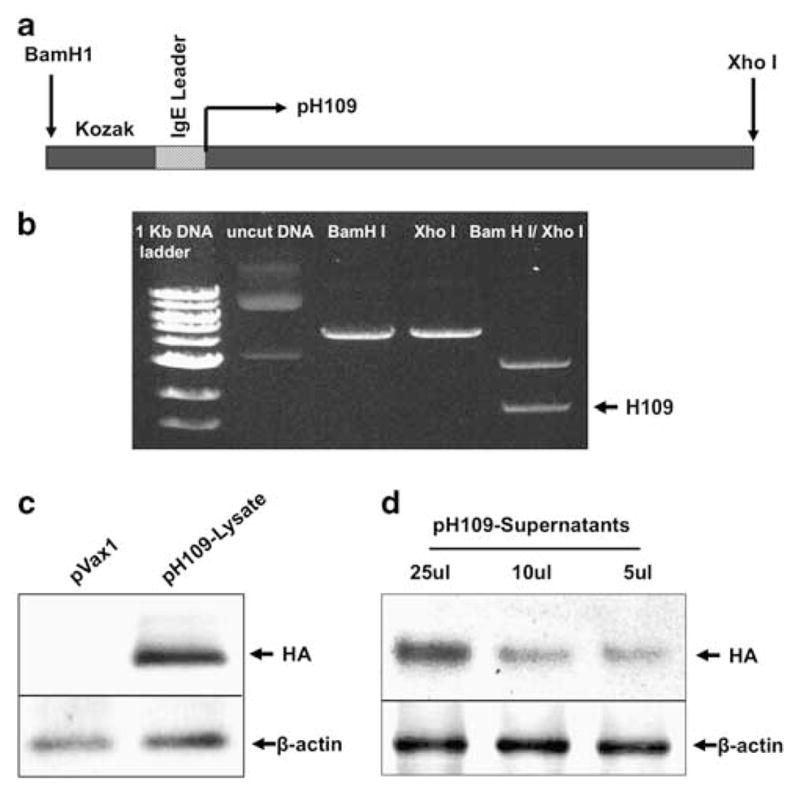
Construction and expression of the HS09 DNA vaccine. (a) Schematic representation of the strategy for cloning the human swine flu consensus gene into the pVax1 vector (pH109). Location of the Kozak sequence, the IgE leader peptide, enzyme restriction sites and the pH109 initiation site are displayed. (b) Gel photograph showing fragments of the pH109 plasmid following restriction digestion with Xho1 and BamHI enzymes. (c, d) Western blot analysis of pH109 expression in human 293T cells transfected with 10 μg pVax1 control vector or the pH109 as indicated. Lysates (c) or cell supernatants (d) were extracted at 48 h post transfection and immunoblotting was performed using anti-HA Abs.
Next, to determine whether pH109 in combination with pHMGB1 could induce a potent immune response against synthetic DNA Ags, we immunized and electroporated BALB/c mice with each of the individual vaccine candidates (Figure 6a). Similarly to what was observed for NP, pHMGB1 increased cellular immunity to pH109 vaccination by 59%, the count being -840 spot-forming unit versus -493 spot-forming unit in the pH109 group as measured by IFN-γ ELISPOT (Figure 6b). Splenocytes from control pVax1-accinated mice stimulated with these Ags and negative media control were all well below the 40-spot assay threshold.
Figure 6.

Immunogenicity of pH109 with and without pHMGB1 adjuvant. The immunogenicity of pH109 and the effects of HMGB1 adjuvant on the induction of cellular responses were measured. (a) Immunization schedule for the murine study is shown. (b) Splenocytes were stimulated with multiple pools of overlapping peptides spanning the entire length of the HA antigen, standard IFN-γ was performed and IFN-γ spot-forming unit (SFU) were enumerated. The results are representative of three independent experiments. (c) Total anti-HA IgG responses in the blood serum induced with vaccinations using either pH109 or pH109+pHMGB1 DNA vaccine in BALB/c mice as measured by endpoint Ab enzyme-linked immunosorbent assay. Error bars represent±s.d. from the mean and are representative of three independent experiments. (d) To measure type-specific IgG responses, mice were bled and pooled sera were diluted to 1:50 for reaction with HA. The assay was performed in triplicate and values represent mean (n=3) and bars s.d. (e) The nAb titers against an influenza (A/Mexico/InDRE4487/2009) virus infection was measured for Madin–Darby canine kidney cells and data are shown as the geometric means from each group (n=4).
Enhancement of neutralizing Ab responses by addition of pHMGB1
Previously we have shown that i.m. vaccination of mice and non-human primates can stimulate measurable and protective Ab responses.15 To determine whether co-injection of the pH109 vaccine with pHMGB1 might influence humoral immune responses against HA, sera obtained at 1 week after the final DNA inoculation were tested by end point Ab enzyme-linked immunosorbent assay. As shown in Figure 6c, co-immunization with HMGB1 adjuvant induced HA-specific IgG. These responses could be further broken down into particular IgG subclasses, which showed that co-injection with HMGB1 adjuvant induced a significant increase in IgG1 levels as compared with IgG2a levels (Figure 6d). Therefore, pHMGB1 was capable of enhancing HA-specific IgG, namely IgG1, responses when co-administered with pH109 expressing a novel consensus H1N1 HA protein.
Lastly, owing to the robust levels of HA specific IgG driven by the HMGB1 adjuvant, the protective efficacy of these responses were measured by a micro neutralization assay.10 HMGB1-adjuvanted responses elicited a much higher neutralizing Ab titer against the autologous H1N1 virus (A/Mexico/InDRE4487/2009) that was about twofold greater than did the pH109 alone (Figure 6e). These results indicated that co-administration of this potent immunostimulatory adjuvant strongly enhanced the humoral immune response compared with pH109 alone, and that this strong adjuvant activity may be an effective immunological adjuvant in DNA vaccination against other infectious pathogens and diseases.
DISCUSSION
We have previously shown that HMGB1 DNA could induce the recruitment of DCs to the injection site and favor the acquisition of a mature DCs phenotype, which in turn could result in enhanced cellular and humoral responses to an HIV plasmid vaccine in vivo.11 It is likely that DCs have a key role in the stimulation of immune responses to DNA vaccination, which may explain why several approaches to stimulate the maturation and recruitment of professional APCs have been developed.4,17,18 In this study, we expanded on these previous results using the HMGB1-encoding plasmid as an adjuvant in a DNA vaccine against influenza. Although NP-based DNA vaccines have been shown to trigger significant levels of immune responses and protection against influenza challenge, several studies have explored different adjuvants to increase the potency of such vaccines.19–21
For the first time, we show herein that an HMGB1-encoding plasmid could enhance the immunogenicity and protective efficacy of a DNA vaccine against influenza. It is likely that the increased immunogenicity of the DNA vaccine administered in conjunction with pHMGB1 and EP was because of the recruitment of higher numbers of APCs to the electroporated muscle, inguinal lymph nodes and the spleen (Figure 1). As a result, this recruitment and activation of APCs may have helped to bolster NP-specific T- and B-cell responses. Plasmid HMGB1-adjuvanted NP-vaccinated mice showed an increase in the T-cell response by 59% as measured by ELISPOT (Figure 2) and 30% for the CD8+ response by carboxyfluorescein succinimidyl ester proliferation assay (Figure 3). However, addition of HMGB1 adjuvant induced an increase of 46% in the CD4+ T-cell proliferative response on average, compared with only a 30% increase in the CD8+ T-cell response, which may ultimately contribute to the overall quality of the memory CD8+ T-cell response.22,23
Although we were not able to reach complete protection from mortality in the BALB/c mouse strain, we observed that the increased response induced by pHMGB1 to the vaccination with pNP reduced the morbidity associated with the infection as shown by reduced weight loss, increased speed of recovery as well as delayed damage of lung tissue and protection from pneumonia (Figure 4). This finding is in accordance with the fact that pHMGB1 influences cellular immunity when delivered along with different plasmid-encoded Ags inducing higher number of IFN-γ secreting cells and increasing the Ag-specific CD8+ proliferation rate. Therefore, increased protective efficacy of DNA vaccination in combination with the pHMGB1 adjuvant may be the result of increased infiltration of activated APCs, namely DCs, into the muscle and spleen following i.m. immunization. Indeed, considering the structural and internal nature of the NP protein, adaptive immune responses against this Ag are mainly Th1-based. Thus, better survival against lethal flu challenge was likely a result of enhanced NP-specific T-cell responses, but not B-cell responses. Our results show that co-immunization with HMGB1 has strong adjuvant activity, driving stronger cellular immune responses to DNA vaccination directed against a broad range of pathogens.
To study the adjuvanting effects of pHMGB1 on humoral immunity, we developed a novel consensus vaccine expressing the HA from pandemic human swine 1909 influenza A type H1N1 (Figure 5). Adjuvanted mice displayed an increase in the HA-specific Ab response, which was represented by significantly higher levels of IgG1 levels than of IgG2 levels (Figure 6). As a result, these levels were likely responsible for the increased protective Ab responses as measured in the neutralization assay. These data demonstrate that adjuvanting the pH109 DNA vaccine with a plasmid expressing HMGB1 drove higher numbers and activation levels of APCs to the site of injection and major lymph areas, which likely was responsible for driving enhanced B- and T-cell effector and memory responses. The data suggest that the pHMGB1 adjuvant could be useful in vaccine strategies aiming to achieve enhanced Ab responses. This is particularly important when considering that an overwhelming majority of licensed vaccines in the United States mediate protection via the generation of pathogen-specific Abs. We and others are currently pursuing the precise mechanisms associated with EP-adjuvanted immunogenicity. Although the contribution of tissue damage at the immunization site to vaccine immunogenicity is not to be underestimated, it is important to note that we and others have identified a direct relationship between voltage and tissue damage in which higher levels decrease in vivo immunogenicity of plasmid-encoded Ag.22,24–26 Thus, it is likely that many factors in addition to tissue damage contribute to the potency of DNA vaccination in combination with EP. For example, we have also compared EP versus no EP delivery of both Ag and HMGB-1 (Figures 2b and c) and in the absence of EP very poor adjuvanted effects were observed. These data suggest that EP-enhanced delivery and expression levels were also very critical in the observed effects. Furthermore, it is also possible that EP stimulates a greater inflammatory environment that facilitates the function of the HMGB1 adjuvant.27,28 However, additional experiments will be needed to clarify the precise role of EP in this process and are thus beyond the scope of the current investigation. Altogether, although these results are promising, the role of using molecular adjuvants, such as pHMGB1, for the induction of protective Abs during DNA vaccination strategies is an important area for further investigation.
In summary, we have established the validity of the adjuvant effect of HMGB1 herein in an important flu Ag system and studied its ability to confer protective immunity against a viral challenge model. We hypothesized that molecular adjuvants, including chemokines and cytokines, can be incorporated into a vaccine strategy to bias the immune response toward cellular or humoral immunity. In particular, HMGB1 was shown to be involved in the expansion of strong humoral cellular immunity when used in this context.11 We found that HMGB1 did augment the protective effect of the DNA vaccine, as shown by a significant increase in the survival rate in the co-immunized animals compared with the controls. Therefore, studies should continue in investigating the adjuvanting effects of APC activating molecular adjuvants, such as pHMGB1, in increasing the potency on currently developing DNA vaccine strategies.
MATERIALS AND METHODS
DNA constructs and in vitro protein expression
Plasmids expressing mouse HMGB1 (pHMGB1), influenza A (H1H5) virus NP (pNP) HIV-1 Gag (pGag) and a consensus human swine flu HA sequence from pandemic influenza A subtype H1N1/09 virus (pH109) were designed and constructed in a similar manner and some have been previously described.11,15,24,29,30 Briefly, influenza A NP and HA novel H1N1/09 sequences were downloaded from the Los Alamos National Laboratory Influenza Sequence Database. Sequences were chosen from geographically diverse locations. MegAlign (DNASTAR, Madison, WI, USA) was used to align the sequences and generate consensus sequences. The full-length consensus constructs were optimized for expression, including codon and RNA optimization (GeneArt, Regensburg, Germany), and synthesized and inserted into the pVAX1 expression vector (Invitrogen, Carlsbad, CA, USA) as previously described.11,15,30,31 In vitro expression of pH109 was confirmed by transfecting the DNA into 293T cells (106) using the Fugene transfection method (Roche, Nutley, NJ, USA) as previously described.11 At 72 h post transfection, supernatants or protein lysates (50 μg) were harvested and fractioned on 10% SDS polyacrylamide gels and transferred to a poly(vinylidene difluoride) membrane (Bio-Rad, Hercules, CA, USA). Immunoblot analyses were performed with anti-HA antiserum (mouse monoclonal Ab to (monoclonal Ab) to HA; clone12CA5; Abcam, MA, USA) and visualized using horseradish peroxidase-coupled goat anti-rabbit IgG using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for visualization.32
Animals and immunizations
Female BALB/c mice (6–8 week old; The Jackson Laboratory, Bar Harbor, ME, USA) were housed in a temperature-controlled, light-cycled facility at the University of Pennsylvania and cared for under the guidelines of the National Institute of Health and the University of Pennsylvania. All animal experiments were performed in accordance with the national and institutional guidelines for animal care and were approved by the Review Board of the University of Pennsylvania. For immunizations, the quadriceps muscles were injected three times, 2 weeks apart, with 25 μg of pNP, pH109 or empty control expression vector pVax1 alone, or in combination with 10 μg of pHMGB1 and were immediately followed by in vivo EP as previously described.8,30,31 Briefly, square-wave pulses were delivered through a triangular three-electrode array consisting of 26-gauge solid stainless steel electrodes. Two constant-current pulses of 0.1 Amps were delivered for 52 msec per pulse separated by a 1 sec delay using the CELLECTRA adaptive constant current device (Inovio Pharmaceuticals, Blue Bell, PA, USA).
Fluorescence-activated cell sorting analysis and immunohistochemical analysis of APCs activation and maturation
Mice were injected i.m. with 25 μg of HIV-1-pGag or Flu-pNP-encoding plasmid alone or in combination with 10 μg of pHMGB1, and electroporated as previously described three times, 2 weeks apart.8,30,31 Empty pVax1 vector was used as a control. At 1 week after the final pDNA delivery, mice were killed and inguinal lymph nodes or spleens were collected, crushed using a stomacher machine and filtered through 40 μm cell strainers. Cells were then stained for the activation markers CD80 and CD86 and samples were acquired and analyzed as described above. For immunohistochemical staining, quadriceps muscles were collected and tissues were washed with cold phosphate-buffered saline (PBS) then fixed overnight in 4% formaldehyde. After dehydration, samples were embedded in paraffin wax and sectioned at 5 μm. CD83+ infiltrating cells were visualized through the standard avidin biotin complex immunohistochemistry staining method using affinity purified anti-mouse CD83 (eBioscience, San Diego, CA, USA) and Histostain-Plus kits (Invitrogen, Camarillo, CA, USA). A positive reaction for the CD83 surface marker was represented by development of a brown color. Splenic sections samples were collected from pHMGB1-adjuvanted mice (with pNP or pGag) and fixed in 10% buffered formalin solution and were stained for CD11c+.
Splenocyte isolation and ELISPOT assay
Mice were killed at 1 week following the third immunization and the samples were pooled according to group. Spleens were crushed using a Stomacher machine (Seward Laboratory Systems Inc., Bohemia, NY, USA), and the resulting product was filtered using a 40 μm cell strainer for splenocyte isolation. The cells were then treated for 5 min with ACK lysis buffer (Invitrogen) to lyse red blood cells and then the splenocytes were resuspended in RPMI medium supplemented with 10% fetal bovine serum. An ELISPOT assay was conducted as previously described.31,33 Briefly, 96-well ELISPOT plates (Millipore, Billerica, MA, USA) were coated with anti-mouse IFN-γ capture Ab and incubated for 24 h at 4 °C (R&D Systems, Minneapolis, MN, USA). The following day, plates were washed and blocked for 2 h with 1% bovine serum albumin. In all, 20 000 splenocytes from each group of mice were stimulated overnight at 37 °C in 5% CO2 in the presence of RPMI 1640 (negative control), Con A (positive control) or specific peptides (10 μg ml−1; NIH-AIDSRRP). Peptide pools consisted of 15-mer peptides overlapping by 11 amino acids and spanning the entire plasmid encoded genes. After 18–24 h of stimulation, the cells were washed and incubated for another 24 h at 4 °C with biotinylated anti-mouse IFN-γ monoclonal Ab (R&D Systems). The plates were then washed, and streptavidin–alkaline phosphatase was added to each well and incubated for 2 h at room temperature. The plates were washed again, and 5-bromo-4-chloro-3′-indolylphosphate p-toluidine salt and nitro blue tetrazolium chloride (chromogen color reagent) were added to each well (R&D Systems). The plates were then rinsed with distilled water and dried at room temperatrure. Spots were counted with an automated ELISPOT reader (Cellular Technology Ltd., Shaker Heights, OH, USA).6
Proliferation assay and flow cytometry
Mice were killed at month following the third immunization, splenocytes were harvested as described above, and then pelleted and resuspended in 1 ml of carboxyfluorescein diacetate succinimyl ester (2.5 μM; Molecular Probes, Eugene, OR, USA) in PBS and incubated for 10 m at 37 °C. Cells were washed twice with complete media and plated in 96-well round bottom plates along with 10 μml−1 peptide pools. Concavalin A (5 μg ml−1) and complete media were used as controls and cultures were incubated for 4 days. At the end of this period, cells were stained with LIVE/DEAD Violet Viability Dye (Invitrogen), anti-CD3 PE-Cy5 (BD Bioscience, San Jose, CA, USA), anti-CD4 PE-Cy7 (BD Bioscience) and anti-CD8 APC-Cy7 (BD Bioscience). In all, 50 000 viable CD3+ events were acquired on a LSRII flow cytometer (BD Immunocytometry Systems, San Jose, CA, USA) and data was analyzed using FlowJo version 7.5 (TreeStar, San Carlos, CA, USA).
End point Ab enzyme-linked immunosorbent assay detecting HA-specific Abs
HA-specific Ab levels following the third injection of pH109 was determined as previously described.11 Briefly, 96-well high-binding polystyrene plates (Costar, Corning Incorporated, Corning, NY, USA) were coated overnight at 4 °C with HA protein (5 μg ml−1; recombinant H1N1; Protein Science corporation, Meriden, CT, USA) diluted in PBS. The next day, plates were washed with PBS containing 0.05% Tween-20, blocked for 1 h with 3% bovine serum albumin in PBS containing 0.05% Tween-20, and then incubated with different dilutions of serum as indicated from immunized and naive mice for 1 h at 37 °C. Bound IgG was detected using goat anti-mouse IgG–horseradish peroxidase (GE Healthcare, formerly Amersham Biosciences, Piscataway, NJ, USA) at a dilution of 1:5000. Bound enzyme was detected by the addition of the chromogen substrate solution tetramethyl benzidine (R&D Systems), and read at 450 nm on a Biotek EL312e Bio-Kinetics reader (Bio-Tek Instruments, Winooski, VT, USA). All serum samples were tested in triplicate.
Viral challenge and histopathology
At 1 week after the last immunization, mice were intranasally challenged with 10 lethal dose 50 (2000TCID50) of live influenza A/PR8/34 in 30 μl of PBS. The animals were temporally anesthetized with anesthetic (Avertin, Sigma-Aldrich, St Louis, MO, USA) given by i.p. injection and subsequently processed for influenza A/PR8/34 infection. In total, 30 μl of dosed virus diluted in PBS was applied to the nares of anesthetized mice, resulting in the aspiration of the virus into upper and lower airways.9 After challenge, weight, clinical signs and mortality were recorded daily for 14 days. In accordance with the Institutional Animal Care and Use Committee guidelines, mice that lost 30% of the initial body weight were immediately killed. For histopathological analysis, on day 4 post challenge mouse lungs were explanted and fixed with 4% formaldehyde. Lung tissues were embedded in paraffin and cut into 5 μm sections. The tissue sections were mounted on glass slides, stained with hematoxylin and eosin and inspected microscopically.
Neutralization assay using influenza (A/Mexico/InDRE4487/2009) virus
The HA-specific neutralization assay was performed in microtiter plates as previously described.10,34 The influenza (A/Mexico/InDRE4487/2009) virus was cultured in the allantoic cavities of 10-day-old embryonated hen eggs and incubated for 2 days at 37 °C. The allantoic fluid was collected and stored at 80 °C. The virus was titrated in Madin–Darby canine kidney cell cultures to determine the plaque-forming units per milliliter. Titers of neutralizing antibodies were determined essentially as described. In brief, 50 μl of influenza virus containing 100 plaque-forming units of virus was incubated with 50 μl of twofold dilutions of the specific receptor-destroying enzyme-treated serum for 1 h at 37 °C in a 96-well round bottom plate. After 1 h, the virus–serum samples were transferred to a 96-well flat-bottom plate containing an Madin–Darby canine kidney cell monolayer and incubated for 5–10 min at 37 °C. Following the incubation, 100 μl of additional minimum essential medium (supplemented with 0.1% bovine serum albumin and 1 μg ml−1 of phenylalanyl chloromethyl ketone trypsin) was added to each well and incubated for 2 days at 37 °C. The microneutralization titer was defined as the reciprocal of the highest dilution of serum that neutralized 100 plaque-forming units of virus in Madin–Darby canine kidney cell cultures (as detected by absence of cytopathic effects).34
Statistical analysis
Data is presented as the mean±s.d. from the mean as calculated from the data collected from at least three independent experiments. Graphpad Prism 5 (Graphpad software Inc., San Diego, CA, USA) was used for analysis.
Acknowledgments
We thank the University of Pennsylvania, Department of Pathology Histology Core and Daniel Martinez for their help with immunohistochemical analyses. We also acknowledge Dr Niranjan Y Sardesai and Dr J Joseph Kim from Inovio Pharmaceuticals, PA, for their research expertize. This research was supported by the Inovio Pharmaceuticals, PA, under a collaborative research and development agreement with University of Pennsylvania (KM and DBW).
Footnotes
CONFLICT OF INTEREST
The DBW laboratory notes possible commercial conflicts associated with this work, which may include the following: Inovio, BMS, Virxsys, Ichor, Merck, Althea and Aldevron, and possibly others. The funders had no role in the study design, data collection, analysis, decision to publish or preparation of the manuscript.
References
- 1.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurunathan S, Wu CY, Freidag BL, Seder RA. DNA vaccines: a key for inducing long-term cellular immunity. Curr Opin Immunol. 2000;12:442–447. doi: 10.1016/s0952-7915(00)00118-7. [DOI] [PubMed] [Google Scholar]
- 3.Chattergoon MA, Muthumani K, Tamura Y, Ramanathan M, Shames JP, Saulino V, et al. DR5 activation of caspase-8 induces DC maturation and immune enhancement in vivo. Mol Ther. 2008;16:419–426. doi: 10.1038/sj.mt.6300373. [DOI] [PubMed] [Google Scholar]
- 4.Pinto AR, Reyes-Sandoval A, Ertl HC. Chemokines and TRANCE as genetic adjuvants for a DNA vaccine to rabies virus. Cell Immunol. 2003;224:106–113. doi: 10.1016/j.cellimm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Khan AS, Smith LC, Abruzzese RV, Cummings KK, Pope MA, Brown PA, et al. Optimization of electroporation parameters for the intramuscular delivery of plasmids in pigs. DNA Cell Biol. 2003;22:807–814. doi: 10.1089/104454903322625019. [DOI] [PubMed] [Google Scholar]
- 6.Boyer JD, Robinson TM, Kutzler MA, Vansant G, Hokey DA, Kumar S, et al. Protection against simian/human immunodeficiency virus (SHIV) 89. 6P in macaques after coimmunization with SHIV antigen and IL-15 plasmid. Proc Natl Acad Sci USA. 2007;104:18648–18653. doi: 10.1073/pnas.0709198104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muthumani K, Kudchodkar S, Zhang D, Bagarazzi ML, Kim JJ, Boyer JD, et al. Issues for improving multiplasmid DNA vaccines for HIV-1. Vaccine. 2002;20:1999–2003. doi: 10.1016/s0264-410x(02)00086-5. [DOI] [PubMed] [Google Scholar]
- 8.Hirao LA, Wu L, Khan AS, Hokey DA, Yan J, Dai A, et al. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine. 2008;26:3112–3120. doi: 10.1016/j.vaccine.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kutzler MA, Kraynyak KA, Nagle SJ, Parkinson RM, Zharikova D, Chattergoon M, et al. Plasmids encoding the mucosal chemokines CCL27 and CCL28 are effective adjuvants in eliciting antigen-specific immunity in vivo. Gene Therapy. 2010;17:72–82. doi: 10.1038/gt.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JJ, Bagarazzi ML, Trivedi N, Hu Y, Kazahaya K, Wilson DM, et al. Engineering of in vivo immune responses to DNA immunization via codelivery of costimulatory molecule genes. Nat Biotechnol. 1997;15:641–646. doi: 10.1038/nbt0797-641. [DOI] [PubMed] [Google Scholar]
- 11.Muthumani G, Laddy DJ, Sundaram SG, Fagone P, Shedlock DJ, Kannan S, et al. Co-immunization with an optimized plasmid-encoded immune stimulatory interleukin, high-mobility group box 1 protein, results in enhanced interferon-gamma secretion by antigen-specific CD8T cells. Immunology. 2009;128:e612–e620. doi: 10.1111/j.1365-2567.2009.03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 2005;106:609–616. doi: 10.1182/blood-2004-10-3906. [DOI] [PubMed] [Google Scholar]
- 13.Laddy DJ, Yan J, Kutzler M, Kobasa D, Kobinger GP, Khan AS, et al. Heterosubtypic protection against pathogenic human and avian influenza viruses via in vivo electro-poration of synthetic consensus DNA antigens. PLoS ONE. 2008;3:e2517. doi: 10.1371/journal.pone.0002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JJ, Nottingham LK, Sin JI, Tsai A, Morrison L, Oh J, et al. CD8 positive T cells influence antigen-specific immune responses through the expression of chemokines. J Clin Invest. 1998;102:1112–1124. doi: 10.1172/JCI3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laddy DJ, Yan J, Khan AS, Andersen H, Cohn A, Greenhouse J, et al. Electroporation of synthetic DNA antigens offers protection in nonhuman primates challenged with highly pathogenic avian influenza virus. J Virol. 2009;83:4624–4630. doi: 10.1128/JVI.02335-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shedlock DJ, Shen H. Requirement for CD4T cell help in generating functional CD8T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 17.Morrow MP, Weiner DB. Cytokines as adjuvants for improving anti-HIV responses. AIDS. 2008;22:333–338. doi: 10.1097/QAD.0b013e3282f42461. [DOI] [PubMed] [Google Scholar]
- 18.Hung CF, Wu TC. Improving DNA vaccine potency via modification of professional antigen presenting cells. Curr Opin Mol Ther. 2003;5:20–24. [PubMed] [Google Scholar]
- 19.Morrow MP, Pankhong P, Laddy DJ, Schoenly KA, Yan J, Cisper N, et al. Comparative ability of IL-12 and IL-28B to regulate Treg cell populations and enhance adaptive cellular immunity. Blood. 2009;113:5868–5877. doi: 10.1182/blood-2008-11-190520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayak BP, Sailaja G, Jabbar AM. Augmenting the immunogenicity of DNA vaccines: role of plasmid-encoded Flt-3 ligand, as a molecular adjuvant in genetic vaccination. Virology. 2006;348:277–288. doi: 10.1016/j.virol.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 21.Lena P, Villinger F, Giavedoni L, Miller CJ, Rhodes G, Luciw P. Co-immunization of rhesus macaques with plasmid vectors expressing IFN-gamma, GM-CSF, and SIV antigens enhances anti-viral humoral immunity but does not affect viremia after challenge with highly pathogenic virus. Vaccine. 2002;20(Suppl 4):A69–A79. doi: 10.1016/s0264-410x(02)00391-2. [DOI] [PubMed] [Google Scholar]
- 22.Broderick KE, Kardos T, McCoy JR, Fons MP, Kemmerrer S, Sardesai NY. Piezoelectric permeabilization of mammalian dermal tissue for in vivo DNA delivery leads to enhanced protein expression and increased immunogenicity. Hum Vaccin. 2011;7:22–28. doi: 10.4161/hv.7.0.14559. [DOI] [PubMed] [Google Scholar]
- 23.Sun JC, Bevan MJ. Defective CD8T cell memory following acute infection without CD4T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin F, Shen X, McCoy JR, Mendoza JM, Yan J, Kemmerrer SV, et al. A novel prototype device for electroporation-enhanced DNA vaccine delivery simultaneously to both skin and muscle. Vaccine. 2011 doi: 10.1016/j.vaccine.2010.12.057. e-pub ahead of print 1 January 2011. [DOI] [PubMed] [Google Scholar]
- 25.Heller LC, Cruz YL, Ferraro B, Yang H, Heller R. Plasmid injection and application of electric pulses alter endogenous mRNA and protein expression in B16. F10 mouse melanomas. Cancer Gene Ther. 2010;17:864–871. doi: 10.1038/cgt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferraro B, Heller LC, Cruz YL, Guo S, Donate A, Heller R. Evaluation of delivery conditions for cutaneous plasmid electrotransfer using a multielectrode array. Gene Therapy. 2011 doi: 10.1038/gt.2010.171. e-pub ahead of print 23 December 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 28.Luke JM, Simon GG, Soderholm J, Errett JS, August JT, Gale M, Jr , et al. Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J Virol. 2011;85:1370–1383. doi: 10.1128/JVI.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan J, Harris K, Khan AS, Draghia-Akli R, Sewell D, Weiner DB. Cellular immunity induced by a novel HPV18 DNA vaccine encoding an E6/E7 fusion consensus protein in mice and rhesus macaques. Vaccine. 2008;26:5210–5215. doi: 10.1016/j.vaccine.2008.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muthumani K, Lankaraman KM, Laddy DJ, Sundaram SG, Chung CW, Sako E, et al. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine. 2008;26:5128–5134. doi: 10.1016/j.vaccine.2008.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laddy DJ, Yan J, Corbitt N, Kobasa D, Kobinger GP, Weiner DB. Immunogenicity of novel consensus-based DNA vaccines against avian influenza. Vaccine. 2007;25:2984–2989. doi: 10.1016/j.vaccine.2007.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muthumani K, Choo AY, Zong WX, Madesh M, Hwang DS, Premkumar A, et al. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat Cell Biol. 2006;8:170–179. doi: 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutzler MA, Robinson TM, Chattergoon MA, Choo DK, Choo AY, Choe PY, et al. Coimmunization with an optimized IL-15 plasmid results in enhanced function and longevity of CD8T cells that are partially independent of CD4T cell help. J Immunol. 2005;175:112–123. doi: 10.4049/jimmunol.175.1.112. [DOI] [PubMed] [Google Scholar]
- 34.Kobinger GP, Meunier I, Patel A, Pillet S, Gren J, Stebner S, et al. Assessment of the efficacy of commercially available and candidate vaccines against a pandemic H1N1 2009 virus. J Infect Dis. 2010;201:1000–1006. doi: 10.1086/651171. [DOI] [PMC free article] [PubMed] [Google Scholar]