

Abstract
Background
Adoptive Immunotherapy using chimeric antigen receptor (CAR) engineered T cells specific for CD19 has shown promising results for the treatment of B cell lymphomas and leukemia. This therapy involves the transduction of autologous T cells with a viral vector and the subsequent cell expansion. Here, we describe a new, simplified method to produce anti-CD19-CAR T cells.
Methods
T cells were isolated from peripheral blood mononuclear cell (PBMC) with anti-CD3/anti-CD28 paramagnetic beads. After 2 days, the T cells were added to culture bags pre-treated with RetroNectin and loaded with the retroviral anti-CD19 CAR vector. The cells, beads and vector were incubated for 24 hours and then a second transduction was performed. No spinoculation was used. Cells were then expanded for an additional 9 days.
Results
The method was validated using 2 PBMC products from a patient with B-CLL and one PBMC product from a healthy subject. The 2 PBMC products from the B-CLL patient contained 11.4% and 12.9% T cells. The manufacture process led to final products highly enriched in T cells with a mean CD3+ cell content of 98%, a mean expansion of 10.6 fold and a mean transduction efficiency of 68%. Similar results were obtained from the PBMCs of the first 4 ALL patients treated at our institution.
Discussion
We developed a simplified semi-closed system for the initial selection, activation, transduction and expansion of T cells using anti-CD3/anti-CD28 beads and bags, to produce autologous anti-CD19 CAR transduced T cells to support an ongoing clinical trial.
Keywords: Adoptive Cellular Immunotherapy, CD19 Antigen, Genetic Engineering, Genetic Transduction, Precursor Cell Lymphoblastic Leukemia-Lymphoma
Introduction
Cancer is the leading cause of disease related death in U.S. pediatric patients [1] and acute lymphoblastic leukemia (ALL) represents the most common pediatric malignancy. Although disease-free survival rates are high, approximately 20% of patients dies of their underlying disease, making ALL the leading cause of cancer death in childhood [2]. For those who relapse, long-term prognosis is poor. Even when a second remission is achieved, long-term disease-free survival rates below 50% are expected [3]. New treatment approaches are therefore needed.
Adoptive immunotherapy is considered an attractive way to treat cancer [4-10] and genetic modification of T cells with genes encoding tumor specific chimeric antigen receptors (CARs) represents a novel strategy to obtain large quantities of tumor-reactive T cells to be used for the treatment of adult [11-13] and pediatric cancer patients [14].
CD19 has been chosen as a target for CAR T cells because it is universally expressed on ALL tumor cells, and not expressed on pluripotent hematopoietic stem cells [15-17]. Anti-CD19-CAR T cells are under investigation in several institutions using different viral constructs and various manufacturing processes and have shown promising clinical results [18-21].
For the production of CAR engineered T cells, procedures and techniques enabling ex vivo gene modifications of cells followed by expansion to clinically relevant cell numbers are of key importance. For this purpose cell culture systems have been developed in compliance of good manufacture practice criteria (GMP). Current manufacturing processes rely on the use of recombinant human fibronectin fragment CH296 (RetroNectin®) that has demonstrated to improve transduction efficiency bringing together retroviral particles and cells. This has often been paired with spinoculation, a procedure that promotes gene transfer by pre-loading the retroviral vector stocks on the RetroNectin by a 2-hour centrifugation, followed by a second centrifugation of target cells. Some of the processes use bags and are semi-closed, but others use plates and flasks and are open, presenting a large risk of contamination and spilling of vector.
In the present study we describe the process that has been developed in our institution for the production of therapeutic doses of autologous anti-CD19-CAR T cells to support phase I clinical trial for the treatment of B cell malignancies in pediatric patients, using a replication defective MSGV1-based retroviral vector (MSGV-FMC63-28z) encoding a chimeric receptor containing the signaling domains of CD28 and CD3-zeta. The goal of this study was to test the level of the transduction efficiency we could achieve using tissue culture bags as vessel for transduction and cell expansion process, avoiding spinoculation. We made use of GMP paramagnetic beads coated with anti-CD3 and anti-CD28 to isolate CD3+ cells from peripheral blood mononuclear cell products (PBMCs) collected by apheresis and at the same time stimulate sufficient CD3+ cell proliferation to facilitate transduction and subsequent expansion. The process described allowed us to generate anti-CD19-CAR T cells using a semi-closed system without the spinoculation step maintaining acceptable transduction efficiency.
Materials and Methods
Construction and GMP production of the MSGV-FMC63-28z recombinant retroviral vector
The MSVG-FMC63-28z recombinant retroviral vector was prepared and cryopreserved following GMP conditions in the Surgery Branch, NCI, NIH Vector Production Facility (SBVPF). The construction and production of the MSGV-FMC63-28z vector has been described elsewhere by Kochenderfer et al. [22]. These studies were approved by an NIH institutional review board.
Culture media
T cell initiation medium
AIM V medium (Gibco, Grand Island, NY), supplemented with 5% heat-inactivated human AB Serum (Valley Biomedical, Winchester, VA), 1% Gluta-Max (Gibco, Grand Island, NY), 40 IU/mL IL-2 (Novartis Vaccines and Diagnostics, Inc. Emeryville, CA).
T cell expansion medium
AIM V medium (Gibco, Grand Island, NY), supplemented with 5% heat-inactivated human AB Serum (Valley Biomedical, Winchester, VA), 1% Gluta-Max (Gibco, Grand Island, NY), 300 IU/mL IL-2 (Novartis Vaccines and Diagnostics, Inc. Emeryville, CA).
Generation of clinical grade anti-CD19-CAR transduced T cells
The process was optimized using PBMC products collected by apheresis from healthy subjects. The final process for the manufacture of clinical grade anti-CD19-CAR T cells was validated using a PBMC product from one healthy subject (VR1) and two PBMC products from a patient with B-CLL (VR2 and VR3). Clinical anti-CD19-CAR T cell products were generated to treat four ALL patients enrolled in the phase I clinical study NCT01593696 sponsored by the NCI.
On day 0, fresh PBMC products collected by apheresis were enriched for CD3+ cells using anti-CD3 and anti-CD28 antibodies bound to paramagnetic beads (Dynabeads ClinExVivo CD3/CD28, Invitrogen, Camarillo, CA) at a ratio of 3:1 (beads:cells). The enrichment was performed using cells diluted to a total nucleated cells (TNC) concentration of 20-30×106/ml in PermaLife bags made of FEP (OriGen Biomedical, Austin, TX). The cells and beads were co-incubated for 2 hours at room temperature and CD3+ cell enrichment performed using Dynal ClinExVIVO MPC magnet (Invitrogen, Camarillo, CA). Cells in the CD3+ fraction were resuspended in initiation media at a concentration of 1×106 cells/ml in PermaLife bags (OriGen Biomedical, Austin, TX).
On day 1, clinical grade RetroNectin (Takara Bio Division, Shiga, Japan) was reconstituted in sterile water at 1 mg/ml and used to coat PermaLife bags (OriGen Biomedical, Austin, TX) at a concentration of 2 μg/cm2 in a solution of 10 μg/ml in PBS (Lonza, Walkerville, MD). The coated bags were incubated overnight at 4°C. On day 2, the RetroNectin solution was aspirated from bags and same volume of blocking solution, consisting of 2.5% human serum albumin (HSA) (Baxter, Westlake Village, CA) in PBS, was added to each bag and incubated at room temperature (RT) for 30 minutes. The blocking solution was aspirated and each bag washed with a solution of 2.5% HEPES (Lonza, Walkerville, MD) diluted in Hank’s balanced salt solution (HBSS) (Lonza, Walkerville, MD).
On day 2 retroviral supernatant was rapidly thawed and added to each bag at the final volume that was half of the one specified by the manufacturer for each culture bag. In addition, a RetroNectin coated bag was filled with expansion media and used as negative untransduced control (UT CNTR bag). The bags were then incubated for 2 hours at 37°C, 5% CO2. At the end of the incubation time, activated CD3+ cells in expansion media were mixed 1:1 (v/v) with the clinical grade vector contained in the RetroNectin coated bags; at this point IL-2 concentration was raised to 300 IU/ml. The cultures were placed back to the incubator and left for at least 24 hours. A second transduction was performed on day 3 using newly coated bags as previously described. An untransduced control was processed in parallel; the cells were treated the same except for the lack of exposure to the viral vector.
On day 4, the transduction was stopped; transduced and untransduced cells were counted, transferred in a new culture bag and resuspended in fresh T cell expansion medium at a concentration of 0.4×106 cells/ml. The cultures were maintained until day 11+2 and fed every other day with fresh expansion media to maintain cell concentration at 0.4×106 cells/ml.
On day 7 quality controls were performed, transduction efficiency was evaluated and in process mycoplasma assessed.
On day 11+2, cells were harvested. The anti-CD3/anti-CD28 paramagnetic beads were removed using the Dynal ClinExVIVO MPC magnet (Invitrogen, Camarillo, CA), washed, concentrated and quality control assessment was performed. The cells were packaged for fresh infusion at a concentration of 2.5×106 cells/mL in infusion media which contained plasmalyte A (Baxter, Deerfield, IL) plus 4% HSA (Baxter, Westlake Village, CA). Leftover cells were cryopreserved according to our internal procedures and used for post-thaw stability testing.
Flow cytometry analysis
On day 7, 9 and on final harvest the expression of anti-CD19-CAR T cell was evaluated by flow cytometry using a Protein L assay. Biotinylated Protein L was purchased from Thermo Scientific (Rockford, IL), reconstituted in sterile water at a stock concentration of 50ng/μl, and stored at 4°C. 5 μg was used to label 1×106 cells. The cells and Protein L were incubated for 30 minutes at 4°C in the dark. The Protein L final concentration was determined using titration experiments performed during assay development. Excess reagents were removed by washing the tubes twice using cold FACS buffer. After 20 minutes 30 μl of a solution of PE-labeled Streptavidin (BD Biosciences, San Jose, CA), pre-diluted 1:120 in PBS, was added to each tube.
7-AAD (Immunotech, Marseille, FR) was used to determine viability, and the final product was characterized for expression of CD3, CD8 (Invitrogen, Camarillo, CA), CD4, and CD19 (BD Biosciences, San Jose, CA). The untransduced control cells were assessed in the same way. Stained cells were acquired with a BD FacsCanto II (BD Biosciences, San Jose, CA) and analyzed with FlowJo (Treestar Inc, Ashland, OR).
Quality control analysis and PCR for testing replication competent retrovirus (RCR)
The final product was evaluated for sterility, endotoxin and mycoplasma following standard operation procedures optimized and validated in our institution according to regulatory agencies standards. For RCR-PCR 1% of final product was centrifuged and resulting pellet frozen at -80°C. Total DNA was extracted using Qiagen-QIAamp DNA Blood Midi Kit (Qiagen, Valencia, CA); content and purity of the extracted DNA were evaluated by NanoDrop Spectrophotometer. 500 ng of DNA was used for each PCR reaction, using the following primers (IDT, Coralville, IA): hActin FWD: 5- ACA CTG TGC CCA TCT ACG AGG -3 hActin REV: 5- AGG GGC CGG ACT CGT CAT ACT -3 GALV FWD: 5- ACC ACA GGC GAC AGA CTT TT -3 GALV REV: 5- TGA GAC AGC CTC TCT TTT AGT CCT -3.
As negative control DEPC water was used and a GALV plasmid from Lofstrand (Rockville, MD) as positive control.
Each PCR reaction was set up as follows: 25 μl of HotStarTaq Master Mix (2X) (Qiagen, Valencia, CA), 5 μl of each primer from a 10X working solution at 4 μM and brought to the final volume of reaction of 50 μl with DEPC water.
The reaction was run on a DNA Engine (BioRad, Hercules, CA) with the following program: a) 95° C for 15′, b) 94° C for 1′, c) 70° C for 30″, d) 72° C for 1′, repeat from b) to d) 30 times and final 72° C for 5′. PCR product was run on electrophoresis gel.
Cytotoxicity assays
Anti-CD19-CAR T cells were incubated at various effector-to-target (E:T) ratios with 5000 51Cr labeled targets in 96-well plates. Targets were labeled by incubating them with 100 μCi of 51Cr (Perkin Elmer, Boston, MA) at 37° C for 1 hour then washed three times with media immediately prior to mixing with effector cells. K562 cells stably transfected with CD19 (K562-CD19) served as a positive control while K562 cells stably transfected with nerve growth factor receptor (NGFR) served as a CD19 negative control. Each E:T ratio for each effector and target pair and all controls were performed in triplicate. Spontaneous 51Cr release was determined by incubating labeled targets alone while maximum 51Cr release was determined by adding 100 uL of 0.1 N HCl to labeled targets. After 4 hours of incubation at 37° C, 25 μL of supernatant from each well was transferred to corresponding 96-well LumaPlates-96 (Perkin-Elmer, Boston, MA). 51Cr activity in the supernatant was determined using a Top Count Reader (Packard, Location). Percent Cytotoxicity was defined by the formula: % Cytotoxicity = [(experimental release) − (spontaneous release)] ÷ [(maximum release) − (spontaneous release)] × 100.
Results
Initial selection and activation of CD3+ cells
We performed 3 validation runs after completing the process optimization phase. The validation was performed using one PBMC product collected by apheresis from one healthy subject (VR1) and two separate PBMC collections from one patient with B-CLL (VR2 and VR3). The validation study is summarized in Fig. 1.
Figure 1.

Scheme of the manufacturing process. On day 0, the apheresis product was incubated with anti-CD3/anti-CD28 paramagnetic Dynabeads at a ratio of beads:CD3+ cells of 3:1. The suspension of cells and beads was then exposed to a magnet (ClinExVivo MPC) to select the CD3+ cell fraction. The selected cells were washed and resuspended in initiation medium, with low IL-2 concentration. After 2 days of culture the cells and beads were added to culture bags (Permalife) that had been treated with RetroNectin and loaded with anti-CD19 CAR viral vector and incubated for at least 24 hours. The transduction step was repeated the day after and then the cells and beads were transferred to new culture bags and were expanded for 9 more days. At the completion of the cell expansion, the beads were removed and discarded with the ClinExVivo MPC magnet and the cells were washed and prepared for the infusion.
During our pre-validation studies, we tested two different methods to activate the T cells: OKT3 (30ng/ml) and anti-CD3/anti-CD28 paramagnetic beads. We decided to use the anti-CD3/anti-CD28 paramagnetic beads due to their capability for enriching the culture with CD3+ cells, avoiding the need for an intermediate selection step. The enrichment process debulked the PBMC product of CD19+ blasts typical of the patient population to be enrolled in the clinical study. Moreover, the total fold expansion was much higher in the culture treated with the anti-CD3/anti-CD28 beads than with OKT3 (data not shown). Characteristics of the initial PBMC products are shown in Table 1. As shown, the CD3+ content in both apheresis products from the patient with B-CLL was less than that of the product from the healthy donor (11.4% and 12.9% for the patient products VR2 and VR3 and 49.6% for healthy subject VR1) whereas the CD19+ content in the patient’s samples was more than 6-fold greater than the basal level of the healthy donor (80.7% for patient product VR3 and 12.9% for the healthy subject product VR1).
Table 1.
Characteristics of the apheresis and final anti-CD19-CAR products: one from a healthy subject and two from a patient with B-CLL
| Apheresis Products | Final Products | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Healthy Donor VR1 (%) |
Patient VR2 (%) |
Patient VR3 (%) |
Healthy Donor VR1 (%) |
Patient VR2 (%) |
Patient VR3 (%) |
|
| CD3+ | 49.6 | 11.4 | 12.9 | 95.0 | 98.2 | 99.7 |
| CD3+ CD4+ | 32.1 | NT | 6.5 | 38.4 | NT | 66.1 |
| CD3+ CD8+ | 15.9 | NT | 5.5 | 43.8 | 31.0 | 31.9 |
| CD19+ | 12.9 | NT | 80.7 | 0.3 | NT | 0.0 |
| CD56+ | 8.2 | 0.9 | 1.1 | 0.1 | NT | 0.1 |
NT = not tested
T-cells transduction and expansion
On days 2 and 3, T cells were transduced with anti-CD19-CAR vector. During preclinical development, we tested several conditions to optimize the transduction steps while moving the process from open plates to bags and removing the spinoculation step with its inherent risk of bag or plate breakage. We obtained similar results to spinoculation by incubating T cells in bags with retroviral vector in solution for 24 hours at 37°C, 5% CO2 and >90% humidity (data not shown). We therefore elected to use the latter method of T cell transduction.
At the completion of transduction on day 4, the cells were evaluated for total nucleated cell number and viability by manual count and trypan blue exclusion. Based on the TNC count, T cells were maintained at 0.4×106 TNC/ml in T-cell expansion medium until day 13. On day 7 and every other day up to the day of harvest the culture was evaluated for TNC count and viability by manual count and trypan blue exclusion and transduction efficiency (Fig. 2). The cumulative fold TNC expansion of each of the 3 samples was comparable and similar to the expansion of the UT CNTR. There was a slight increase at the end of the culture period in the quantity of TNCs in patient sample VR3 compared with the healthy subject’s sample (VR1) (Panel A). In contrast, the viability of cells from the healthy subject was greater than that of the patient’s cells early in the culture period but not at the end of the process. Towards the end of the process the viability of the healthy subject cells, VR1, decreased slightly, whereas that of the patient cells remained stable or increased (Panel B). The transduction efficiency for all the samples ranged from 50% to 80% through the cell expansion phase, and was similar for healthy subject and patient cells (Panel C). The average transduction efficiency was approximately 60% and remained quite stable through the cell expansion process (Panel D).
Figure 2.

Viability, expansion and transduction efficiency of cultured T cells.
Panel A) The cumulative fold expansion of T cells isolated from one healthy donor PBMCs concentrate (VR1) and two different PBMCs concentrates from a B-CLL patient (VR2 and VR3). The average expansion of untransduced control T cells is also shown (UT). Panel B) The viability of cultured transduced T cells from a healthy donor (VR1) and two different PBMCs concentrates from a B-CLL patient (VR2 and VR3). The average viability of untransduced control T cells is also shown (UT). Panel C) The transduction efficiency on days 7, 9, and 13 of cultured transduced T cells from a healthy donor (VR1) and two different PBMCs concentrates from a B-CLL patient (VR2 and VR3). The transduction efficiency of untransduced control T cells is also shown (UT), to assess the staining specificity. Panel D) The average transduction efficiency on days 7, 9, and 13 for the three validation runs. The transduction efficiency of untransduced control T cells is also shown (UT), to assess the staining specificity.
Final product characterization
The final products showed a mean fold increase of 10.6 (Supplemental Table 1), a viability of 70.4% and a transduction efficiency of 68.4%. Therefore, our release criterion for the viability was set at 60%. All three final products were highly pure, since for all samples ≥95% of the cells expressed CD3 (Table 1). Finally, to test whether B-CLL tumor cells can persist through the manufacturing process, we evaluated the final products from 1 patient and 1 healthy subject for the presence of CD19+ cells and did not find a significant quantity of CD19+ cells in either product (Table 1). Bacterial and fungal cultures performed on all 3 final products showed no growth and PCR analysis of the products for mycoplasma was negative. In addition, analysis of the products for replication competent retrovirus (RCR) by PCR, using the GaLV envelope as target sequence was negative (Fig. 3).
Figure 3.
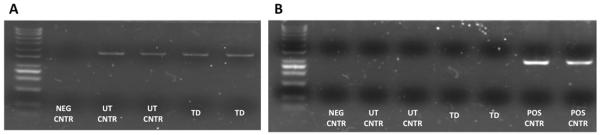
RCR PCR results. 500 ng of DNA from each sample were used for each PCR reaction, using either Actin primers (Panel A) at a final concentration of 0,4 μM (FWD: 5- ACA CTG TGC CCA TCT ACG AGG -3; REV: 5- AGG GGC CGG ACT CGT CAT ACT -3) as a positive control or GalV primers (Panel B) at a final concentration of 0,4 μM (FWD: 5- ACC ACA GGC GAC AGA CTT TT -3; GALV REV: 5- TGA GAC AGC CTC TCT TTT AGT CCT -3). As negative control DEPC water was used instead of DNA. As a positive control a GALV plasmid was used instead of the sample’s DNA. PCR products were run on electrophoresis gel.
In vitro cytotoxicity studies of anti-CD19-CAR T cells
To test the functionality of the final product we performed cytotoxic T lymphocyte (CTL) assays against the CD19+ ALL cell lines, NALM6, REH, SEM, and KOPN8. CD19-CAR T cells generated from two separate manufacturing runs of the B-CLL patient (VR2 and VR3) were incubated for 4 hours with 51Cr labeled targets. K562 cells stably transfected with the CD19 antigen or the nerve growth factor receptor (NGFR) served as positive and negative controls, respectively. Manufactured anti-CD19-CAR T cells were able to kill their targets in a CD19-specific manner (Fig. 4, Panel A and C). More impressively, very high levels of cytotoxicity were appreciated even at very low effector-to-target (E:T) ratios. In contrast, the corresponding untransduced controls demonstrated little cytotoxicity (Figure 4, Panel B and D). Therefore, anti-CD19-CAR T cells generated from patient apheresis have the potential for significant and specific in vivo lysis of ALL blasts.
Figure 4.

In vitro activity of manufactured anti-CD19-CAR T cells. Anti-CD19-CAR transduced T cells (Panels A and C) generated from two different apheresis from the same B-cell CLL patient, VR2 (Panel A) and VR3 (Panel C), kill CD19+ ALL cell lines (NALM6, KOPN8, REH, SEM) and the positive control, K562-CD19, but not the CD19 negative targets, K562-NGFR. Percent cytotoxicity is shown at various effector-to-target (E:T) ratios. In contrast, cells prepared in similar manner but not transduced with the CAR have little non-specific cytotoxicity (Panels B and D, VR2 and VR3 respectively).
Stability of the final product
To test the stability of the final products, we determined cell recovery, viability and transduction efficiency in both fresh and cryopreserved products. Final fresh products were evaluated after 3 hours (T=3) of storage at room temperature at a concentration of 2.5×106 TNC/mL in infusion medium, compared to the moment of the final suspension of the product in infusion medium (T=0) (Figure 5). All products were stable in terms of number (Panel A), (TNC at T=0 was set as 100%), viability (Panel B) and transduction efficiency (Panel C).
Figure 5.

Stability of harvested anti-CD19-CAR transduced T cells after 3 hours of room temperature storage. The average viable cells recovery (panel A), cell viability (panel B) and transduction efficiency (panel C) of three products (VR1, VR2 and VR3) immediately after harvest and after 3 hours of storage in infusion medium at room temperature are shown, together with the data from the correspondent untransduced controls (UT).
Final cryopreserved products were evaluated immediately after thawing from storage under liquid nitrogen and resuspending in infusion medium (T=0), and after 3 hours of room temperature storage (T=3). The TNC recovery at T=0 was similar to the precryopreservation TNC, but after 3 hours of post-thaw storage cell loss was significant (approximately 20%) (Figure 6, Panel A). In contrast, viability was stable after 3 hours, with only a slight decrease from T=0 (80% at T=0 vs. 70% at T=3) (Panel B). It is noteworthy that the transduction efficiency increased after thawing and remained stable after 3 hours of post-thaw storage at room temperature (Panel C).
Figure 6.

Stability of harvested anti-CD19-CAR transduced T cells that had been cryopreserved and thawed. The average viable cells recovery (panel A), cell viability (panel B) and transduction efficiency (panel C) of three products (VR1, VR2 and VR3) immediately after thawing and after 3 hours of storage at room temperature are shown, together with the data from the correspondent untransduced controls (UT).
Clinical product characterization
Finally, to prove the robustness of our method, we monitored fold expansion, viability and transduction efficiency of four clinical products derived from as many ALL patients. Cells were processed as described; harvest was performed on Day 11 of culture. The four final products showed a median fold increase of 21 (Supplemental Figure 1), a viability of 87.5% and a transduction efficiency of 39.6%. All four final products were highly pure, since for all samples ≥95% of the cells expressed CD3. In addition, to test whether tumor cells can persist through the manufacturing process, we evaluated the final products for the presence of CD19+ cells and did not find a significant quantity of CD19+ cells in the products (<0.5%). Bacterial and fungal cultures performed showed no growth and mycoplasma testing was negative. In addition, analysis of the products for replication competent retrovirus (RCR) by PCR, using the GaLV envelope as target sequence was negative.
Discussion
Adoptive immunotherapy represents a safe and promising treatment for patients with B cell leukemia and lymphomas who fail standard therapy. There are several potential adoptive cellular therapies that can be used to treat cancer. The specificity of polyclonal T cells can be reassigned by gene transfer of either peptide-specific high affinity T cell receptors (TCRs) or CARs. Promising clinical results with genetically engineered high affinity TCRs have been achieved in melanoma patients [23], however CAR-based methods offer several advantages over the transfer of TCRs. CARs, like monoclonal antibodies, don’t rely on MHC molecules for the recognition of target cells, so a single receptor can be used to treat patients with any HLA type. Moreover, one of the immune evasion strategies exhibited by tumors is the down-regulation of MHC molecule expression and/or the deregulation of antigen-presentation machinery, but CARs are not affected by this strategy because they bind directly to native proteins expressed on the target cell’s surface. Finally, CARs can also recognize non-protein antigens unlike TCRs [24].
CD19 currently represents the ideal molecule to target B cell malignancies for adoptive immunotherapy; its expression is restrictively limited to normal and malignant B cells and not expressed by other normal cells, including pluripotent hematopoietic stem cells [15-17]. The depletion of all CD19-expressing cells has been shown not to be a lethal condition [25]. Several clinical trials with CAR-expressing T cells targeting tumor cells including anti-CD19-CAR have reported very promising preliminary clinical results [18-20]. This therapy has, however, been associated with prolonged depletion of B cells [26].
Most CAR clinical trials have typically used gammaretrovirus- or lentivirus-based vectors which stably express the CAR constructs in T cells. Gammaretroviruses have shown acceptable efficacy for expressing CARs in T lymphocytes and have proved to be safe [27-28].
Previous studies using retroviral anti-CD19-CAR vectors have transduced cells in open systems using flasks, spinoculation and soluble anti-CD3 for T cell stimulation and feeder cells for T cell expansion [29]. Other studies have used bags, spinoculation and anti-CD3/anti-CD28 beads for T cell stimulation and expansion. Spinoculation is a procedure that facilitates gene transfer by cocentrifugation of target T cells with viral vector stocks, and it has been part of processes for gene transfer to improve transduction efficiency. However, in a GMP compliant setting, the removal of spinoculation from the manufacture process offers several advantages, reducing time and labor, and more important possible contamination of products due to the use of open systems or breakage of containers. The process we have optimized and validated in the present study is able to achieve high transduction efficiencies without the need of spinoculation, representing an advantage compared to previous described processes. We also confirmed what has been previously showed by other groups that the anti-CD3/anti-CD28 beads can be used to enrich PBMC apheresis products for CD3+ cells. This T cell enrichment is able to initiate culture with a higher purity starting material even in patients with high concentrations of circulating malignant B cells.
The results of stability testing of the final product showed that fresh products maintained their characteristics even after 3 hours from final preparation in infusing medium. Stability of the final products after a freeze/thaw cycle was less encouraging. Cryopreserved cells should also be effective, but the product might have to be infused immediately after it is thawed because after 3 hours of post-thaw room temperature storage the cell recovery and viability were lower. This is an important point to consider if in the future CAR therapies will be available as “off the shelf” drugs, but further testing and optimizations may be needed.
A limitation of our validation study was the unavailability of PBMCs from patients with hematologic disease, our experience was based mainly on normal subjects and two PBMC products from a B-CLL patient. The feasibility and consistency of the optimized procedure was evaluated once we manufactured the first clinical products. Four anti-CD19-CAR T cells were generated from as many ALL patients and final results were similar to validation runs. In particular, with the exception of one technical failure, the other three showed high transduction efficiency, high fold increase and viability. Based on these facts, we are confident that the behavior shown by cultures in this study will be similar for other samples, although some inter-donor variability likely will be observed.
Conclusion
We have developed a simplified semi-closed system for the initial selection, activation, transduction and expansion of T cells, using anti-CD3/anti-CD28 beads (for selection/activation) and bags (as vessel), to produce autologous anti-CD19-CAR-transduced T cells, without the tedious step of spinoculation. We consider our simplified process an easy and scalable platform that should also be effective for the production of other CAR T cells that make use of gammaretroviral vectors.
Supplementary Material
Supplemental Figure 1. Viability, expansion and transduction efficiency of four ALL patient cultured T cells.
Panel A) cumulative fold expansion of T cells isolated from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel B) viability of cultured transduced T cells from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel C) transduction efficiency on days 7, 9, and 11 of cultured transduced T cells from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel D) average transduction efficiency on days 7, 9, and 11 for the four clinical products.
{kind=link}
Supplemental Table 1. Number of cells (x10e6) during the Expansion Phase of manufacture
Acknowledgement
This research was supported by the Intramural Research Program of the NIH, Clinical Center and National Cancer Institute.
Abbreviations
- ALL
Acute Lymphoblastic Leukemia
- B-CLL
B-cell Chronic Lymphoblastic Leukemia
- CAR
Chimeric Antigen Receptor
- CTL
Cytotoxic T Lymphocyte
- DEPC
DiEthylPyroCarbonate
- E:T
Effector to Target
- FEP
Fluorinated Ethylene Propylene
- GMP
good manufacture practice
- HBSS
Hank’s Balanced Salt Solution
- HSA
Human Serum Albumin
- IL-2
InterLeukine 2
- MHC
Major Histocompatibility Complex
- MPC
Magnetic Particles Concentrator
- MSGV1
Mouse Stemcell Virus-based Gamma-retroviral Vector1
- NGFR
Nerve Growth Factor Receptor
- PBMC
peripheral blood mononuclear cell
- PBS
Phosphate Buffer Solution
- RCR
Replication Competent Retrovirus
- REV
Reverse
- SBVPF
Surgery Branch, NCI, NIH Vector Production Facility
- TCR
T Cell Receptor
- TNC
Total Nucleated Cells
- UT CNTR
UnTransduced Control
- VR
Validation Run
- 7-AAD
7-AminoActinomycin D
Footnotes
Disclosure of Interest The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.NIH (National Institutes of Health), National Cancer Institute A snapshot of pediatric cancers. 2011 Retrieved from http://www.cancer.gov/PublishedContent/Files/aboutnci/servingpeople/snapshots/2011_Pediatric_snapshot.508.pdf.
- 2.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, Edwards BK, editors. SEER Cancer Statistics Review, 1975-2008. National Cancer Institute; Bethesda, MD: [based on November 2010]. 2011. http://seer.cancer.gov/csr/1975_2008/ SEER data submission, posted to the SEER web site. [Google Scholar]
- 3.Hahn T, Wall D, Camitta B, Davies S, Dillon H, Gaynon P, Larson RA, Parsons S, Seidenfeld J, Weisdorf D, McCarthy PL., Jr The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant. 2005;11:823–61. doi: 10.1016/j.bbmt.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula US, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2344–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenberg PD, Riddell SR, Press OW. Adoptive immunotherapy for indolent non Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA. 2004;101:14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadelain M, Riviére I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 12.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cells receptors. Proc Natl acad Sci USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, Rosenberg SA, Eshhar Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178:361–366. doi: 10.1084/jem.178.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Can Res. 2012;18:2780–90. doi: 10.1158/1078-0432.CCR-11-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, Schlossman SF. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131:244–250. [PubMed] [Google Scholar]
- 16.Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18:385–397. doi: 10.3109/10428199509059636. [DOI] [PubMed] [Google Scholar]
- 17.Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, Wick MR, Myers DE, Waddick K, Ledbetter JA. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 1988;71:13–29. [PubMed] [Google Scholar]
- 18.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P. A phase I study on adoptive immunotherapy using gene modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12:6105–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically-engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with Chimeric Antigen Receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska M, Hosey J, Sadelain M, Brentjens RJ, Riviére I. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, Wilson WH, Rosenberg SA. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T cell receptor genes. Int J Cancer. 2001;94:228–236. doi: 10.1002/ijc.1457. [DOI] [PubMed] [Google Scholar]
- 25.Rafailidis PI, Kakisi OK, Vardakas K, Falagas ME. Infectious complications of monoclonal antibodies used in cancer therapy: a systematic review of the evidence from randomized controlled trials. Cancer. 2007;109:2182–2189. doi: 10.1002/cncr.22666. [DOI] [PubMed] [Google Scholar]
- 26.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, Dinauer M, Sadat M, Aiuti A, Deola S, Radrizzani M, Hagenbeek A, Apperley J, Ebeling S, Martens A, Kolb HJ, Weber M, Lotti F, Grande A, Weissinger E, Bueren JA, Lamana M, Falkenburg JH, Heemskerk MH, Austin T, Kornblau S, Marini F, Benati C, Magnani Z, Cazzaniga S, Toma S, Gallo-Stampino C, Introna M, Slavin S, Greenberg PD, Bregni M, Mavilio F, Bordignon C. Safety of retroviral gene marking with a truncated NGF receptor. Nat. Med. 2003;9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 28.Brenner MK, Heslop HE. Is retroviral gene marking too dangerous to use? Cytotherapy. 2003;5:190–193. doi: 10.1080/14653240310001307. [DOI] [PubMed] [Google Scholar]
- 29.Numbenjapon T, Serrano LM, Chang WC, Forman SJ, Jensen MC, Cooper LJ. Antigen-independent and antigen-dependent methods to numerically expand CD19-specific CD8+ T cells. Exp. Hematol. 2007;35:1083–1090. doi: 10.1016/j.exphem.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 costimulation on long-term proliferation of CD4+ T Cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Viability, expansion and transduction efficiency of four ALL patient cultured T cells.
Panel A) cumulative fold expansion of T cells isolated from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel B) viability of cultured transduced T cells from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel C) transduction efficiency on days 7, 9, and 11 of cultured transduced T cells from four ALL patients PBMCs concentrate (P1, P2, P3, P4). Panel D) average transduction efficiency on days 7, 9, and 11 for the four clinical products.
Supplemental Table 1. Number of cells (x10e6) during the Expansion Phase of manufacture