

Abstract
The RecA protein is the main bacterial recombinase and the activator of the SOS system. In Escherichia coli and Salmonella enterica sv. Typhimurium, RecA is also essential for swarming, a flagellar-driven surface translocation mechanism widespread among bacteria. In this work, the direct interaction between RecA and the CheW coupling protein was confirmed, and the motility and chemotactic phenotype of a S. Typhimurium ΔrecA mutant was characterized through microfluidics, optical trapping, and quantitative capillary assays. The results demonstrate the tight association of RecA with the chemotaxis pathway and also its involvement in polar chemoreceptor cluster formation. RecA is therefore necessary for standard flagellar rotation switching, implying its essential role not only in swarming motility but also in the normal chemotactic response of S. Typhimurium.
Introduction
RecA is a DNA-dependent ATPase [1], [2] present in almost all members of the Domain Bacteria [3], [4]. As a protein that is highly conserved among bacterial species, it is commonly used in phylogenetic studies [5]. In addition, RecA is the main bacterial recombinase involved in the central steps of homologous recombination and recombinational DNA repair [6]–[8]. It is also the activator of the DNA damage response known as the SOS system [9]. In this system, RecA acts as a DNA damage sensor by binding to single-stranded DNA, which activates the protein and thereby its co-protease activity. Activated RecA (RecA*) prompts autocleavage of the LexA repressor, thus inducing its expression and that of other SOS genes, mostly those involved in DNA recombination and repair [9]. RecA* is also able to induce the autocleavage of other serine proteases such as UmuD [10] and several repressors of the bacteriophage lytic cycle [11]–[13]. Furthermore, RecA is directly associated with other repair pathways such as the activated error-prone DNA polymerase V [10], [14], excision base repair [15], and RecN, involved in the repair of double-stranded DNA breaks [16].
Increasing its activity profile, a new role for RecA in swarming motility, has been added to its functional catalog [17], [18]. Swarming is a specialized and highly coordinated form of flagellar-driven multicellular surface translocation [19], [20] and the fastest mode of bacterial surface navigation [21]. In the absence of RecA, the swarming ability of both Escherichia coli and Salmonella enterica sv. Typhimurium is impaired [17], [18]; interestingly, the same phenotype is observed in S. Typhimurium strains overexpressing RecA protein [18]. Thus, none of the as yet known RecA activities, i.e., SOS response activation, recombinational DNA repair, or genetic recombination, seem to be necessary for the control of swarming motility [17]. Nevertheless, a possible link between RecA and the chemotaxis pathway through the CheW protein has been suggested. The CheW coupling protein is essential for the formation of the ternary signaling complex that also contains the CheA autokinase and MCPs (methyl-accepting chemotaxis proteins) [22]. The in vitro interaction of RecA and CheW was shown in a large-scale genome-wide screen assay [23] and a balance between the intracellular concentrations of these two proteins was shown to be essential for swarming [18].
Swarming is not governed by chemotaxis, nevertheless it has been described that the chemosensory pathway is essential for the motility on solid surfaces [24]. Mutants in the chemotaxis (che) pathway fail to swarm because of defective colony hydration [25], which in turn is associated with the flagellar rotation bias present in che mutants. Thus, in the wild-type strain flagellar rotation switches from clockwise (CW) to counterclockwise (CCW), whereas che mutants have CW or CCW biases depending on the specific mutation [26]. During CCW rotation, all flagella of the bacterium form a bundle that falls apart when one or more flagellar motors turn in the CW direction. This switch promotes lubrication of the cell–surface interface, which is needed to overcome surface friction, both of which are critical requirements for swarming motility in temperate swarmers such as S. Typhimurium and E. coli [27]. The impaired swarming phenotype of che mutants can be restored by adding water or osmolytes to the semi-solid surface [25], [27] or by restoring the normal flagellar rotation bias [28].
To further investigate the role of RecA in the control of swarming motility, in this work we examined the relationship between RecA and CheW in two-hybrid experiments and on co-immunoprecipitation assays, which confirmed the interaction of these two proteins. In addition, we explored the possibility that RecA affects the bacterial flagellar rotation pattern, by studying the swimming profile, flagellar motor rotation, and the chemotactic response of a recA knockout S. Typhimurium mutant (ΔrecA). The results confirmed a role for RecA in polar chemoreceptor cluster formation and therefore in flagellar rotation switching.
Materials and Methods
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. Except when indicated, all strains of bacteria were grown at 37°C in Luria–Bertani (LB) broth or on LB plates. When necessary, ampicillin (100 µg/ml) or chloramphenicol (34 µg/ml) was added to the culture. The same cell growth conditions were used for the microfluidics and optical trap assays (described below). The bacteria were grown overnight in 2 ml of LB broth supplemented, when needed, with the appropriate antibiotic. Each culture was diluted 1∶10 into LB broth without antibiotics but containing an oxygen scavenging system consisting of 100 µg glucose oxidase/ml and 20 µg catalase/ml (final concentrations) [29]. The added glucose is a substrate for the oxygen scavenging system and provides the energy needed for swimming under anaerobic conditions [29]. The cells were incubated at 37°C for 1 h and then diluted 100-fold in measuring medium (1% Bacto Tryptone, 0.8% NaCl, 2% glucose, 100 mM Tris-Cl, pH 7.5) containing the oxygen scavenging system. In all cases the scavenging system was added at least 2 h before the medium was used, to ensure a stable low level of oxygen.
Table 1. Bacterial strains and plasmids used in this work.
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
| Strains | ||
| DH5α | E. coli supE4 ΔlacU169 (φ80 ΔlacZ ΔM15) hsdR17, recA1, endA1, gyrA96, thi-1, relA1 | Clontech |
| MC1061 | E. coli F− Δ(ara-leu)7697 [araD139] B/r Δ(codB-lacI)3 galK16 galE15 λ− e14− mcrA0 relA1 rpsL150 (StrR) spoT1 mcrB1 hsdR2(r−m+) | CGSC |
| BL21 (DE3) pLysS | E. coli F− dcm ompT lon hsdS(rB−mB−) galλ(DE3) carring pLysS plasmid, CmR | Stratagene |
| LT2 | S. Typhimurium wild type strain | ATCC |
| UA1907 | S. Typhimurium ΔcheWΩcat. CmR | This work |
| UA1908 | S. Typhimurium ΔcheW | This work |
| UA1910 | S. Typhimurium ΔcheR | This work |
| UA1915 | S. Typhimurium ΔcheR ΔcheW | This work |
| UA1913 | S. Typhimurium ΔcheR ΔrecA | This work |
| UA1927 | S. Typhimurium ΔrecAΩ,cat. CmR | This study |
| UA1928 | S. Typhimurium ΔcheB | [29] |
| UA1929 | S. Typhimurium ΔcheY | [29] |
| UA1930 | S. Typhimurium ΔcheW ΔrecA | This work |
| UA1931 | S. Typhimurium ΔrecA | This work |
| Plasmids | ||
| pKOBEGA | Vector containing the λ Red recombinase system, Ampr, temperature sensitive | Generous gift of Prof. G. M. Ghigo, [33] |
| pKD3 | Vector carrying FRT-Cm construction, AmpR, CmR | [31] |
| pCP20 | Vector carrying FLP system, OriVts, AmpR | [31] |
| pGEM-T | Cloning Vector; AmpR | Promega |
| pGEX 4T-1 | Expression vector carrying the Ptac IPTG - inducible promoter and the lacIq gene; GST fusion tag, AmpR | Amersham Biosciences |
| pUA1108 | pGEX 4T-1 derivative plasmid carrying without the GST fusion tag, carrying only the Ptac promoter and the lacIq gene; used as overexpression vector, AmpR | This work |
| pUA1109 | pUA1108 derivative containing the native S. Typhimurium recA gene under the control of the Ptac promoter, AmpR. | This work |
| pUA1127 | pUA1108 derivative vector carrying the eYFP::cheR fusion, AmpR | This work |
| pUA1130 | pUA1108 derivative containing the native S. Typhimurium recA gene under the control of the Ptac promoter, AmpR. | This work |
| pUA1131 | pUA1108 derivative overexpression vector carrying the cheW-FLAG gene | This work |
| pB2HΔα | pACYCDuet-1 derivative vector with the E. coli Δα β-galactosidase fragment under the control of the Ptac promoter | BCCM/LMBP, [36] |
| pB2HΔω | pACYCDuet-1 derivative vector with the E. coli Δω β-galactosidase fragment under the control of the Ptac promoter | BCCM/LMBP, [36] |
| pUA1114 | pB2HΔα derivative vector contaning the Δα-recA fusion | This work |
| pUA1115 | pB2HΔω derivative plasmid contaning the Δω-recA fusion | This work |
| pUA1116 | pB2HΔα derivative vector contaning the Δα-cheW fusion | This work |
| pUA1117 | pB2HΔω derivative plasmid contaning the Δω-cheW fusion | This work |
| pUA1118 | pB2HΔα derivative plasmid contaning the Δα-amyA fusion, used as a control in two hybrid assays. | This work |
| pUA1119 | pB2HΔω derivative plasmid contaning the Δω-dnaE fusion, used as a control in two hybrid assays. | This work |
Dead cells used for optical trap assays were prepared by the addition of 2% formaldehyde to the culture, with subsequent dilution steps carried out following the same protocol used for live bacterial cultures.
For chemotactic assays, overnight cultures of S. Typhimurium were grown in tryptone broth (1% Bacto Tryptone, 0.5% NaCl) supplemented, when required, with the appropriate antibiotics [30]. All strains used in the chemotactic assays (LT2, UA1928 (ΔcheB), UA1931 (ΔrecA) and UA1931/pUA1130) had similar growth kinetics in tryptone broth medium (data not shown). The cultures were then diluted 1∶100 in the same medium but without antibiotics and incubated at 30°C with constant shaking until an OD600 of approximately 0.5 was reached. The culture was then harvested by centrifugation at 4500 g for 10 min at room temperature. The obtained cell pellet was washed twice in 1 ml of tempered tethering buffer (10 mM potassium-phosphate pH 7, 67 mM NaCl, 10 mM Na-lactate, 0.1 mM EDTA, and 0.001 mM l-methionine) and the resuspended cells were diluted to approximately 6×107 colony-forming units (cfu)/ml.
Construction of the S. Typhimurium mutant strains
The S. Typhimurium mutants were constructed using the one-step PCR-based gene replacement method [31], except when indicated. All DNA techniques were performed as described elsewhere [32]. The chloramphenicol resistance cassette from the plasmid pKD3 was amplified using suitable oligonucleotides containing 80-nucleotide stretches homologous to each of the insertion sites and the corresponding P1 and P2 sites of pKD3 (Table S1). The PCR products were digested with DpnI and used to transform S. Typhimurium electrocompetent cells containing the pKOBEGA plasmid [33]. Following selection of the transformant clones, the latter plasmid was eliminated by taking advantage of its temperature sensitivity, incubating the clones at 42°C. Gene substitution was confirmed by PCR and sequencing using the appropriate primers.
To construct the ΔcheRΔcheW and ΔcheRΔrecA mutant strains, the chloramphenicol resistance cassette present in the ΔcheR strain was eliminated as previously described using the pCP20 plasmid [31]. Afterwards, either ΔrecA or ΔcheW mutations from UA1927 and UA1907 were transferred to the ΔcheR chloramphenicol-sensitive strain (UA1910) by transduction, using the P22 HT bacteriophage [34]. The same procedure was used to construct the ΔrecA ΔcheW strain. In this case, the chloramphenicol resistance cassette present in ΔcheW strain was removed and the ΔrecA mutation was transducted from the UA1927 strain. In all cases, the absence of the prophage in the selected clones was determined by streaking them onto green plates as previously described [35]. All of the resulting strains were verified by PCR and sequencing.
β-Galactosidase-based two-hybrid system
The two-hybrid assay was performed as described [36]. The recA and cheW genes were PCR-amplified using suitable oligonucleotides (Table S1) that included SphI and BamHI restriction sites in the amplicon. After their release from the plasmids by endonuclease digestion, the amplified genes were cloned in both pB2HΔα and pB2HΔω vectors. The same procedure was used to clone the amyA and dnaE genes into pB2HΔα and pB2HΔω, respectively. These constructs served as the non-interaction assay controls, as previously described [36]. All of the constructs were confirmed by sequencing.
To simultaneously express the two fusion proteins within a cell, electrocompetent E. coli MC1061 cells were co-transformed with the two plasmids of interest and the transformants were selected by adding chloramphenicol and ampicillin to the solid medium. The presence of both fusions was confirmed by PCR and sequencing.
For β-galactosidase assays, the selected clones were grown in LB supplemented with ampicillin and chloramphenicol at 37°C until an OD550 of 0.2 was reached. IPTG was then added to the culture to a final concentration of 20 nM and the cultures were incubated at 37°C. Samples were taken 5 h after IPTG addition, and β-galactosidase activity was assayed as described by Miller (1991) [37]. The relative expression of β-galactosidase in each strain was calculated as the enzyme's activity with respect to that of the non-interaction control strain, which expressed the ΔαAmyA and ΔωDnaE proteins [36]. The reported results are the means of at least three independent assays, each performed in triplicate.
Construction of RecA and CheW tagged proteins
Co-immunoprecipitation assays were carried out using RecA-6×His and CheW-FLAG tagged proteins. The recA and cheW genes were PCR-amplified using the appropriate oligonucleotide pair (Table S1). In both cases, the corresponding tag sequence was included at the 5′ end of the suitable oligonucleotides that also contained a 3× Gly linker between the tag and the corresponding gene sequence (Table S1). The PCR products were digested with NdeI and BamHI and cloned into pUA1108, with each tagged protein under the control of the Ptac promoter. The plasmids were transformed into E. coli DH5α and confirmed by sequencing. The confirmed plasmids were used in the electrotransformation of the S. Typhimurium ΔrecA ΔcheW strain, thereby ensuring that every RecA and CheW protein produced by that strain carried the specific tag. The selected transformants were confirmed again by PCR and sequencing.
Co-immunoprecipitation assays
The assays were performed as described by D'Ulisse and others [38] with modifications. Briefly, cultures of S. Typhimurium ΔrecA ΔcheW harboring the plasmids with constructs encoding the tagged proteins were grown in LB broth supplemented with ampicillin to an OD550 of 0.2. Expression of the tagged genes was induced by the addition of 1 mM IPTG. After 3 h of growth, the cultures were centrifuged and the resulting pellet was washed in TBS 1× buffer (1.5 M NaCl, 250 mM Tris, pH 7.3) and then resuspended in cold IP lysis buffer (1× TBS, 15% glycerol, and 1% Triton X-100). The samples were incubated at 4°C for 40 min, with vortexing every 5 min. Finally, the samples were centrifuged at 4°C and the supernatant was collected. The protein concentration was determined by the Bradford method. As a control, cell lysates of S. Typhimurium ΔrecA ΔcheW containing the empty pUA1108 were also obtained following the same procedure.
Pure Proteome Protein A magnetic beads (Millipore) were used for immunoseparation. Either anti-6×His mouse IgG (Roche) or anti-FLAG mouse IgG (Acris) antibody was attached to the beads according to the manufacturer's instructions.
For co-immunoprecipitation the corresponding cell lysates were mixed at a ratio of 1∶1 and incubated at 30°C for 1 h without shaking, to allow interaction of the proteins. The appropriate antibody-coated magnetic beads (either anti-6×His or anti-FLAG IgGs) were added to the mixture and the samples were incubated overnight at 4°C with gentle mixing on a shaker. The beads were recovered and washed three times with wash buffer (1×TBS, 15% glycerol, and 1% Triton X-100) and finally resuspended in 45 µl of Laemmli sample buffer and heated for 10 min at 90°C. The samples were separated by SDS-PAGE on a 15% polyacrylamide gel and analyzed by western blotting using as primary antibody either anti-6×His or anti-FLAG mouse IgG and as secondary antibody horseradish peroxidase (HRP)-coupled anti-mouse IgG goat IgG antibody (Acris) together with Luminata western HRP chemiluminescence substrates (Millipore). HRP-coated Precision Plus protein western C standard (BioRad) was used as the molecular mass marker.
Microfluidics assays
Microfluidics experiments were performed for each bacterial strain as previously described by Ahmed et al. [39]. After overnight incubation, cultures in log phase were diluted 1∶50, to a final volume of 2 mL, in fresh medium and grown to an optical density OD600 = 0.45±0.02. In order to optimize trajectory identification, cells were then diluted 1∶4 before injecting them in a 60 µm thick microfluidic channel hosting a microwell (∅/◯ 2 mm) at its center. The focal plane was set to 30 µm above the glass bottom, at channel mid-depth, to minimize the interaction of bacteria with surfaces. Cells were imaged with a 20× objective (Nikon Pan Fluor ELWD, NA 0.45, WD 7.4 mm) in phase contrast at 25 frames per second, for a total of 20 s per experiment. Three biological replicates, each with 5 technical replicates, were carried out for each strain. All frames were segmented to obtain the cells' coordinates and cells were then tracked using in-house developed tracking routines based on the nearest neighbor method, implemented in MATLAB (The Mathworks, Natick, MA). Manual selection of high-quality tracks followed trajectory identification and allowed 300–500 trajectories per strain to be used for the analysis of motility.
Optical trapping assays
Optical trapping was carried out as previously described using a 1064-nm optical beam from a laser coupled to a single-mode fiber (Avanex) expanded up to 10 mm and then highly focused by an immersion oil objective (Nikon, CFI PL FL 100× NA 1.30 WD 0.16 mm) [29]. The oxygen scavenging system guaranteed a constant low level of oxygen and hence cell survival during the measurements [40].
As previously described [29], data for each bacterial strain were obtained from ten different randomly chosen cells of four distinct biological replicates; thus 40 cells per strain were analyzed.
The forward scattered light is collected by a 40X objective and projected into a quadrant photo diode (New Focus 2911). By this technique, the position of each trapped cell was acquired for 1000 s at 2 kHz of acquisition rate. The entire set of acquired data (1000 s) was then divided into 1-s-blocks. For each data block the angular velocity of the cell around the optical axis (Θ value) was calculated as described [29]. About 80% of the histograms showed very similar patterns. All plots shown below for the wild-type, mutant strains, and dead bacteria present the Θ histogram of one trapped cell either from the corresponding bacterial strain or from a dead cell control. In all cases, the selected histograms were within the above-mentioned 80%. During the experiments, videos were recorded using a CCD camera at the beginning (capture) and end (liberation) of the measurements.
Chemotaxis capillary assays
Chemotaxis assays were conducted as described by Adler [30], with some modifications. The chemotaxis chamber set was formed by placing three V-shaped bent needles (40 mm 18 G needle, Nipro) on the surface of an aseptic 140-mm Petri dish (Deltalab) and then covering them with a 24×65 mm microscope cover slip (Menzel-Glässer).
One-ml capillary tubes, 3 cm long (Microcaps, Drummond Scientific Co.), were used. One end of each tube was heat-sealed in a flame. After autoclaving, the sealed capillaries were filled with either tethering buffer or 10 mM l-aspartate dissolved in tethering buffer [41].
Approximately 2 ml of each cell suspension was placed in the chemotaxis chambers, which were then incubated for 1 h at 30°C. After the incubation, the exterior of the capillaries was rinsed under a stream of sterile double-distilled water. The sealed end of the capillaries was then broken off and the contents of the tube were emptied into a 1.5-ml microcentrifuge tube containing 0.9% NaCl. Suitable dilutions were plated on LB plates; after an overnight incubation, the cfu/ml were calculated.
Construction of the eYFP::cheR fusion
The enhanced yellow fluorescent protein (eYFP)::cheR fusion was constructed by the overlap extension procedure as follows. The cheW and eYFP genes (Clontech) were amplified using CheRstmF/CheRstmBamHI and eYFPNdeI/eYFPR oligonucleotide pairs, respectively, with eYFPR and cheRstmF containing complementary overhangs and a 3× Gly linker (Table S1). The resulting DNA fragments were annealed and amplified in a second round of PCR using eYFPNdeI and CheRstmBamHI to form the corresponding eYFP::cheR fusion. These outer primers contained NdeI and BamHI restriction sites that were used to clone the fragments into the IPTG-inducible pUA1108 expression vector, giving rise to plasmid pUA1127, in which the eYFP::cheR fusion is under Plac promoter control. The fusion was confirmed by sequencing and the pUA1127 plasmid was transformed into several genetic backgrounds (ΔcheR, ΔcheRΔrecA, and ΔcheRΔcheW) to obtain the bacterial strains used in the chemoreceptor clustering assays.
Chemoreceptor clustering assay
Receptor clustering experiments were performed as described [22], [42], [43] with modifications. Briefly, overnight cultures of S. Typhimurium strains carrying the pUA1127 (eYFP::cheR) plasmid were grown at 30°C in tryptone broth supplemented with ampicillin under constant agitation. After 24 h of incubation, the cultures were diluted 1∶100 in tryptone broth supplemented with ampicillin and 25 mM IPTG to induce eYFP::cheR fusion expression. The cultures were then incubated at 30°C until an OD600 of 0.5 was reached. The cells were harvested by low speed centrifugation (5300 g) for 15 min, washed once in cold tethering buffer, and finally resuspended in 100 ml of ice-cold tethering buffer. The cells were maintained on ice throughout the assay.
For fluorescence microscopy assays, the cells were immobilized and fixed at the same focal plane using thin 1% agarose pads in tethering buffer. Three µl of cells were applied on the pad, which was then covered with a clean cover slip. Fluorescence microscopy was performed using a Zeiss AxioImager M2 microscope (Carl Zeiss Microscopy) equipped with a Zeiss AxioCam MRm monochrome camera (Carl Zeiss Microscopy) and a filter set for eYFP (excitation BP 500/25; beam splitter FT 515; emission BP535/30). Cell fields were photographed and at least 250 cells were inspected by eye to determine the presence and type of clusters. All fluorescence images were obtained at 1000× magnification and were acquired under identical conditions. Each experiment was performed in triplicate using independent cultures. The images presented in the corresponding figure are representative of the entire images that are included as (Files S1, S2 and S3). ImageJ software (National Institutes of Health) was used to either quantify the number of clusters or to prepare images for publication.
Statistical analysis
The chemotaxis capillary and chemotaxis clustering assays were statistically evaluated using, respectively, a two-way or one-way analysis of variance (ANOVA) with Prism (GraphPad), as previously described [44], [45]. In all cases, the analyses were followed by the Bonferroni multiple comparison post-hoc test, with p<0.05 defined as statistically significant. In all of the figures, the error bars indicate the standard deviation.
Results
The interaction of RecA and CheW proteins
Large-scale protein-protein in vitro interaction studies had previously identified RecA as a prey protein when CheW was used as bait, but not vice versa [23]. Thus, to ascertain the RecA and CheW interaction two-hybrid assays and co-immunoprecipitation experiments were carried out.
In the two-hybrid assay, the previously described pB2HΔα/pB2HΔω system [36] was used. RecA and CheW proteins were fused to the two non-functional but complementary β-galactosidase truncations (Δα and Δω) in the system. In the reporter strain, β-galactosidase activity is driven by protein-protein recognition between both non-β-galactosidase parts of the chimeras. As shown in Fig. 1A, relative β-galactosidase expression by strains co-expressing either the ΔαRecA/ΔωCheW or the ΔαCheW/ΔωRecA chimera pair was >7, indicating significantly higher β-galactosidase activity in these strains than in the non-interaction assay control strain [36].
Figure 1. RecA protein directly interacts with CheW.

A) Two-hybrid assay. Measurement of the β-galactosidase activity of strains co-expressing the chimera protein pairs ΔαRecA/ΔωCheW or ΔαCheW/ΔωRecA. The results are expressed relative to those obtained with the non-interacting control strain expressing ΔαAmyA and ΔωDnaE [36]. Measurements were made 5 h after the addition of 20 nM IPTG to the culture. In each case the mean value from three independent experiments (performed in triplicate) is shown. Error bars indicate the standard deviation. B) Co-immunoprecipitation assay. Lysates prepared from cells overexpressing RecA-6×His and CheW-FLAG tagged proteins were mixed to allow the proteins to interact. Immunoprecipitation (IP) was performed by adding magnetic beads coated with either anti-6×His or anti-FLAG antibodies to the mixture and the attached proteins were recovered and separated by SDS-PAGE electrophoresis. The presence of the recombinant protein in the supernatants was assessed by western blotting (WB). As a control, co-IP assays were conducted using lysates from a ΔrecAΔcheW S. Typhimurium strain carrying an empty overexpression plasmid, thus expressing neither RecA-6×His nor CheW-FLAG proteins. The presence (+) or absence (−) of RecA, CheW, or both tagged proteins in the corresponding lysate mixture is indicated. Black and white arrows show the position of RecA-6×His and CheW-FLAG, respectively. IP indicates the antibody attached to the beads and WB the primary antibody used in western blotting. MW indicate the molecular mass marker.
To further confirm the two-hybrid assay results and thus obtain additional evidence for the interaction between RecA and CheW, co-immunoprecipitation assays were carried out using S. Typhimurium ΔrecA ΔcheW strains carrying the corresponding plasmids that overexpress either the RecA-6×His or the CheW-FLAG tagged proteins (Table 1). The immunoprecipitation was performed by using magnetic beads coated with either anti-6×His or anti-FLAG mouse IgG antibodies that specifically interact with the corresponding tagged protein and the recovered proteins were detected by Western blot. As seen in Fig. 1B, when both recombinant proteins were present in the protein mixture, and anti-6×His antibody coated beads were used, CheW-FLAG proteins were observed in the recovered supernatants. The same results were observed when anti-FLAG antibody coated beads were added to the mixture, then RecA-6×His proteins were recovered. All together, these data indicate that RecA-6×His was able to pull down CheW-FLAG and vice versa. Thus, the results of the two assays together confirm RecA–CheW pair formation and suggest the association of RecA with the chemotaxis pathway through its interaction with CheW.
The absence of RecA causes a decrease in swimming speed
To further understand the role of RecA in motility, the swimming speed of a S. Typhimurium ΔrecA mutant was evaluated through microfluidics assays and compared to that of the wild-type strain. In this assay, swimming speed computed over the whole set of identified trajectories was measured. As shown in Fig. 2, which depicts the relative frequency of swimming speeds for each strain, the absence of RecA prompted a change in the swimming profile. Specifically, at the highest relative frequency, the velocity of the mutant was lower than that of the wild-type strain under the same experimental conditions.
Figure 2. The lack of recA reduces the swimming speed of S. Typhimurium.
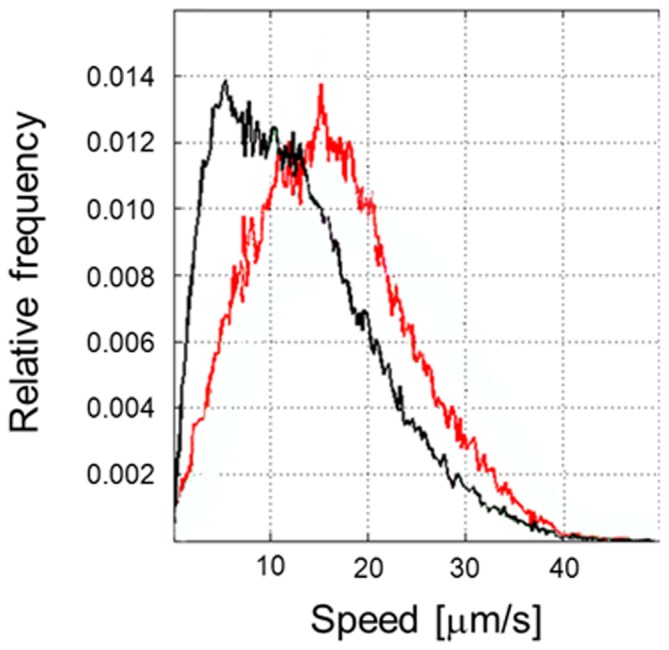
The experimentally observed probability distribution of the swimming speeds within a population of wild-type S. Typhimurium (red line) and of the ΔrecA mutant (black line), assessed using a microfluidics assay [39]. Values are expressed as the relative frequency of a given speed within a cell population. For each strain, the results were obtained from three independent experiments supported by five technical replicates each, for a total of 300–500 cells tracked per strain.
The ΔrecA mutant present a CW-bias of flagella rotation
To determine whether the slower swimming speed prompted by the absence of RecA was due to a bias in flagellar rotation, the flagellar rotation patterns of the ΔrecA mutant was studied using a single optical trap. Optical trapping is an excellent tool for analyzing the dynamic properties of bacteria [46]–[48]. It is based on the ability of an optical beam to trap a single cell because the refractive index of cells (and their constituents) is higher than that of the surrounding medium. Once trapped, the movement of that cell is measured, yielding information on its momentary position. In the case of rod shaped S. Typhimurium, a cell trapped in the single optical beam aligns itself along the optical axis. Thus, besides Brownian motion, the torque produced by flagellar rotation alters the dynamics of the cell [29]. The measurement of the rotation profile for each strain is expressed as the distribution of the change in the mean value of Θ, which is the angular velocity of the cell around the optical axis [29]. Using this technique we were able to distinguish CW to CCW switching of the flagella.
Figure 3 shows the flagellar rotation profile of both the S. Typhimurium ΔrecA mutant and the wild-type strain. Dead wild-type cells and ΔcheB and ΔcheY mutants were used as controls. There is no change in the angular velocity of dead cells, which exhibit only Brownian motion; thus, their Θ distribution pattern is centered at zero. Among the mutants, ΔcheB cells, described by their tumbling motility because of their CW flagellar rotation bias [49], displayed a Θ distribution pattern centered near zero. Thus, there was no change in the average angular velocity of the mutant cells but, as would be expected, the histogram was broader than that of the dead cells. Conversely, the flagella rotation profile of ΔcheY cells had a mean Θ that was not zero and was highly positive, consistent with the smooth swimming ability characteristic of this strain because of its CCW-biased flagellar rotation pattern [50], [51]. In the living wild-type cells, the Θ histogram showed the anticipated two peaks, reflecting normal switching between CCW and CW flagellar rotations. The peak centered at zero along the x axis corresponded to the tumbling state (CW rotation) whereas the peak located around 7.5 was due to the running state (CCW rotation) (Fig. 3). By contrast, but similar to the ΔcheB mutant, ΔrecA cells showed only one peak, centered near zero, in their Θ distribution pattern (Fig. 3). The absence of the second peak was indicative of the CW-biased rotation of ΔrecA cells.
Figure 3. Flagellar rotation is CW-biased in the S. Typhimurium ΔrecA mutant.
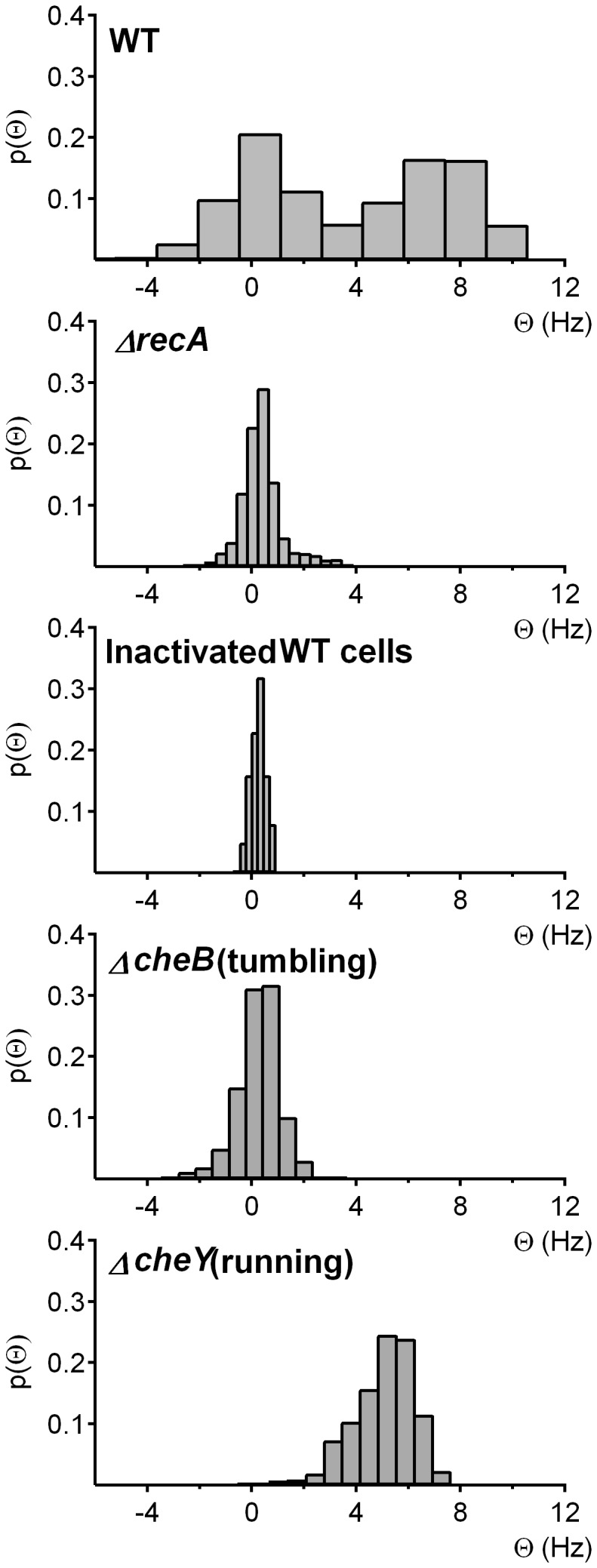
The flagellar switching profiles of S. Typhimurium LT2 wild-type (WT), dead wild-type (dead), ΔrecA, ΔcheB (tumbling), and ΔcheY (running) cells were evaluated. The resulting histograms show the distribution of the change in the mean cellular angular velocity around the optical axis (Θ). A zero-centered peak, as displayed by dead cells and the ΔcheB tumbling mutant, is indicative of CW-biased flagellar rotation, and a peak with positive values, as displayed by the ΔcheY running mutant, CCW-biased rotation. The presence of two peaks, one zero-centered and the other centered at positive values, indicates a mixed population displaying both CW and CCW rotational patterns and thus a non-biased flagellar rotational pattern. For each strain, the results were obtained from four independent experiments of ten cells each.
Chemotaxis response of recA mutants
To further confirm the switching defect of the ΔrecA mutant, its ability to move towards an l-aspartate source was evaluated using a classical capillary assay. The results are shown in Fig. 4. As expected, and in concordance with observations in other tumbling strains such as the ΔcheB mutant [49], the capillary assays clearly demonstrated that cells lacking RecA are unable to respond to the presence of l-aspartate. Furthermore, chemotaxis by the ΔrecA mutant was restored when the RecA deficiency was complemented by the presence of a plasmid containing the recA gene under the control of an IPTG- inducible promoter (Fig. 4) but not by the presence of the empty plasmid (data not shown). These results unequivocally showed that the presence of RecA is essential for a normal chemotactic response.
Figure 4. The chemotactic response of the S. Typhimurium ΔrecA mutant is impaired.

The chemotactic responses of S. Typhimurium wild-type (WT), ΔrecA, ΔrecA complemented (ΔrecA pUA1109), and ΔcheB (tumbling) cells were assessed using Adler's capillary assay [30] with the modifications described in Materials and Methods. Values are expressed as the number of viable cells (in cfu/ml) in a capillary tube containing either 10 mM aspartate (+) or tethering buffer alone (−). The results are the mean of five independent experiments of three capillaries each. Error bars indicate the standard deviation. ***p<0.001 and *p<0.05 as determined by two-way ANOVA with Bonferroni correction.
Chemotaxis receptor clustering
Based on our results, the phenotype of the ΔrecA mutant is similar to that of other che mutants, since in all cases the absence of RecA impairs not only swarming [17], [18] but also the switching of flagellar rotation (Fig. 3) and the chemotactic response (Fig. 4). To elucidate the role of RecA in chemotaxis, and taking into consideration the direct interaction of RecA with the CheW coupling protein, which bridges the MCPs to histidine kinase CheA [43], [52], we asked whether, like CheW, RecA was involved in chemoreceptor clustering. To investigate this possibility fluorescently tagged CheR (eYFP-CheR) was used as a specific reporter for chemoreceptor localization [43].
The eYFP-CheR fusion was constructed and cloned into pUA1108 vector under the control of an IPTG-inducible promoter (Table 1). For correct chemoreceptor localization, native CheR had to be removed; accordingly, the plasmid was included in the S. Typhimurium ΔcheR ΔrecA transformant. Additionally, it was also used to obtain the ΔcheR and ΔcheR ΔcheW mutants. The ΔcheR mutant served as the positive control strain since it exhibited normal polar clusters. The ΔcheR ΔcheW strain was used as the negative control strain since the absence of CheW inhibits polar cluster formation [22]. As expected, and in agreement with previous reports [43], in the positive control (the ΔcheR strain) single tight polar spots were seen in ∼70% of the observed cells. These spots corresponded to the clustering of thousands of chemoreceptors at the cell pole (Fig. 5). However, in agreement with previous data [43], in the absence of CheW compact polar clusters were formed in only ∼10% of the cells; instead, the presence of diffuse clusters (known as caps) was observed (Fig. 5A). Thus, according to our findings, the absence of RecA significantly impairs normal polar cluster formation, which occurred in only ∼50% of the cells, and increases the presence of caps. Nonetheless, neither the reduction in polar spot formation nor the increase in caps was as high in the absence of RecA as in the absence of CheW.
Figure 5. The formation of polar chemoreceptor clusters is altered in the absence of RecA protein.

A) A representative fluorescence microscopy image of the ΔcheR ΔrecA strain harboring plasmid pUA1127, containing the inducible eYFP::cheR fusion. Images of the ΔcheR and ΔcheRΔcheW strains containing the gene fusion were also included as positive and negative controls of polar chemoreceptor cluster structuring, respectively. B) The fraction of cells with well-structured polar chemoreceptor clusters. The percentage of cells showing polar, round, and diffraction-limited spots (previously referred to as clusters; [43]) was quantified in each strain. The results are the mean of three independent imaging experiments. Error bars represent standard deviation. ***p<0.001 and *p<0.01 as determined by one-way ANOVA with Bonferroni correction.
Discussion
A role for RecA in controlling the swarming motility of both E. coli and S. Typhimurium was clearly shown in previous studies [17], [18]. However, besides its possible connection to CheW [18], [23], nothing was known about the mechanisms that link RecA to motility. To determine whether RecA is associated with the chemotaxis pathway and, specifically, with flagellar function, we examined its putative direct interaction with CheW, its role in swimming motility and in chemotaxis, as well as the flagellar switching pattern of cells lacking RecA.
Our results support a tight relationship between RecA and the chemotactic response. First, our results unequivocally confirmed the interaction of RecA with CheW through two widely used techniques. The results of the two-hybrid assays were in concordance with those previously obtained in a large-scale genome-wide screening assay [23] and suggested an interaction between RecA and CheW (Fig. 1A), which was definitively shown in the co-immunoprecipitation assays (Fig. 1B). Second, the motility phenotype of cells lacking RecA, as determined herein, was similar to that of some che mutants. The latter finding was supported by microfluidics assays, in which the average swimming speed of S. Typhimurium ΔrecA was lower than that of the wild-type strain (Fig. 2), and by the observed differences in the flagellar rotation patterns of these two strains (Fig. 3). Thus, the absence of RecA impaired flagellar switching, leading to a CW bias similar to that of other tumbling strains, like the ΔcheB mutants [49]. Furthermore, consistent with the tumbling phenotype of the ΔrecA mutant, our results demonstrate that RecA is essential for a normal chemotactic response. Specifically, in quantitative chemotaxis assays the ΔrecA mutant was unable to detect the presence of l-aspartate, a well-known chemoattractant (Fig. 4); instead, chemotaxis was restored only when the recA deficiency was complemented by a plasmid carrying a copy of the recA gene (Fig. 4). The slower-moving phenotype of the RecA-deficient mutants can be explained by the CW bias displayed by these cells. By being anchored in a tumbling state, without normal running, the ΔrecA mutant was slower than the wild-type strain. In a previous study, the inability to switch the direction of flagellar rotation was linked to defects in chemotaxis and to improper colony hydration, leading to an inability to swarm [25], [27], [28]. It was previously reported that an E. coli recA1 mutant did not exhibit any apparent alterations in chemotaxis [53]. Nevertheless, this recA1 strain was not a knockout mutant, as was the ΔrecA mutant used in this work, in which the recA gene was completely removed. Furthermore, the recA1 allele is a single amino acid missense mutation that prevents RecA recombinatorial activity [54] but still allows normal binding to ssDNA as well as ATP-independent renaturation of complementary ssDNA molecules [55]. Thus, the results obtained with the two mutants cannot be compared.
Nevertheless, how RecA modulates flagellar rotation was unclear. In an earlier study, the absence of RecA had no effect on the expression of genes involved in either flagellar biosynthesis or chemotaxis [17], as shown for other proteins such as H-NS, which is required not only for flagellar motor function but also for flagellar biogenesis [56]. The direct association of RecA with the CheW coupling protein led us to ask whether RecA, like CheW, plays a role in the architecture of chemoreceptor arrays. CheW tethers CheA kinase to the MCPs forming the MCP-CheW-CheA ternary complexes and chemoreceptor arrays, enabling MCPs to modulate CheA autokinase activity [22], [57] which, in turn, controls the level of phosphorylated CheY (CheY-P). Once activated by the MCPs, phosphorylated CheA transfers its phosphoryl group to CheY. CheY-P then promotes a switch in the direction of flagellar rotation, from CCW to CW. According to our observations, the RecA protein is necessary for the formation of normal polar chemoreceptors arrays. Although its role may not be the same as that of CheW, the absence of RecA significantly reduces the polar clustering of chemoreceptors (Fig. 5). The absence of MCPs, CheW, or CheA is known to impair chemoreceptor array formation, leading cells to run constantly because of the CCW bias of their flagellar rotation [51]. Conversely, and in addition to the demonstrated effect of RecA on chemoreceptor array formation, our results show that the absence of this protein results in a CW bias, similar to that observed in cheZ, cheR, or cheB null mutants. All of these Che proteins are associated with chemotactic response adaptation: CheZ phosphorylase returns phosphorylated Che (CheY-P) to its non-phosphorylated state (CheY), and CheB methylesterase and CheR methyltransferase control the MCP methylation state, adjusting it to the presence and concentration of external stimuli. Therefore, a similar function can be hypothesized for RecA in the chemotactic response adaptation. It is worth noting that although CheZ, CheR, and CheB co-localize with MCPs-CheW-CheA complexes at the cell poles [42], none of these proteins have an effect on polar cluster formation [43], unlike RecA. While further work is needed to determine the exact role of RecA in the chemotaxis pathway, our results clearly reveal previously unknown functions of RecA: its involvement in the control or modulation of flagellar rotation, and thus not only with swarming but also with swimming and chemotaxis, and its role in the architecture of polar chemoreceptor arrays.
Supporting Information
Oligonucleotides used in this work.
(DOCX)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1910 ( ΔcheR ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1915 ( ΔcheRΔcheW ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1913 ( ΔcheRΔrecA ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
Acknowledgments
IN MEMORIAM
We mourn the loss of Dmitri Petrov (1947–2014) a great colleague and friend.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors acknowledge financial support from the Spanish Ministry of Science and Innovation (grant BFU2011-23478 to JB and FIS200800114 and FIS2011-24409 to DP), Generalitat de Catalunya (grant 2009SGR159 to DP and 2014SGR572 to JB), Fundació Privada Cellex Barcelona and the National Institute of Health (grant 1R01GM100473 to RS). The funders had no role in the design of the study, in data collection and analysis, in the decision to publish, or in the preparation of the manuscript.
References
- 1. Barbe J, Villaverde A, Cairo J, Guerrero R (1986) ATP hydrolysis during SOS induction in Escherichia coli. J Bacteriol 167: 1055–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bedale WA, Cox M (1996) Evidence for the coupling of ATP hydrolysis to the final (extension) phase of RecA protein-mediated DNA strand exchange. J Biol Chem 271: 5725–5732. [DOI] [PubMed] [Google Scholar]
- 3. Moran NA, Wernegreen JJ (2000) Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol Evol 15: 321–326. [DOI] [PubMed] [Google Scholar]
- 4. Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS, et al. (2002) 50 million years of genomic stasis in endosymbiotic bacteria. Science 296: 2376–2379. [DOI] [PubMed] [Google Scholar]
- 5. Eisen JA (1995) The RecA protein as a model molecule for molecular systematic studies of bacteria: comparison of trees of RecAs and 16S rRNAs from the same species. J Mol Evol 41: 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roca AI, Cox MM (1997) RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol 56: 129–223. [DOI] [PubMed] [Google Scholar]
- 7. Cox MM (1999) Recombinational DNA repair in bacteria and the RecA protein. Prog Nucleic Acid Res Mol Biol 63: 311–366. [DOI] [PubMed] [Google Scholar]
- 8. Lusetti SL, Cox MM (2002) The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem 71: 71–100. [DOI] [PubMed] [Google Scholar]
- 9. Sassanfar M, Roberts JW (1990) Nature of the SOS-inducing signal in Escherichia coli. The involvement of DNA replication. J Mol Biol 212: 79–96. [DOI] [PubMed] [Google Scholar]
- 10. Jiang Q, Karata K, Woodgate R, Cox MM, Goodman MF (2009) The active form of DNA polymerase V is UmuD'(2)C-RecA-ATP. Nature 460: 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roberts JW, Roberts CW (1975) Proteolytic cleavage of bacteriophage lambda repressor in induction. Proc Natl Acad Sci U S A 72: 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sauer RT, Ross MJ, Ptashne M (1982) Cleavage of the lambda and P22 repressors by recA protein. J Biol Chem 257: 4458–4462. [PubMed] [Google Scholar]
- 13. Eguchi Y, Ogawa T, Ogawa H (1988) Cleavage of bacteriophage phi 80 CI repressor by RecA protein. J Mol Biol 202: 565–573. [DOI] [PubMed] [Google Scholar]
- 14. Patel M, Jiang Q, Woodgate R, Cox MM, Goodman MF (2010) A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit Rev Biochem Mol Biol 45: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reuven NB, Arad G, Maor-Shoshani A, Livneh Z (1999) The mutagenesis protein UmuC is a DNA polymerase activated by UmuD', RecA, and SSB and is specialized for translesion replication. J Biol Chem 274: 31763–31766. [DOI] [PubMed] [Google Scholar]
- 16. Keyamura K, Sakaguchi C, Kubota Y, Niki H, Hishida T (2013) RecA protein recruits structural maintenance of chromosomes (SMC)-like RecN protein to DNA double-strand breaks. J Biol Chem 288: 29229–29237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomez-Gomez JM, Manfredi C, Alonso JC, Blazquez J (2007) A novel role for RecA under non-stress: promotion of swarming motility in Escherichia coli K-12. BMC Biol 5: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Medina-Ruiz L, Campoy S, Latasa C, Cardenas P, Alonso JC, et al. (2010) Overexpression of the recA gene decreases oral but not intraperitoneal fitness of Salmonella enterica. Infect Immun 78: 3217–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harshey RM (2003) Bacterial motility on a surface: many ways to a common goal. Annu Rev Microbiol 57: 249–273. [DOI] [PubMed] [Google Scholar]
- 20. Kearns DB (2010) A field guide to bacterial swarming motility. Nat Rev Microbiol 8: 634–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henrichsen J (1972) Bacterial surface translocation: a survey and a classification. Bacteriol Rev 36: 478–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cardozo MJ, Massazza DA, Parkinson JS, Studdert CA (2010) Disruption of chemoreceptor signalling arrays by high levels of CheW, the receptor-kinase coupling protein. Mol Microbiol 75: 1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, et al. (2006) Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res 16: 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burkart M, Toguchi A, Harshey RM (1998) The chemotaxis system, but not chemotaxis, is essential for swarming motility in Escherichia coli. Proc Natl Acad Sci U S A 95: 2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q, Suzuki A, Mariconda S, Porwollik S, Harshey RM (2005) Sensing wetness: a new role for the bacterial flagellum. EMBO J 24: 2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manson MD (1992) Bacterial motility and chemotaxis. Adv Microb Physiol 33: 277–346. [DOI] [PubMed] [Google Scholar]
- 27. Partridge JD, Harshey RM (2013) More than motility: Salmonella flagella contribute to overriding friction and facilitating colony hydration during swarming. J Bacteriol 195: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mariconda S, Wang Q, Harshey RM (2006) A mechanical role for the chemotaxis system in swarming motility. Mol Microbiol 60: 1590–1602. [DOI] [PubMed] [Google Scholar]
- 29. Martinez IA, Campoy S, Tort M, Llagostera M, Petrov D (2013) A simple technique based on a single optical trap for the determination of bacterial swimming pattern. PLoS One 8: e61630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adler J (1973) A method for measuring chemotaxis and use of the method to determine optimum conditions for chemotaxis by Escherichia coli. J Gen Microbiol 74: 77–91. [DOI] [PubMed] [Google Scholar]
- 31. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green MR, Sambrook J, Sambrook J (2012) Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
- 33. Chaveroche MK, Ghigo JM, d'Enfert C (2000) A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 28: E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Campoy S, Jara M, Busquets N, Perez De Rozas AM, Badiola I, et al. (2002) Role of the high-affinity zinc uptake znuABC system in Salmonella enterica serovar typhimurium virulence. Infect Immun 70: 4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis RW, Bostein D, Roth JR (1980) Advanced bacterial genetics. A manual for genetic engineering. In: Cold Spring Harbor Laboratory Press CSH, NY, editor.
- 36. Borloo J, De Smet L, Vergauwen B, Van Beeumen JJ, Devreese B (2007) A beta-galactosidase-based bacterial two-hybrid system to assess protein-protein interactions in the correct cellular environment. J Proteome Res 6: 2587–2595. [DOI] [PubMed] [Google Scholar]
- 37.Miller JH (1972) Experiments in Molecular Genetics: Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38. D'Ulisse V, Fagioli M, Ghelardini P, Paolozzi L (2007) Three functional subdomains of the Escherichia coli FtsQ protein are involved in its interaction with the other division proteins. Microbiology 153: 124–138. [DOI] [PubMed] [Google Scholar]
- 39. Ahmed T, Shimizu TS, Stocker R (2010) Microfluidics for bacterial chemotaxis. Integr Biol (Camb) 2: 604–629. [DOI] [PubMed] [Google Scholar]
- 40. Landry MP, McCall PM, Qi Z, Chemla YR (2009) Characterization of photoactivated singlet oxygen damage in single-molecule optical trap experiments. Biophys J 97: 2128–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Block SM, Segall JE, Berg HC (1983) Adaptation kinetics in bacterial chemotaxis. J Bacteriol 154: 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sourjik V, Berg HC (2000) Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol Microbiol 37: 740–751. [DOI] [PubMed] [Google Scholar]
- 43. Kentner D, Thiem S, Hildenbeutel M, Sourjik V (2006) Determinants of chemoreceptor cluster formation in Escherichia coli. Mol Microbiol 61: 407–417. [DOI] [PubMed] [Google Scholar]
- 44. Brennan CA, DeLoney-Marino CR, Mandel MJ (2013) Chemoreceptor VfcA mediates amino acid chemotaxis in Vibrio fischeri. Appl Environ Microbiol 79: 1889–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raterman EL, Welch RA (2013) Chemoreceptors of Escherichia coli CFT073 play redundant roles in chemotaxis toward urine. PLoS One 8: e54133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Altindal T, Chattopadhyay S, Wu XL (2011) Bacterial chemotaxis in an optical trap. PLoS One 6: e18231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Min TL, Mears PJ, Chubiz LM, Rao CV, Golding I, et al. (2009) High-resolution, long-term characterization of bacterial motility using optical tweezers. Nat Methods 6: 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koch M, Rohrbach A (2012) Object-adapted optical trapping and shape-tracking of energy-switching helical bacteria. 6: 680–686. [Google Scholar]
- 49. DeFranco AL, Parkinson JS, Koshland DE Jr (1979) Functional homology of chemotaxis genes in Escherichia coli and Salmonella typhimurium. J Bacteriol 139: 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warrick HM, Taylor BL, Koshland DE Jr (1977) Chemotactic mechanism of Salmonella typhimurium: preliminary mapping and characterization of mutants. J Bacteriol 130: 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parkinson JS (1978) Complementation analysis and deletion mapping of Escherichia coli mutants defective in chemotaxis. J Bacteriol 135: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sourjik V (2004) Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol 12: 569–576. [DOI] [PubMed] [Google Scholar]
- 53. Smith RA, Parkinson JS (1980) Overlapping genes at the cheA locus of Escherichia coli. Proc Natl Acad Sci U S A 77: 5370–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawashima H, Horii T, Ogawa T, Ogawa H (1984) Functional domains of Escherichia coli recA protein deduced from the mutational sites in the gene. Mol Gen Genet 193: 288–292. [DOI] [PubMed] [Google Scholar]
- 55. Bryant FR, Lehman IR (1986) ATP-independent renaturation of complementary DNA strands by the mutant recA1 protein from Escherichia coli. J Biol Chem 261: 12988–12993. [PubMed] [Google Scholar]
- 56. Ko M, Park C (2000) Two novel flagellar components and H-NS are involved in the motor function of Escherichia coli. J Mol Biol 303: 371–382. [DOI] [PubMed] [Google Scholar]
- 57. Liu JD, Parkinson JS (1989) Role of CheW protein in coupling membrane receptors to the intracellular signaling system of bacterial chemotaxis. Proc Natl Acad Sci U S A 86: 8703–8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides used in this work.
(DOCX)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1910 ( ΔcheR ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1915 ( ΔcheRΔcheW ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
ZIP File containing the entire set of images used to determine the presence and type of clusters found in the S. Typhimurium UA1913 ( ΔcheRΔrecA ) strain carrying the pUA1127 (eYFP:: cheR ) plasmid.
(RAR)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.