

Abstract
Poorly differentiated sinonasal carcinomas are a heterogenous group of aggressive neoplasms that encompasses squamous cell carcinoma including basaloid variant, lymphoepithelial carcinoma, sinonasal undifferentiated carcinoma, and neuroendocrine-type small cell carcinoma. We herein describe 3 cases of a hitherto unreported variant combining features of basaloid carcinoma with variable intermingled rhabdoid cells. Patients were 2 women (aged 28 and 35) and a man (52 y) who presented with sinonasal masses. All had advanced local disease with bone involvement (pT4). None had a history of irradiation or a family history of rhabdoid tumors. Treatment was surgery and adjuvant chemoradiation. One patient developed liver, lung, pleural, and pericardial metastases (63 mo) and is currently (70 mo) alive under palliative treatment. Another developed recurrent cervical lymph node metastases and died of disease 8.5 years later. The youngest patient was disease-free at last follow-up 7 years later. Histologic features were very similar in all 3 cases and showed intimate admixture of compact basaloid cell nests with peripheral palisading, perivascular pseudorosettes, and a few scattered rhabdoid cells. Rhabdoid cells were more extensive in the metastasis in 1 case but formed a minor inconspicuous component in the primary tumors in all cases. Striking features common to all cases were (1) basaloid “blue” appearance at low power, (2) papilloma-like exophytic component, (3) extensive pagetoid surface growth with prominent denuding features, and (4) replacement of underlying mucous glands mimicking an inverted papilloma. Clear-cut origin from benign papilloma and overt squamous differentiation were lacking. Diffuse (2) or partial (1) p16 expression was noted, but all cases lacked human papillomavirus DNA by molecular tests. In situ hybridization was negative for Epstein-Barr virus. Immunohistochemistry showed diffuse expression of pancytokeratin. CK5 and vimentin showed intermingling of CK5+/vimentin− basaloid and CK5−/vimentin+ rhabdoid cells. Complete loss of nuclear SMARCB1 expression was seen in all cases including also the denuding carcinoma in situ–like surface lesions. To our knowledge, this variant of sinonasal carcinoma has not been reported before. The identical features in all 3 cases suggest a specific disease rather than a nonspecific dedifferentiated phenotype. Awareness of this rare variant and thus reporting of additional cases is necessary for defining its full morphologic and biological spectrum.
Key Words: rhabdoid carcinoma, basaloid carcinoma, SMARCB1, INI1, sinonasal tract, hybrid neoplasm
Carcinoma of the sinonasal tract is uncommon with an overall incidence of approximately 0.5 cases per 100,000 population per year and a male to female ratio of 1.8:1.1,2 It represents no more than 3% to 5% of head and neck cancer.2 Several distinctive histologic subtypes have been described on the basis of specific clinical, histopathologic, immunohistochemical, and etiologic features.3 However, occurrence of different types of poorly differentiated neoplasms of different histogenetic derivation seems to be a distinctive and unique feature of the sinonasal tract.4 Classification of this group of poorly differentiated neoplasms might be challenging regarding their distinction from nonepithelial mimics on one hand and recognizing special histologic subtypes with distinctive behavior on the other hand.4,5 Following recognition of sinonasal undifferentiated carcinoma (SNUC) as a distinctive and highly aggressive variant of sinonasal cancer in 1986,6 poorly differentiated sinonasal carcinomas underwent a continuous refinement of their subtyping. Currently recognized subtypes in the poorly differentiated group include basaloid squamous cell carcinoma (SCC), lymphoepithelial carcinoma, SNUC, and small cell carcinoma of neuroendocrine type, in addition to other types.3
Whereas conventional SCC represents the most common carcinoma type encountered in the sinonasal tract (60%), the basaloid variant of SCC is far less common, comprising 5% of head and neck basaloid SCC.7,8 The behavior of basaloid head and neck SCC varied among different studies. Although generally considered a highly aggressive neoplasm compared with conventional SCC,3 more recent larger studies showed survival characteristics that were not significantly different from conventional SCC or even paradoxically better than SCC.8 This suggests that the group of head and neck basaloid SCC is likely heterogenous with different etiology, biology, and behavior. In this study, we describe 3 cases of sinonasal basaloid carcinoma that showed strikingly similar histologies, occurred in relatively younger patients but over a wide age range, and showed a variable but distinctive growth pattern associated with subtle rhabdoid cells and complete loss of nuclear SMARCB1/INI1. We assume that this pattern represents a distinctive variant of sinonasal basaloid carcinoma, for which we suggest the descriptive term “SMARCB1(INI1)-deficient basaloid carcinoma.”
MATERIALS AND METHODS
All 3 cases were identified from the routine surgical pathology files at the Institute of Pathology, University Hospital of Erlangen. After observing clear-cut rhabdoid cells in a recent liver metastasis from case 1, we stained the metastasis and the primary tumor (diagnosed in 2008) for SMARCB1 and found both to be negative. On the basis of the histologic features of this index case, we reviewed 15 sinonasal carcinomas primarily diagnosed as basaloid SCC, SNUC, or carcinoma ex Schneiderian papilloma for similar features and found 2 more identical cases. All 3 cases (all were in-house cases) were initially diagnosed as basaloid SCC in 2002, 2006, and 2008. No further cases could be identified among consecutive 112 sinonasal carcinomas treated between 2002 and 2014 at our department (71 SCCs of different types, 11 intestinal-type, and 3 non–intestinal-type adenocarcinomas, 9 SNUCs, 8 salivary-type carcinomas, 7 carcinomas arising in Schneiderian papillomas, 2 lymphoepithelial carcinomas, and 1 atypical carcinoid). Thus, the 3 patients represent 2.7% of sinonasal carcinomas treated at our Hospital during the same period. Representative paraffin blocks were available from the primary tumors and the metastases in all cases. Tumor specimens were fixed in buffered formalin and embedded for routine histologic examination. Immunohistochemical studies were performed on 3-μm-thick sections cut from paraffin blocks using a fully automated system (“Benchmark XT System”; Ventana Medical Systems Inc., Tucson, AZ) and the following antibodies: pancytokeratin (clone KL-1, 1:200; Immunotech), vimentin (V9, 1:100; Dako), desmin (clone D33, 1:250; Dako), protein S-100 (polyclonal, 1:2500; Dako), CD34 (clone BI-3C5, 1:200; Zytomed), ERG (EPR3864, prediluted; Ventana), CA125 (clone M11, 1:20; Dako), SMARCB1/INI1 (MRQ-27, 1:50; Zytomed), CK7 (OV-TL, 1:1000; Biogenex), CK20 (KS20.8, 1:50; Dako), p63 (4A4, 1:100; Zytomed), CD56 (clone MRQ-42, 1:100; Cell Marque), synaptophysin (clone SY38, 1:50; Dako), E-cadherin (clone 36, 1:20,000; BD Biosciences), p16 (clone JC8, 1:100; Santa Cruz Biotechnology), anti-NUT (clone C52B1, 1:45; Cell Signaling), and Ki67 (MiB1, 1:100; Dako). Epstein-Barr virus (EBV) in situ hybridization (EBER 1/2 probes; ZytoVision, Bremerhaven, Germany) was performed according to the manufacturer’s instructions. In addition, 200 oropharyngeal SCC samples were stained with SMARCB1 on tissue microarrays to assess whether this variant is likely unique to the sinonasal tract.
HPV Testing
For HPV testing, DNA was extracted from formalin-fixed paraffin-embedded tumor tissue. All samples were tested for DNA integrity by polymerase chain reaction (PCR) using primers for the human β globin gene (BG1/BG2). Four PCR assays were performed in parallel to screen for the presence of HPV-DNA. Two sets of consensus primers (located in the highly conserved helicase region of the E1 gene and detecting >60 HPV types including all high-risk types) were used: primers PPF1/PPR2 generate a 254 bp PCR product with a sensitivity of 10 genome copies per sample, and primers CP4/CP5 generate a 459 bp PCR product with a sensitivity of 100 genome copies per sample.9 Two additional primer sets were used for type-specific amplification: primers 6-1/6-2 generate a 413 bp PCR product only from DNA of HPV types 6 and 11, and primers 16-1 and 16-2 generate a 149 bp PCR product only from DNA of HPV type 16. Plasmids containing sequences of HPV types 6, 11, and 16 and HeLa cells containing HPV type 18 served as positive controls (the nucleotide sequence of the primers and cycling conditions are available upon request).
RESULTS
Case Histories
Case 1
A 35-year-old woman presented with nonspecific sinonasal symptoms. Imaging examination showed a mass in the right anterior ethmoid cells extending into both frontal sinuses with destruction of the frontal bone and the right medial orbital wall (Fig. 1). There was no evidence of regional or distant metastasis. She underwent complete resection of the tumor with enucleation of the ocular bulb and resection of the bony wall of the ethmoid sinus followed by adjuvant chemoradiation. She remained disease-free under regular follow-up until she was diagnosed with liver and lung metastasis 63 months later. Biopsies confirmed liver and lung metastases as well as pleural and pericardial carcinomatosis. Shortly thereafter, a life-threatening intrapulmonary bleeding from lung metastasis necessitated emergency lobectomy of the right lower lung lobe. She is currently alive with disease under palliative treatment 70 months after initial diagnosis. Careful analysis of her personal and family history revealed no relevant neoplasms.
FIGURE 1.
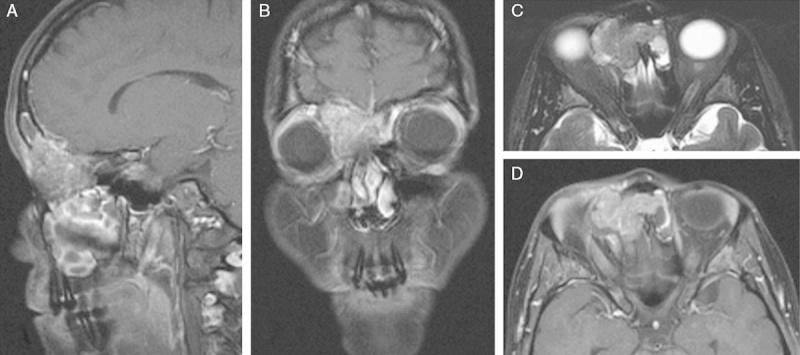
Representative magnetic resonance tomography of case 1 showed a mass in the right anterior ethmoid cells extending into both frontal sinuses with destruction of the frontal bone and the right medial orbital wall. The lesion has a salt and pepper–like appearance on both postcontrast T1w images (A, B, D) and T2w images (C). Infiltration of the extraconal space and displacement of the right eye ball were noted.
Case 2
This 52-year-old man was diagnosed with a sinonasal mass in the left frontal sinus and ethmoid cells, which was biopsied on 2 occasions and diagnosed as inverted Schneiderian papilloma. At the same time, a suspicious nodule was removed from the left parotid gland and showed metastatic poorly differentiated carcinoma within a lymph node. Because no other primary tumor could be found, a radical resection of the sinonasal lesion was performed and showed a poorly differentiated carcinoma. The patient underwent neck dissection, which showed a total of 5 positive nodes. Adjuvant chemoradiation was given. The patient remained disease-free for 6 years after which he presented with contralateral cervical nodal disease and biopsy-proven local recurrence. Again he underwent surgical treatment followed by adjuvant radiochemotherapy. The neck specimen contained 14 positive nodes. He suffered from multiple local recurrences and cervical regional metastases and died of disease 8.5 years from initial diagnosis.
Case 3
This young woman (28 y) was diagnosed with a sinonasal tumor in the right frontal sinus and anterior ethmoidal cells. Following initial biopsy, the tumor was then removed by multiple local radical resections until an R0 status was achieved. There was no evidence of regional or distant metastatic disease at the time of surgery. She received adjuvant radiochemotherapy. Currently (7 y from surgery), she is doing well without local recurrence or metastasis.
Pathologic Findings
The tumors were described grossly as having a prominent exophytic component and diffuse infiltrative growth. All 3 primary tumors showed remarkably similar (almost identical) histologic features and are thus described together. They were composed of superficial papillary fronds (Figs. 2A, B) and compact sheets and nests of medium-sized polygonal basaloid epithelial cells with predominantly cohesive growth and peripheral palisading closely mimicking basaloid SCC (Fig. 2C). In some areas, poorly cohesive growth was seen occasionally associated with stromal hemorrhage. A distinctive pattern was radial growth around variably sized blood vessels mimicking perivascular pseudorosettes (Fig. 2D). The peripheral cells were arranged in a palisading manner with their nuclei being kept away from the basal membrane (Fig. 2D). Scattered among the basaloid cells were isolated small to medium-sized rhabdoid cells with pale-eosinophilic paranuclear rhabdoid inclusions that varied greatly in their number and distribution in the primary tumors (Figs. 2B, E). These rhabdoid cells were abundant in the metastasis from case 1, particularly in areas with poorly cohesive growth and hemorrhages. However, the carcinomatous basaloid aspect still dominated the histologic picture in the metastasis, and the rhabdoid cellular component was generally not a prominent feature at low-power examination. The degree of anaplasia varied greatly from relatively bland-looking monomorphic “blue cells” (Figs. 2C, D) to highly anaplastic nuclear features (Fig. 2F). Clear-cut squamous differentiation (copious eosinophilic cytoplasm, intercellular bridges, dyskeratotic cells, or keratin pearls) was absent.
FIGURE 2.
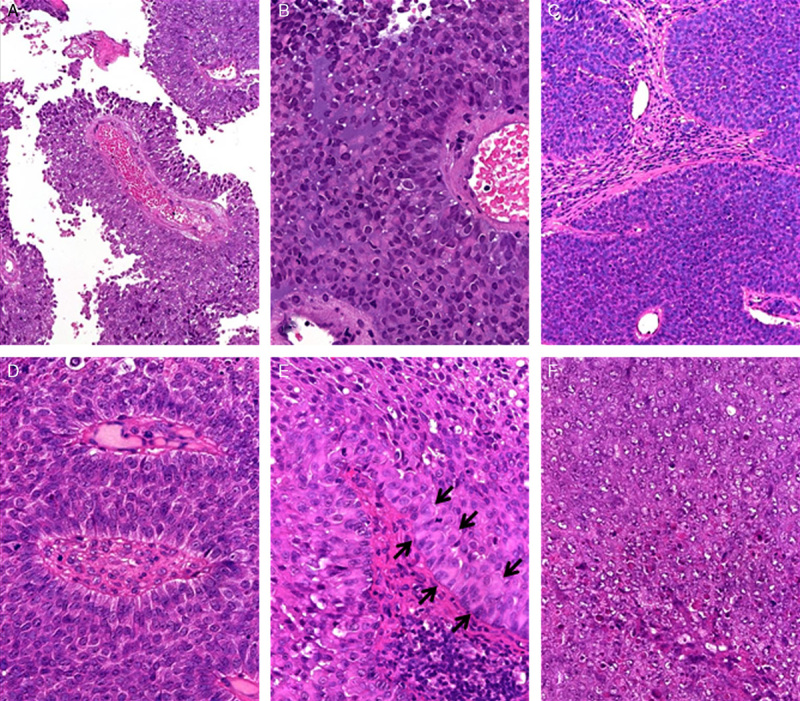
Histologic appearance of SMARCB1-negative sinonasal carcinomas. A, Characteristic papillary superficial pattern in the primary tumor of case 1. B, Same case showed prominent rhabdoid cells amid “blue” basaloid cells. C, Monotonous basaloid pattern in case 2. D, Angiocentric growth with perivascular pseudorosettes was seen in all cases. Note monotonous cell morphology and inversed polarity of nuclei. E, Prominent rhabdoid inclusions were seen in the basal layers in this area from case 3 (arrows; note dsycohesive central areas mimicking squamoid cells). F, Area with high-grade nuclear features and necrosis (case 3).
A distinctive feature seen in all 3 cases was the extensive replacement of the surface epithelium and the adjacent seromucous glands by tumor cells, occasionally with a prominent papillary pattern in the superficial parts and inverted-like growth into the subepithelial lamina propria. This pattern was responsible for misinterpretation of case 2 as inverted papilloma on 2 initial limited biopsies. The superficial spread varied from thin denuding carcinoma in situ (CIS)-like (Fig. 3A and B) and pagetoid nested (Fig. 3C), to flat-papillary (Fig. 3D), inverted (Fig. 3E), and thick compact squamous CIS-like (Fig. 3F) growth. Immunohistochemistry showed strong expression of pancytokeratin (KL-1) with a diffuse cytoplasmic pattern except in the rhabdoid cells, which displayed paranuclear staining (Fig. 4A). Vimentin and CK5 stained 10% to 50% of cells with variable and generally inverse distribution of CK5-positive and vimentin-positive cells (Figs. 4B, C). Similar staining pattern to CK5 was observed for p63. CA125 showed staining in isolated tumor cells (<5%). Cases 1 and 3 strongly expressed p16 (Fig. 3C, inset). Case 2 showed scattered p16-positive cells. E-cadherin showed strong membranous staining in all tumor cells in all cases. The proliferation fraction (Ki67) varied between 20% and >50%. All other markers listed above in the Materials and methods section were negative. SMARCB1 was completely negative in the tumor cells in the primary and in the metastatic lesions in all cases (Fig. 4D). The denuding and papillary surface components were also SMARCB1 negative (Fig. 3B). All 3 cases were negative for high-risk HPV infection by PCR analysis. EBV in situ hybridization was also negative in all cases.
FIGURE 3.
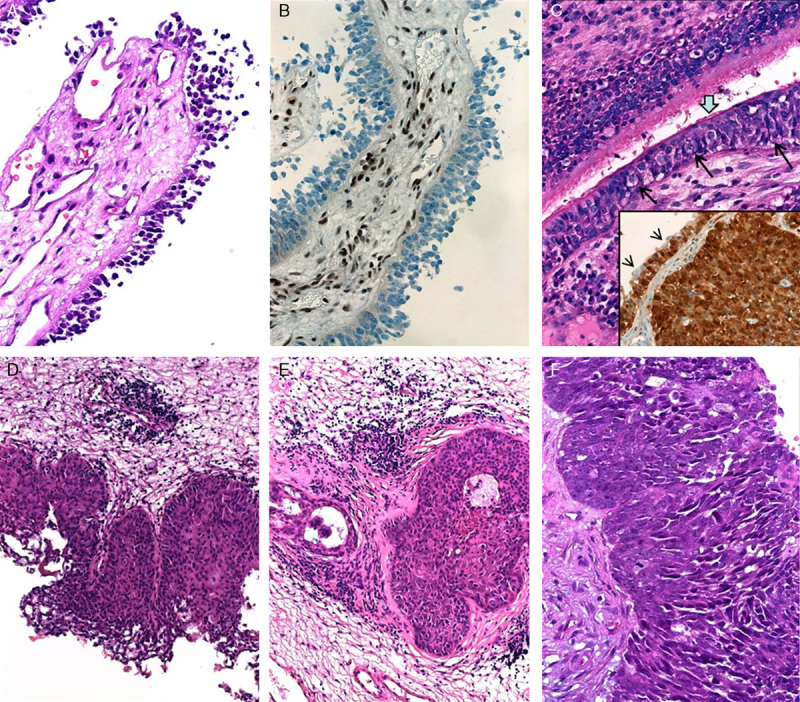
Spectrum of surface changes seen in the mucosa overlying SMARCB1-negative sinonasal carcinomas. A, Dyscohesive “denuding” atypical neoplastic cells covered the papillary fronds (case 3). B, These cells are negative for SMARCB1 (stromal and endothelial cells in the papillary cores stained positive). C, Pagetoid spread (long black thin arrows) was seen beneath ciliated respiratory epithelium (thick arrow; case 1). C inset, This case strongly expressed p16 in the invasive tumor (right) and in the pagetoid surface component (left), note scattered p16-negative residual normal epithelial cells on top of staining cells (arrow heads). D, Case 2 showed superficial flat-papillary growth closely mimicking Schneiderian papilloma with spreading along deep submucosal glands. E, Residual lumen is seen indicating preexistent normal gland (uninvolved part of the gland is also seen on the left). This case was initially misinterpreted as inverted papilloma. F, Case 3 showed areas of plump fused papillae resulting in pleomorphic basaloid CIS-like appearance.
FIGURE 4.
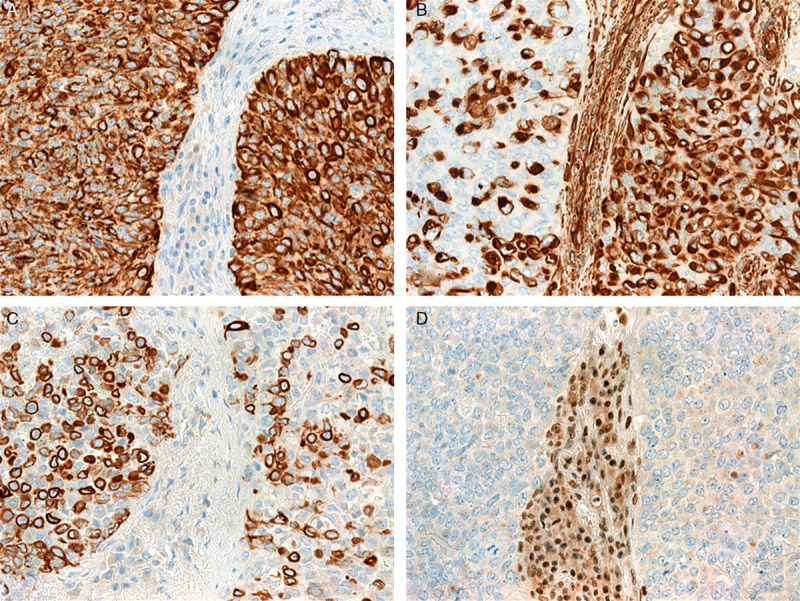
Immunohistochemical findings in SMARCB1-negative sinonasal carcinomas. A, Pancytokeratin (KL-1) was expressed in almost all tumor cells in all cases and showed diffuse or punctate perinuclear cytoplasmic pattern. B, Vimentin expression varied from diffuse (right) to partial/hybrid pattern (left). Note characteristic paranuclear dot-like pattern highlighting rhabdoid cells. C, CK5 showed an inverse pattern compared with that of vimentin indicating a dual population of CK5+/vimentin− (basaloid) and CK5−/vimentin+ (rhabdoid) cells. D, Complete loss of nuclear SMARCB1/INI1 expression (case 1, note strong staining in inflammatory and endothelial cells).
SMARCB1 Status in the Control Groups
To assess for the possible frequency of SMARCB1 loss in sinonasal carcinomas of different types and in Schneiderian papillomas, we stained 40 Schneiderian papillomas and a total of 112 consecutive sinonasal carcinomas of different types treated at our department between 2002 and 2014 (see the Materials and methods section); all showed intact nuclear SMARCB1 expression except the 3 cases presented herein. All 200 oropharyngeal SCC samples showed retained SMARCB1 expression (data not shown).
DISCUSSION
In this study, we presented 3 cases of a distinctive sinonasal carcinoma that showed unusual clinicopathologic features. A review of the literature did not reveal any similar cases reported previously. The 3 cases showed several common features including almost identical histologic appearance, relatively young age at presentation (mean age, 38 y), aggressive growth indicated by frequent presentation as pT4 disease, and propensity for regional and/or distant spread (seen in 2 of 3 cases). Although this small case series does not allow for any generalized conclusion regarding behavior and prognosis, the disease course in our 3 cases is likely different from the highly aggressive SMARCB1-deficient malignant extrarenal rhabdoid tumors. The latter usually present with widespread disease at the time of diagnosis, and they carry a poor prognosis with short survival.10 Likewise, rhabdoid “dedifferentiation” associated with SMARCB1 loss in carcinomas arising in the gastrointestinal tract of adults heralded a highly aggressive course with a mean survival of 4 months (>80% of patients died of disease within 1 y).11 However, more cases need to be analyzed before hard conclusion regarding age range, histologic spectrum, behavior, and prognosis of this rare variant can be made.
We interpreted the extensive CIS-like replacement of the surface epithelium by tumor cells (seen in all cases) as tumor spread along the mucosa and not as a precursor lesion (intraepithelial neoplasia). This view was supported by (1) the similar extensiveness of this finding in all cases, (2) the presence of similar growth along mucosal glands underneath, (3) the presence of prominent areas with clear-cut pagetoid surface spread, (4) the absence of gradual or transitional areas, and (5) the immunophenotypic identity of the surface component with the frankly invasive tumor component. The prominent denuding-like appearance (reminiscent of the denuding CIS seen in the urinary tract) is likely the result of an inherent tendency of the superficial neoplastic component for dyscohesive growth. Notably, this papillary pattern was seen also in the lung metastasis, which highlights the tumor propensity for this type of growth. Although this pattern resulted into formation of papillary fronds suggesting a carcinoma arising from preexisting Schneiderian papilloma,12 we could not detect any clear-cut benign residual papilloma even after extensive search for this, and oncocytic cells were absent as well.
The etiology of the current cases is unclear. All 3 cases were negative for EBV. Their distinctive basaloid and partially papillary histologic appearance at low power may suggest HPV-associated papillary basaloid SCC.13–15 This is further complicated by the strong expression of p16 (diffuse in 2 cases and patchy in 1 case). However, the complete absence of precursor lesions, absence of clear-cut squamous differentiation, and lack of detectable HPV by molecular studies ruled out this possibility. In addition, the papillary structures in our cases are different from the typical plump papillae of SCC related to HPV. It is noteworthy that p16 has not been validated as a surrogate marker for HPV status outside the oropharynx. Our cases also lacked the characteristic growth and cytologic features of NUT midline carcinomas (none expressed NUT by immunohistochemistry, and they lacked abrupt squamous differentiation and diffuse p63 expression).16,17 Rare SMARCB1-deficient neoplasms have been reported to arise as a dedifferentiated component of a well-definable SMARCB1-positive “parent” neoplasm.18–20 However, our cases lacked evidence of a “parent tumor” or unequivocal precursor lesions, even after extensive search. Accordingly, these cases likely do not represent composite malignant extrarenal rhabdoid tumors in adults.21 Sinonasal teratocarcinosarcoma is another rare entity that may display rhabdoid features and mimic our cases.22 This rare entity, however, features mesenchymal and teratomatous elements in addition to epithelial differentiation, features not seen in our cases.
SMARCB1 is a tumor-suppressor gene located at 22q11.2. It is considered an integral component of the chromatin remodeling complex SWI/SNF. Loss of nuclear SMARCB1 expression characterizes a heterogenous family of neoplasms with highly varying histologic appearance and biological behavior.23 Among others, pediatric atypical teratoid/rhabdoid tumors, malignant rhabdoid tumors of the kidney and extrarenal tissues, renal medullary carcinoma, both types of epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumors, and some myoepithelial soft tissue neoplasms are the main types characterized to date.11,23 In addition to these (predominantly pediatric) aggressive neoplasms, which are defined by primary loss of SMARCB1 expression as a consequence of deletions or mutations involving the SMARCB1 locus, secondary SMARCB1 loss has been reported in carcinomas arising in different visceral organs including the gastrointestinal tract,11 pancreas,18 uterus,19 and nervous system.20 Common to most of these entities are: loss of SMARCB1 as a key alteration, rhabdoid cell component that varies from subtle (represented by a few scattered cells) to extensive comprising almost 100% of the neoplasm, and a frequently (but not obligatory) aggressive clinical course with widespread metastasis at time of diagnosis.23 Although it is well known that a rhabdoid phenotype in several tumors does not represent a specific nosologic entity,24,25 it is clear from this and other previous studies that the presence of even a minor rhabdoid cell population in an otherwise conventional neoplasm should warrant investigation of the SMARCB1 status.11,23
In contrast to the well-established and accepted proximal-type epithelioid sarcoma of soft tissue,26 rare examples of SMRACB1-deficient neoplasms have been reported as proximal-type epithelioid sarcoma in visceral organs including 1 recent skull base tumor involving the sinonasal tract.27–29 All these cases, however, were more similar to malignant rhabdoid tumors and proximal-type epithelioid sarcoma being characterized by uniformly undifferentiated dyscohesive large rhabdoid cells and absence of clear-cut epithelial pattern of growth or differentiation. In the current study the epithelial nature of the neoplasms was evident from the characteristic cohesive uniform basaloid tumor appearance, infiltrative growth similar to SCC at this site, abundance of cytokeratin expression as opposed to limited vimentin expression, prominent association with the surface epithelium, and metastatic pattern that is not different from conventional sinonasal SCC. Also unusual for the prototypical SMARCB1-deficient rhabdoid neoplasms is the retention of the site-specific basaloid tumor appearance in all cases resulting in initial diagnosis of basaloid SCC in all 3 cases. Thus, the terms malignant extrarenal rhabdoid tumor and proximal-type epithelioid sarcoma seem inappropriate for these cases.
SMARCB1-deficient neoplasms in children30 and very rarely in adults31,32 have been linked to germline SMARCB1 mutations. None of our cases had a personal or family history of SMARCB1-related rhabdoid or other relevant neoplasms, but germline investigation, although recommended in 1 case, was not performed. Thus, the remote possibility of an undetected germline mutation cannot be definitely excluded. Although our cases have not been analyzed by molecular studies for the underlying SMARCB1 alterations, recent studies have appreciated the central role of SMARCB1 loss by immunohistochemistry as the defining and most consistently reproducible feature of these neoplasms.23,33 Notably, the frequency and types of the genetic alterations have varied greatly from tumor to tumor, with some cases featuring wild-type SMARCB1 suggesting epigenetic or other yet unknown molecular mechanisms in the pathogenesis of the nuclear SMARCB1 loss.34 Given that we identified these cases retrospectively from among a total of >100 sinonasal carcinomas, this variant seems to be rare comprising <3% of sinonasal carcinomas in our files. However, paucity of rhabdoid cells as the “hallmark cells” of this variant might be responsible for it being underreported.
In summary, we herein described 3 cases of a distinctive variant of basaloid sinonasal carcinoma featuring variable rhabdoid cell elements and complete loss of SMARCB1. This variant needs to be distinguished from carcinoma arising in Schneiderian papilloma, NUT midline carcinoma, HPV-associated basaloid, or papillary sinonasal SCC, SNUC, and metastatic atypical teratoid/rhabdoid tumors of the central nervous system. The molecular pathogenesis and etiology of this highly distinctive neoplasm remains to be further investigated.
Footnotes
Conflicts of Interest and Source of Funding: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: a historical analysis of population-based data. Head Neck. 2012;34:877–885 [DOI] [PubMed] [Google Scholar]
- 2.Haerle SK, Gullane PJ, Witterick IJ, et al. Sinonasal carcinomas: epidemiology, pathology, and management. Neurosurg Clin N Am. 2013;24:39–49 [DOI] [PubMed] [Google Scholar]
- 3.Barnes L, Eveson JW, Reichart P, et al. World Health Organization Classification of Tumours. Pathology and Genetics of Head and Neck Tumours. 2005Lyon:IARC Press [Google Scholar]
- 4.Mills SE, Fechner RE. “Undifferentiated” neoplasms of the sinonasal region: differential diagnosis based on clinical, light microscopic, immunohistochemical, and ultrastructural features. Semin Diagn Pathol. 1989;6:316–328 [PubMed] [Google Scholar]
- 5.Stelow EB, Jo VY, Mills SE, et al. A histologic and immunohistochemical study describing the diversity of tumors classified as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol. 2011;35:971–980 [DOI] [PubMed] [Google Scholar]
- 6.Frierson HF, Jr, Mills SE, Fechner RE, et al. Sinonasal undifferentiated carcinoma. An aggressive neoplasm derived from schneiderian epithelium and distinct from olfactory neuroblastoma. Am J Surg Pathol. 1986;10:771–779 [PubMed] [Google Scholar]
- 7.Wieneke JA, Thompson LD, Wenig BM. Basaloid squamous cell carcinoma of the sinonasal tract. Cancer. 1999;85:841–854 [PubMed] [Google Scholar]
- 8.Fritsch VA, Lentsch EJ. Basaloid squamous cell carcinoma of the head and neck: location means everything. J Surg Oncol. 2014;109:616–622 [DOI] [PubMed] [Google Scholar]
- 9.Petry KU, Menton S, Menton M, et al. Inclusion of HPV testing in routine cervical cancer screening for women above 29 years in Germany: results for 8466 patients. Br J Cancer. 2003;88:1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanburg-Smith JC, Hengge M, Hengge UR, et al. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol. 1998;2:351–362 [DOI] [PubMed] [Google Scholar]
- 11.Agaimy A, Rau TT, Hartmann A, et al. SMARCB1 (INI1)-negative rhabdoid carcinomas of the gastrointestinal tract: clinicopathologic and molecular study of a highly aggressive variant with literature review. Am J Surg Pathol. 2014[Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Ward BE, Fechner RE, Mills SE. Carcinoma arising in oncocytic Schneiderian papilloma. Am J Surg Pathol. 1990;14:364–369 [DOI] [PubMed] [Google Scholar]
- 13.Jo VY, Mills SE, Stoler MH, et al. Papillary squamous cell carcinoma of the head and neck: frequent association with human papillomavirus infection and invasive carcinoma. Am J Surg Pathol. 2009;33:1720–1724 [DOI] [PubMed] [Google Scholar]
- 14.Bishop JA, Guo TW, Smith DF, et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am J Surg Pathol. 2013;37:185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehrad M, Carpenter DH, Chernock RD, et al. Papillary squamous cell carcinoma of the head and neck: clinicopathologic and molecular features with special reference to human papillomavirus. Am J Surg Pathol. 2013;37:1349–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelow EB, Bellizzi AM, Taneja K, et al. NUT rearrangement in undifferentiated carcinomas of the upper aerodigestive tract. Am J Surg Pathol. 2008;32:828–834 [DOI] [PubMed] [Google Scholar]
- 17.Bishop JA, Westra WH. NUT midline carcinomas of the sinonasal tract. Am J Surg Pathol. 2012;36:1216–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho YM, Choi J, Lee OJ, et al. SMARCB1/INI1 missense mutation in mucinous carcinoma with rhabdoid features. Pathol Int. 2006;56:702–706 [DOI] [PubMed] [Google Scholar]
- 19.Donner LR, Wainwright LM, Zhang F, et al. Mutation of the INI1 gene in composite rhabdoid tumor of the endometrium. Hum Pathol. 2007;38:935–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinschmidt-DeMasters BK, Birks DK, Aisner DL, et al. Atypical teratoid/rhabdoid tumor arising in a ganglioglioma: genetic characterization. Am J Surg Pathol. 2011;35:1894–1901 [DOI] [PubMed] [Google Scholar]
- 21.Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol. 1995;12:233–248 [PubMed] [Google Scholar]
- 22.Kim JH, Maeng YH, Lee JS, et al. Sinonasal teratocarcinosarcoma with rhabdoid features. Pathol Int. 2011;61:762–767 [DOI] [PubMed] [Google Scholar]
- 23.Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–e63 [DOI] [PubMed] [Google Scholar]
- 24.Bittesini L, Dei Tos AP, Fletcher CD. Metastatic malignant melanoma showing a rhabdoid phenotype: further evidence of a non-specific histological pattern. Histopathology. 1992;20:167–170 [DOI] [PubMed] [Google Scholar]
- 25.Perry A, Fuller CE, Judkins AR, et al. INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol. 2005;18:951–958 [DOI] [PubMed] [Google Scholar]
- 26.Guillou L, Wadden C, Coindre JM, et al. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 1997;21:130–146 [DOI] [PubMed] [Google Scholar]
- 27.Maggiani F, Debiec-Rychter M, Ectors N, et al. Primary epithelioid sarcoma of the oesophagus. Virchows Arch. 2007;451:835–838 [DOI] [PubMed] [Google Scholar]
- 28.Venizelos I, Anagnostou E, Papathomas TG, et al. Proximal-type epithelioid sarcoma of the uterine corpus. Histopathology. 2011;58:321–323 [DOI] [PubMed] [Google Scholar]
- 29.Frank R, Sadri N, Bhatti T, et al. Proximal-type epithelioid sarcoma of the head and neck (HN): a study with immunohistochemical and molecular analysis of SMARCB1. J Clin Exp Oncol. 2013;2:pii: 1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eaton KW, Tooke LS, Wainwright LM, et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter JM, O’Hara C, Dundas G, et al. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol. 2012;36:154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hulsebos TJ, Kenter S, Siebers-Renelt U, et al. SMARCB1 involvement in the development of leiomyoma in a patient with schwannomatosis. Am J Surg Pathol. 2014;38:421–425 [DOI] [PubMed] [Google Scholar]
- 33.Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol. 2007;14:335–339 [DOI] [PubMed] [Google Scholar]
- 34.Tsai CY, Wong TT, Lee YH, et al. Intact INI1 gene region with paradoxical loss of protein expression in AT/RT: implications for a possible novel mechanism associated with absence of INI1 protein immunoreactivity. Am J Surg Pathol. 2012;36:128–133 [DOI] [PubMed] [Google Scholar]