

Abstract
Objective:
To establish the phenotypic spectrum of KIF5A mutations and to investigate whether KIF5A mutations cause axonal neuropathy associated with hereditary spastic paraplegia (HSP) or typical Charcot-Marie-Tooth disease type 2 (CMT2).
Methods:
KIF5A sequencing of the motor-domain coding exons was performed in 186 patients with the clinical diagnosis of HSP and in 215 patients with typical CMT2. Another 66 patients with HSP or CMT2 with pyramidal signs were sequenced for all exons of KIF5A by targeted resequencing. One additional patient was genetically diagnosed by whole-exome sequencing.
Results:
Five KIF5A mutations were identified in 6 unrelated patients: R204W and D232N were novel mutations; R204Q, R280C, and R280H have been previously reported. Three patients had CMT2 as the predominant and presenting phenotype; 2 of them also had pyramidal signs. The other 3 patients presented with HSP but also had significant axonal neuropathy or other additional features.
Conclusion:
This is currently the largest study investigating KIF5A mutations. By combining next-generation sequencing and conventional sequencing, we confirm that KIF5A mutations can cause variable phenotypes ranging from HSP to CMT2. The identification of mutations in CMT2 broadens the phenotypic spectrum and underlines the importance of KIF5A mutations, which involve degeneration of both the central and peripheral nervous systems and should be tested in HSP and CMT2.
Spastic paraplegia type 10 (SPG10) is a rare form of autosomal dominant hereditary spastic paraplegias (HSPs) caused by mutations of the kinesin family member 5A (KIF5A) gene (OMIM: *602821).1 Axonal neuropathy is frequently associated with SPG10 and has been documented by neurophysiologic studies in two-thirds of patients.2–5 Interestingly, 2 mutations in KIF5A (p.Glu251Lys [E251K] and p.Gly235Glu [G235E]) have been identified in patients with an isolated axonal neuropathy but no pyramidal signs, compatible with a clinical diagnosis of Charcot-Marie-Tooth disease type 2 (CMT2).5,6 These findings have raised the hypothesis that KIF5A mutations have a broad phenotypic spectrum and that SPG10 and CMT2 may be allelic disorders. However, there is a lack of large-scale studies to address this question. The aim of this study was to establish whether KIF5A mutations can be responsible for both HSP associated with neuropathy and CMT2.
METHODS
Standard protocol approvals, registrations, and patient consents.
This work was carried out with informed consent and ethics approval from the Joint Ethics Committee of UCL Institute of Neurology and The National Hospital for Neurology and Neurosurgery (UCLH ethics/IRB 99/N103).
Patients.
The diagnosis of HSP or CMT was based on clinical features, imaging, and neurophysiology. Acquired causes of spasticity and neuropathy were excluded. The majority of patients were assessed by M.M.R. and H.H. The vast majority of mutations have been identified mainly in the motor domain1–9; therefore, this domain was screened in 2 series of patients. The first series included 186 unrelated patients with a clinical diagnosis of HSP with or without neuropathy (HSP series) and the second included 215 unrelated patients with axonal neuropathy consistent with CMT2 (CMT2 series). Patients in the HSP series were negative for mutations in SPAST, REEP1, NIPA1, ATL1, and BSCL2 and patients in the CMT2 series were usually negative for mutations in GJB1, MFN2, MPZ, GDAP1, BSCL2, TRPV4, NEFL, HSPB1, and HSPB8. A high-throughput targeted resequencing panel covering all coding exons of KIF5A was developed to screen an additional 66 unrelated patients with HSP or CMT2 with pyramidal signs. Most of these patients (58/66) were negative for ATL1 and SPAST mutations. One patient was analyzed by whole-exome sequencing (WES). As part of the diagnostic workup, patients carrying KIF5A mutations had neurologic examination and neurophysiologic assessment and one patient had a sural nerve biopsy. In patients with an identified KIF5A mutation, DNAs from affected family members were also tested for segregation where available.
Sanger sequencing.
Genomic DNA was extracted from peripheral blood using Flexigene extraction kit and Autopure LS (Qiagen, Venlo, the Netherlands) extraction system. Coding regions and flanking introns of exons 1 to 12, which encode the motor domain of KIF5A (NM_004984), were amplified using Roche FastStart PCR Master Kit (Roche, Basel, Switzerland) according to manufacturers' instructions. Primers and conditions of PCR reactions are listed in table e-1 on the Neurology® Web site at Neurology.org. Sequence reactions were performed using Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA), and products were cleaned using BigDye Terminator removal plates (Applied Biosystems). PCR products were resolved on Applied Biosystems 3730XI Sequencer. The resulting sequences were aligned and analyzed with Sequencher (Gene Codes Corporation, Ann Arbor, MI) software.
Targeted resequencing.
A high-throughput targeted resequencing panel with a library of oligonucleotide probes covering all exons of KIF5A was designed through the Web-based Agilent Halo Design Wizard (http://www.halogenomics.com/haloplex/custom-reagent-kits). Genomic DNA of 250 ng was used for library preparation and then hybridized to exome-capture probes with the HaloPlex system (Agilent, Santa Clara, CA) following the manufacturer's standard protocol. The final HaloPlex enrichment pool was sequenced using 150 × 150 base paired-end chemistry on the Illumina (San Diego, CA) MiSeq. Before aligning reads to the reference genome, reads were trimmed from HaloPlex adaptor sequences with NextGene software (Softgenetics, State College, PA). Sequence alignment and variant calling were then performed against the reference human genome (UCSC hg19). Variants were filtered for presence of nonsynonymous heterozygous variants with a minor allele frequency <1% in the 1,000 Genomes and the National Heart, Lung, and Blood Institute Exome Sequencing Variant database (EVS database; http://evs.gs.washington.edu/EVS/). All mutations identified by the panel were validated by Sanger sequencing.
Whole-exome sequencing.
Enrichment of coding exons and flanking intronic regions was performed using the SureSelect Human All Exon 50 Mb kit (Agilent) following the manufacturer's standard protocol. Enriched DNA was subjected to standard sample preparation for the Hiseq2000 instrument (Illumina). BWA and GATK software packages were used to align sequence reads to the NCBI hg19 reference and to call variant positions.10,11 All data were then annotated and imported into GEnomes Management Application (GEM.app) for data analysis.12 Variants were filtered with the same criteria applied in targeted resequencing.
Pathogenicity evaluation of novel mutations.
For identified novel mutations, in silico prediction of the functional impact of the mutation was performed using Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.bii.a-star.edu.sg/). A search of these mutations was performed in exome sequences from 221 healthy controls, the EVS database, and the Genome Variant Database for Neuromuscular Diseases (NMD database; http://hihg.med.miami.edu/gvd-nmd/).
RESULTS
Genetic results.
Five different heterozygous missense KIF5A mutations were identified in 6 unrelated patients. Mutations in 2 patients were identified by the targeted resequencing panel, 3 were detected with Sanger sequencing, and in 1 patient, the mutation was identified by WES. Pedigree information and nucleotide changes of the mutations are illustrated in figure 1A. R204Q (c.611G>A; p.Arg204Gln), R280H (c.839G>A; p.Arg280His), and R280C (c.838C>T; p.Arg280Cys) have been previously reported in patients with SPG10,5,6,8 whereas R204W (c.611C>T; p.Arg204Trp) and D232N (c.694G>A; p.Asp232Asn) are novel mutations. The novel mutations were not present in 221 exomes from healthy controls or the EVS and NMD databases. Furthermore, they were predicted to be probably damaging and not tolerated by Polyphen-2 and SIFT and each mutation changed a highly conserved amino acid residue (figure 1B). The above evidence supports the pathogenicity of these novel mutations.
Figure 1. KIF5A mutations.

(A) Chromatographs of the identified mutations and pedigree of the 6 patients in this study. The phenotypes illustrated in different colors refer to (1) classical Charcot-Marie-Tooth disease type 2 (CMT2): classical CMT2 phenotype without pyramidal sign; (2) CMT2 + pyramidal signs: CMT2 is the main manifestation but there are associated pyramidal signs; (3) hereditary spastic paraplegia (HSP) + neuropathy: complex spastic paraplegia associated with peripheral neuropathy; (4) HSP, no neuropathy: spastic paraplegia without peripheral neuropathy. Parents of K-2 and K-3 were clinically unaffected at least until their sixth decades, though they were unavailable for genetic testing. Parents of K-5 died early due to non-neurologic diseases. (B) The amino acid residues where 2 novel missense mutations (R204W and D232N) occurred were highlighted on kinesin orthologues in various species. The alignment showed their strict conservation during evolution from zebrafish to human beings.
Clinical and pathologic findings.
Patients with KIF5A mutations presented with a variable phenotype and age at onset. The clinical features of the patients with KIF5A mutations are summarized in table 1. Among the 3 patients who had CMT2 as the predominant phenotype, only K-1 initially presented with a classical CMT2 phenotype, whereas the remaining 2 (K-2, K-3) had some involvement of the pyramidal tracts. The other 3 patients had spastic paraplegia as the main manifestation and 2 of them (K-4, K-6) also had peripheral neuropathy. Moreover, some patients had additional features: K-3, K-6, and other affected family members of K-6 had cognitive dysfunction and learning difficulties since their childhood; K-5 and her sister had marked cerebellar ataxia complicating the spasticity.
Table 1.
Clinical features of the patients with KIF5A mutations
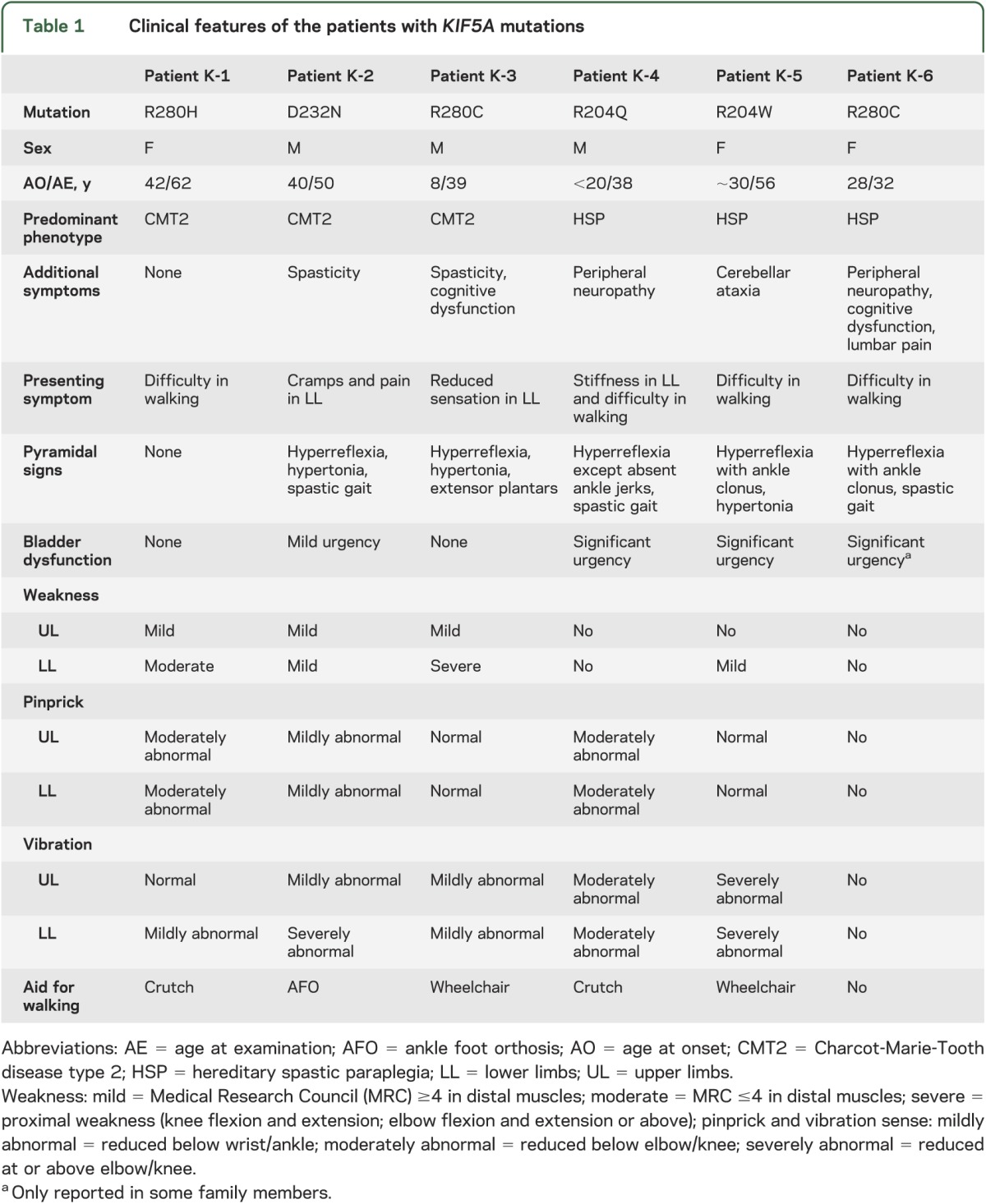
R280C mutation was identified in 2 unrelated patients with different phenotypes: K-3 was of British origin and had the clinical diagnosis of CMT2 with a slight involvement of the corticospinal tract, whereas K-6, who had lived in Austria for several generations, presented with severe spastic paraplegia and mild peripheral neuropathy (figure 1A; K-3, K-6). Variable phenotypes in individuals within the same family were also observed. K-2, who had D232N mutation, presented with complex CMT2 associated with spasticity, whereas his brother had pure HSP (figure 1A; K-2).
Features demonstrating the simultaneous involvement of the peripheral nervous system and CNS in our patients are shown in figure 2. A sural nerve biopsy from K-3 revealed markedly reduced numbers of large myelinated fibers, compatible with axonal degeneration (figure 2, A–D). Meanwhile, spasticity with hyperreflexia was observed on examination. K-2 had wasting of intrinsic foot muscles and pes cavus while the worn-out areas of the shoes indicated a long-standing spastic gait (figure 2, G–H).
Figure 2. Pathologic and clinical evidence of peripheral neuropathy in patients with KIF5A mutations.

Sural nerve biopsy of patient K-3: (A, B) semi-thin resin section stained with methylene blue azure–basic fuchsin shows markedly reduced numbers of large myelinated fibers and frequent small myelinated fibers often associated with regeneration clusters (arrow in B). (C, D) Ultrastructural assessment further confirms frequent regeneration with numerous unmyelinated axonal sprouts associated with regeneration clusters (arrows in C and D) but does not reveal any evidence of active or chronic demyelination. (E, F) Sural nerve from a healthy age-matched control. Scale bar: 90 μm (A), 45 μm (B, E), 5 μm (C, D, F). Patient K-2: (G) wasted foot with pes cavus. (H) The worn-down areas of the patient's shoes are suggestive of a long-existing spastic gait.
Neurophysiologic findings.
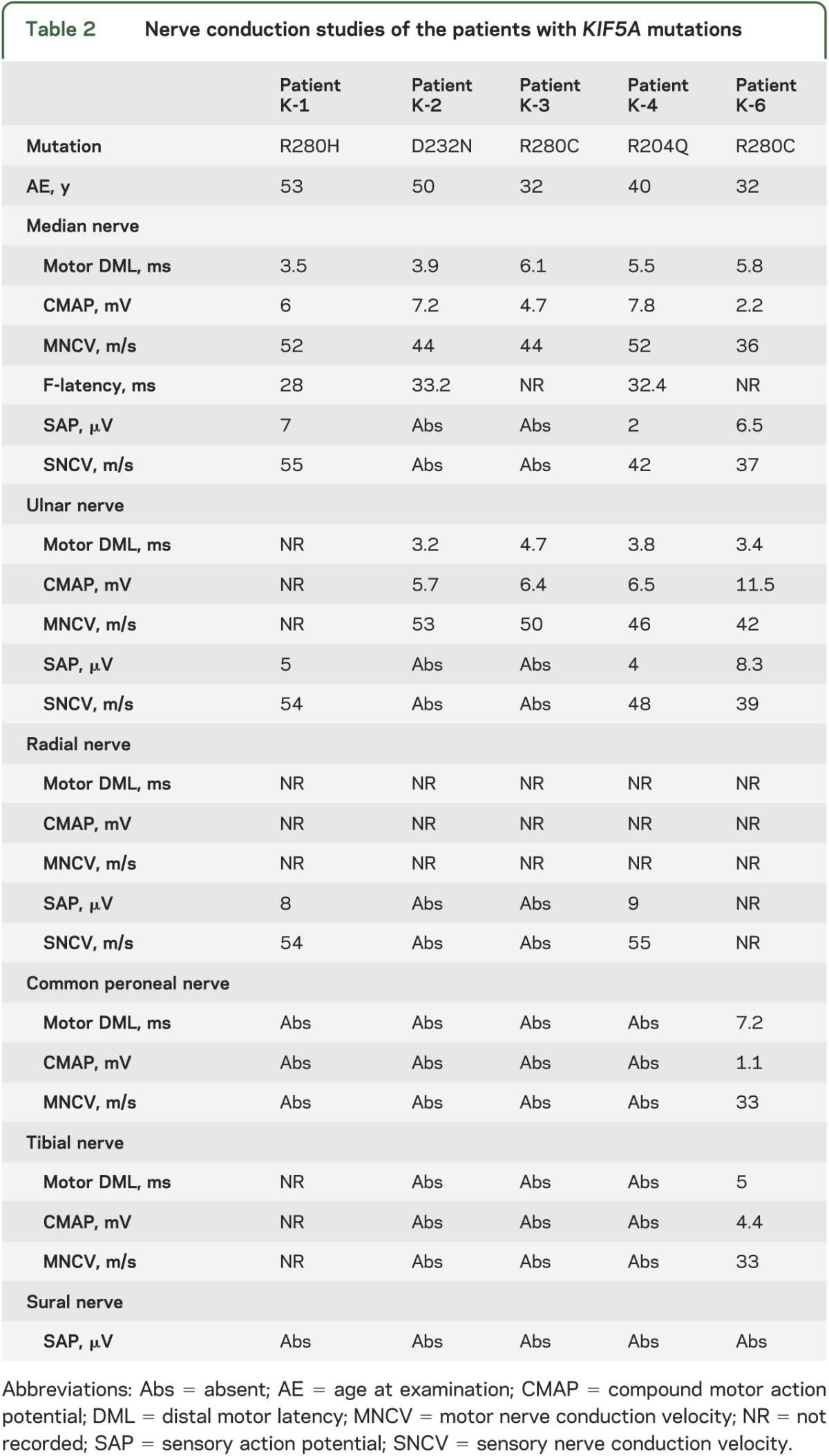
Nerve conduction studies of the 5 patients who had a clinically recognizable peripheral neuropathy are presented in table 2. Neurophysiology showed symmetrically absent or reduced compound muscle action potential and sensory action potentials with relatively preserved nerve conduction velocities in upper and lower limbs, consistent with sensorimotor axonal polyneuropathy.
Table 2.
Nerve conduction studies of the patients with KIF5A mutations
DISCUSSION
This study reports the largest series of HSP and CMT2 patients screened for KIF5A mutations. The identification of 5 different mutations confirms that KIF5A is associated with both HSP and CMT2 phenotypes. We also report the first family with a KIF5A mutation who has associated cerebellar ataxia, broadening the phenotypic spectrum of KIF5A mutations.
The identified KIF5A mutations and their associated phenotypes are presented in figure 3. HSP and CMT2 represent 2 different ends of the spectrum of KIF5A-related disorders: classical or complex CMT2 is relatively rare, while HSP with axonal neuropathy is the commonest phenotype associated with KIF5A mutations. However, a specific phenotype–genotype correlation for KIF5A mutations could not be defined as some mutations (R280C, R280H, and D232N) have been associated with more than one phenotype. The intrafamilial phenotypic variability within families with the D232N mutation provides the most straightforward evidence of the allelic relationship of HSP and CMT2.
Figure 3. Phenotypic spectrum of KIF5A mutations.

Top: Genomic organization of the human KIF5A gene and its mutations. Most of the KIF5A mutations are localized in exon 8∼10 and many are clustered within or around the switch I region (SWI) and the switch II region (SWII), which carry γ-phosphate-sensing activity to convert adenosine-5′-triphosphate into required energy. KIF5A mutations were labeled with the corresponding phenotypes: classical Charcot-Marie-Tooth disease type 2 (CMT2), CMT2 + pyramidal signs, hereditary spastic paraplegia (HSP) + neuropathy, and HSP, no neuropathy (definitions of the phenotypes described in figure 1). The mutations identified in this study were flanked by the novel mutations in pink. The reference for each mutation is listed in table e-2. Bottom left: Venn diagram represents the overlap and distinct phenotypes of KIF5A mutations. The majority of these mutations presented with both spastic paraplegia and neuropathy.
KIF5A encodes the neuronal kinesis heavy chain (KHC) subunit of kinesin-1, a motor protein that plays a role in axonal transport. Kinesin-1 relies on the highly conserved motor domain of KHC to bind to the microtubules and converts the chemical energy of adenosine-5′-triphosphate into mechanical energy required for intracellular trafficking.13–18 With both CMT2 and SPG10 phenotypes, the mutations are localized in the motor domain in the majority and clustering around the 2 switch regions (SWI and SWII in figure 3) that carry γ-phosphate-sensing activity and are particularly important in the interaction with microtubules (figure 3). Half of the families in this study have mutations at codon R280 and 25% (7/28) of all reported families with a KIF5A mutation have an amino acid substitution at this position. Although these families are from different regions and continents, it would be of interest to carry out a haplotype study to investigate a founder effect. The CpG dinucleotide of codon 280 had been proposed as a possible hot spot for KIF5A mutations.17 Our findings support the critical role of this codon in the function of KIF5A.
A small number of SPG10-associated KIF5A mutations (K253N, N256S, R280C, and A361V) have been investigated using in vitro and Drosophila models. The results support a loss of endogenous kinesin-1 function due to a selective dominant-negative mechanism of action on the kinesin complexes, rather than a gain of toxicity.15,19 Only mutations located in the motor domain changed the kinesin gliding properties but the mutation in the neck (A361V) did not. This suggests that mutations outside of the motor domain may have a different disease mechanism.15
CMT2 and HSP have different target cells: the first primarily affects the peripheral nerves and the latter principally affects the corticospinal tract. Both the peripheral nerve axons and the long corticospinal tract rely heavily on axonal transport of appropriate cargoes to maintain normal axonal function. This may explain why KIF5A mutations can cause both HSP and CMT2 and the overlapping phenotypes. KIF5A is another example of a gene in which mutations can cause axonal neuropathy in association with pyramidal signs and is involved in axonal transport similar to BSCL2, REEP1, ATL1, SPAST, and NIPA1 genes.13,14,18,20 Likewise, CCT5, KIF1A, and DYNC1H1 are other genes encoding motor proteins that have an important role in axonal transport and their mutations have been identified in patients with complex syndromes involving both the peripheral nervous system and CNS.21–24 Thus axonal transport represents a potential common pathway in the pathogenesis of both axonal neuropathy and spastic paraplegia.20 Mutant motor proteins affecting axonal transport may lead to a neurodegenerative process. However, the mechanism by which mutations of the same gene can either selectively or simultaneously affect the upper motor neurons and the peripheral axons is still not well understood. Other genetic or environmental factors may influence the phenotype. Further studies are needed to understand the function of KIF5A and its roles in the peripheral nervous system and CNS.
The reported frequency of KIF5A mutations may be an underestimate because of the expense of sequencing this large gene, which comprises 28 coding exons. The application of targeted resequencing for screening KIF5A provides a more comprehensive and cost-effective method. In addition, next-generation targeted sequencing and WES are powerful tools for identifying mutations in patients with atypical complex symptoms. We have demonstrated that these approaches have effectively facilitated the detection of KIF5A mutations and helped broaden the phenotypic spectrum of this gene.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families.
GLOSSARY
- CMT2
Charcot-Marie-Tooth disease type 2
- HSP
hereditary spastic paraplegia
- KHC
kinesis heavy chain
- SPG10
spastic paraplegia type 10
- WES
whole-exome sequencing
Footnotes
Editorial, page 580
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Liu drafted the manuscript and acquired, analyzed, and interpreted data. Dr. Laura acquired, analyzed, and interpreted data and revised the manuscript. Dr. Hersheson acquired, analyzed, and interpreted data and revised the manuscript. Dr. Horga acquired, analyzed, and interpreted data and revised the manuscript. Dr. Jaunmuktane acquired, analyzed, and interpreted neuropathology data. Dr. Brandner acquired, analyzed, and interpreted neuropathology data. Dr. Pittman acquired, analyzed, and interpreted data and revised the manuscript. D. Hughes acquired, analyzed, and interpreted data and revised the manuscript. Dr. Polke acquired, analyzed, and interpreted data and revised the manuscript. M. Sweeney acquired, analyzed, and interpreted data and revised the manuscript. Dr. Proukakis acquired, analyzed, and interpreted data and revised the manuscript. Dr. Janssen acquired, analyzed, and interpreted data and revised the manuscript. Dr. Auer-Grumbach acquired, analyzed, and interpreted data and revised the manuscript. Dr. Zuchner acquired, analyzed, and interpreted data and revised the manuscript. Dr. Shields acquired, analyzed, and interpreted data and revised the manuscript. Dr. Reilly contributed to design and conceptualization of the study, interpreted data, revised the manuscript, and supervised the study. Dr. Houlden contributed to design and conceptualization of the study, interpreted data, revised the manuscript, and supervised the study.
STUDY FUNDING
Supported by the Medical Research Council (MRC) and The Wellcome Trust (H.H.); MRC Centre grant G0601943 (M.M.R.); the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) (M.M.R., M.L.); and the Austrian Science Fond (FWF, P23223-B19) (M.A.). This work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Reid E, Kloos M, Ashley-Koch A, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 2002;71:1189–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schule R, Kremer BP, Kassubek J, et al. SPG10 is a rare cause of spastic paraplegia in European families. J Neurol Neurosurg Psychiatry 2008;79:584–587 [DOI] [PubMed] [Google Scholar]
- 3.Musumeci O, Bassi MT, Mazzeo A, et al. A novel mutation in KIF5A gene causing hereditary spastic paraplegia with axonal neuropathy. Neurol Sci 2010;32:665–668 [DOI] [PubMed] [Google Scholar]
- 4.Tessa A, Silvestri G, de Leva MF, et al. A novel KIF5A/SPG10 mutation in spastic paraplegia associated with axonal neuropathy. J Neurol 2008;255:1090–1092 [DOI] [PubMed] [Google Scholar]
- 5.Goizet C, Boukhris A, Mundwiller E, et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat 2009;30:E376–E385 [DOI] [PubMed] [Google Scholar]
- 6.Crimella C, Baschirotto C, Arnoldi A, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet 2012;82:157–164 [DOI] [PubMed] [Google Scholar]
- 7.Blair MA, Ma S, Hedera P. Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics 2006;7:47–50 [DOI] [PubMed] [Google Scholar]
- 8.Fichera M, Lo Giudice M, Falco M, et al. Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology 2004;63:1108–1110 [DOI] [PubMed] [Google Scholar]
- 9.Lo Giudice M, Neri M, Falco M, et al. A missense mutation in the coiled-coil domain of the KIF5A gene and late-onset hereditary spastic paraplegia. Arch Neurol 2006;63:284–287 [DOI] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez MA, Lebrigio RF, Van Booven D, et al. GEnomes Management Application (GEM.app): a new software tool for large-scale collaborative genome analysis. Hum Mutat 2013;34:842–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunawardena S, Goldstein LS. Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J Neurobiol 2004;58:258–271 [DOI] [PubMed] [Google Scholar]
- 14.Barry DM, Millecamps S, Julien JP, Garcia ML. New movements in neurofilament transport, turnover and disease. Exp Cell Res 2007;313:2110–2120 [DOI] [PubMed] [Google Scholar]
- 15.Ebbing B, Mann K, Starosta A, et al. Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum Mol Genet 2008;17:1245–1252 [DOI] [PubMed] [Google Scholar]
- 16.Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 2009;10:682–696 [DOI] [PubMed] [Google Scholar]
- 17.Xia C, Rahman A, Yang Z, Goldstein LS. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics 1998;52:209–213 [DOI] [PubMed] [Google Scholar]
- 18.Xia CH, Roberts EA, Her LS, et al. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol 2003;161:55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuger P, Sreekumar V, Schule R, et al. Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet 2012;8:e1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol 2012;246:14–25 [DOI] [PubMed] [Google Scholar]
- 21.Bouhouche A, Benomar A, Bouslam N, Chkili T, Yahyaoui M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J Med Genet 2006;43:441–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klebe S, Lossos A, Azzedine H, et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet 2012;20:645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riviere JB, Ramalingam S, Lavastre V, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 2011;89:219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willemsen MH, Vissers LE, Willemsen MA, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet 2012;49:179–183 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.