

Abstract
The identification of biomarkers for post-traumatic stress disorder (PTSD) and resilience/recovery is critical for advancing knowledge about pathophysiology and treatment in trauma-exposed persons. This study examined a series of glucocorticoid-related biomarkers prior to and in response to psychotherapy. Fifty-two male and female veterans with PTSD were randomized 2 : 1 to receive either prolonged exposure (PE) therapy or a weekly minimal attention (MA) intervention for 12 consecutive weeks. Psychological and biological assessments were obtained prior to and following treatment and after a 12-week naturalistic follow-up. Response was defined dichotomously as no longer meeting criteria for PTSD at post-treatment based on the Clinician Administered PTSD Scale for DSM-IV (CAPS). Clinical improvement on the CAPS was apparent for both PE and MA, with no significant difference according to treatment condition. Biomarkers predictive of treatment gains included the BCLI polymorphism of the glucocorticoid receptor gene. Additional predictors of treatment response were higher bedtime salivary cortisol and 24 h urinary cortisol excretion. Pre-treatment plasma dehydroepiandrosterone/cortisol ratio and neuropetide Y (NPY) levels were predictors of reductions in PTSD symptoms, and, for NPY only, of other secondary outcomes as well, including anxiety and depression ratings. Glucocorticoid sensitivity changed in association with symptom change, reflecting clinical state. It is possible to distinguish prognostic and state biomarkers of PTSD using a longitudinal approach in the context of treatment. Identified markers may also be relevant to understanding mechanisms of action of symptom reduction.
Keywords: post-traumatic stress disorder, psychotherapy, glucocorticoid receptor, neuropetide Y, hypothalamic pituitary adrenal axis, biomarkers
1. Introduction
There has been a growing interest in identifying and distinguishing deployment-related functional biomarkers that could be relevant to military personnel and combat veterans [1,2]. Biomarkers associated with risk for the development of post-traumatic stress disorder (PTSD) can ostensibly be measured prior to combat exposure to predict vulnerability for PTSD following a critical incident [3,4]. Although some risk factors would not be subject to change (e.g. genetic polymorphisms, childhood trauma), others, such as those that affect the nature, intensity and duration of the response to a trauma, can change over time (e.g. epigenetic or functional changes associated with an earlier exposure) [5]. Related to risk markers are prognostic measures that are obtained following trauma exposure to permit the prediction of resistance to or emergence, chronicity or recurrence, of symptom development, following biological changes resulting from exposure. Like risk biomarkers, prognostic measures may or may not be subject to further change and are useful in their ability to aid in the prediction of a later outcome [1].
By contrast, diagnostic or state markers of PTSD are measures that distinguish those with and without PTSD at a particular time [6]. In some cases, diagnostic biomarkers would also associate with clinical state or extent of symptom severity among those meeting the diagnostic criteria or in those below the diagnostic threshold. In a cross-sectional sample of people with and without PTSD, risk, prognostic, diagnostic and symptom severity markers can overlap and are not easily distinguished from each other. However, when assessed longitudinally, state markers vary in relation to symptom change while biomarkers indicative of resilience (i.e. recovery, symptom improvement) will either predict symptom improvement or emerge when symptoms decline [7].
Longitudinal assessment of pre- and post-treatment biomarkers can distinguish prognostic from state-related markers of PTSD. The extent to which these markers overlap with those associated with risk for the development of PTSD is of interest because predictors that change over time in association with symptom severity (such as hormonal or gene expression markers) may provide insight into mechanisms related to symptom expression, whereas markers that do not change with clinical state (such as genotype) may have prognostic value [2]. Risk markers that are functionally related to illness onset but also associate with symptom change may be particularly robust targets for treatment development. For example, changes in indices of the hypothalamic pituitary adrenal (HPA) axis have been associated with both risk for PTSD and symptom severity [5,8].
Only a few studies have examined biomarkers before and after psychotherapy in PTSD. These have focused on brain imaging, psychophysiological and endocrine measures, and more recently, epigenetic markers [9]. With respect to brain imaging, findings regarding structural changes in hippocampal volume following treatment are mixed, with one study failing to observe changes [10], and another demonstrating increased hippocampal volume [11]. Amygdala reactivity to laboratory-induced symptom provocation is attenuated following treatment [12]. Increased FKBP5 gene expression has also been observed in association with symptom reduction following psychotherapy for PTSD [11]. Elevated heart rate responses to laboratory stressors diminish following treatment [13]. At post-treatment, responders to psychotherapy have also exhibited increased plasma cortisol [14,15] and decreased salivary cortisol responses immediately following imaginal recall of their trauma [16].
In addition to distinguishing biomarkers, studies examining changes in biological variables following psychotherapy can also inform how biological and psychological mechanisms interact in PTSD. In that psychological factors modulate biological responses to stress and vice versa [17,18], such knowledge will enhance our understanding of mechanisms involved in symptom maintenance and decline. For example, we have previously hypothesized that PTSD occurs because of a failure to properly contain the release of adrenaline in response to threat—an adaptive response that maximizes physiological capacity for fight-or-flight. Sympathetic nervous system responses following threat are contained by the activation of the HPA axis culminating in the release of cortisol [19]. However, persons at risk for PTSD demonstrate attenuated cortisol increases in the acute aftermath of trauma [20], possibly resulting from increased sensitivity of glucocorticoid receptors (GRs) prior to trauma exposure [21]. Premature termination of the HPA axis secondary to enhanced GR sensitivity and negative feedback inhibition could result in sustained elevations of catecholamine levels, and the perpetuation of the intrusive and hyperarousal symptoms of PTSD, leading to the elaboration of avoidance symptoms [19,22].
This study was designed to examine measures of the HPA axis and endocrine markers that have been described in relation to resilience, risk and pathophysiology [23] in association with prolonged exposure (PE) psychotherapy and a minimal attention (MA) condition, described below. Since PE is associated with large effect sizes in civilians [24], the MA condition was included to ensure a sufficient proportion of non-responders and adequate post-treatment variation in clinical severity. We anticipated that PE would result in greater improvement than MA. Randomization to these conditions was designed to yield a sample at post-treatment with a variable degree of symptom improvement, which could be examined using continuous variables, or dichotomously by dividing the sample into treatment responders or non-responders.
Since glucocorticoid sensitivity, levels and circadian rhythmicity have been associated with symptom severity in cross-sectional studies, we hypothesized these measures would reflect state and would change in association with symptom improvement. We further hypothesized that resilience-related markers such as dehydroepiandrosterone (DHEA), the ratio of DHEA/cortisol, and neuropeptide Y (NPY) would be prognostic indicators of recovery, and would associate with or predict PTSD and related secondary markers associated with PTSD (depressive and anxiety symptoms, global mental health functioning) at post-treatment.
Finally, in view of our recent report of methylation status of the 1F promoter region of the GR gene as a predictor of treatment response to PE in a smaller subset of participants to be reported below [9], we added the assessment of a GR gene (NR3C1) BCLI polymorphism that has previously been associated with HPA-axis reactivity generally [25] and in PTSD [26]. Since the BCLI polymorphism has been associated with increased GR sensitivity, we hypothesized that it would be a prognostic indicator of recovery that would associate at post-treatment with other neuroendocrine correlates of response such as glucocorticoid sensitivity and circadian rhythmicity.
2. Subjects and methods
2.1. Subjects
Participants in this study were veterans receiving treatment in the PTSD clinic at the James J. Peters Veterans Affairs Medical Center (JJP VAMC) and were either self-referred or referred by their treating clinician. The study protocol was approved by the Institutional Review Boards of the JJP VAMC and the Icahn School of Medicine at Mount Sinai. Written informed consent and HIPAA authorization were obtained from all participants prior to the initiation of any study procedures, including the assessment of inclusion/exclusion criteria.
A total of 113 veterans (104 men, nine women; 74 who served in Iraq or Afghanistan, 32 from the Vietnam era and seven who served in Gulf War I or other conflicts) provided informed consent. Participants were eligible for randomization if they experienced a Criterion A trauma during military service and met DSM-IV criteria for PTSD with a duration of at least six months. Twenty-eight veterans were excluded following medical and psychological evaluation for one or more of the following reasons: significant illness, such as insulin-dependent diabetes, seizure disorder or disease requiring ongoing treatment with systemic steroids (n = 7); a recent change in medication and unable to wait until stabilization for two months (n = 4); current substance abuse or dependence within the last six months (n = 8); psychosis, bipolar disorder, obsessive compulsive disorder (n = 9). Thirty-three veterans discontinued participation prior to treatment initiation.
Of the 52 that were randomized to condition, 37 completed the trial. Of the 15 who chose to discontinue treatment, 12 (31%) were in the PE group and three (21%) were in the MA group; 13 were OIF/OEF veterans, one served in Vietnam and one in Gulf War I.
2.1.1. Study design
The purpose of the study was to evaluate biological and psychological measures at two time points before and after treatment to evaluate the extent to which biological measures predict or associate with symptom change. Psychotherapy was used rather than biological treatment so that biological change would not be the result of the introduction of medications that might have direct effects on the measures of interest. Given the reported rates of recovery from PTSD following PE in civilian samples [24], an MA condition was introduced so as to ensure a sufficient proportion of ‘non-responders’ and associated range of post-treatment symptom severity. Participants were randomized 2 : 1 to receive either PE or MA. Both PE and MA were administered according to manualized protocols developed by Foa et al. [27] (E. A. Hembree, N. C. Feeny & E. B. Foa 2006, unpublished data).
Veterans underwent a comprehensive psychological evaluation to assess baseline symptom severity. As part of the evaluation, blood, urine and saliva were obtained over a 2-day period prior to randomization. The same psychological and biological assessments were completed after 12 weekly sessions and again after three months. The psychological assessment was performed by an independent (i.e. non-treating) clinical psychologist at post-treatment and follow-up.
PE consists of weekly 90 min sessions [27]. Procedures include education about reactions to trauma, breathing retraining, exposure to trauma memories and situations the patient avoids, and discussion of thoughts and feelings related to exposure exercises. MA consists of weekly 30 min phone calls to the patient by the study therapist to monitor symptoms. The intervention was designed to ensure the safety of the veterans and to prevent loss to follow-up.
At post-treatment, participants were asked not to initiate new therapeutic interventions until after the three-month follow-up so that the stability of biological and psychological changes could be assessed at follow-up. However, in the interests of expediting clinical care, those who had been randomized to MA were permitted the opportunity to engage in PE or other treatment modalities within the PTSD clinic, as desired. Four of the 12 MA participants included in the analyses eventually opted to receive PE. Two received PE after completing their follow-up assessments, and two received PE sooner, and so did not complete follow-up assessments. Therefore, no follow-up scores reflect changes due to an additional intervention.
2.1.2. Clinical outcomes
The primary outcome was PTSD diagnosis (and symptom severity) as measured by the Clinician Administered PTSD Scale for DSM-IV (CAPS) [28]. The PTSD Symptom Scale—Self-Report Version (PSS-SR) was used as a self-report of PTSD symptoms [29]. Three secondary outcomes measured relevant clinical indicators of: (i) depressive symptoms (Beck Depression Inventory (BDI) [30]), (ii) anxiety (Spielberger Trait and State Anxiety Inventory (STAI) [31]), and (iii) general mental health functioning (Medical Outcomes Study 36-item short-form (SF-36) [32]). Other clinical and demographic measures were obtained to (i) characterize the sample, (ii) identify potential clinical predictors of response, and (iii) identify potential covariates for the biological analyses. These included the Deployment Risk and Resiliency Inventory (DRRI) to assess military and civilian life events pre- and post-deployment [33] and the Interpersonal Support Evaluation List (ISEL) [34] to assess social support.
Treatment was provided in the PTSD clinic by licensed or licence-eligible clinical psychologists who had been trained and supervised to criterion by VA certified PE providers on at least two completed PE treatment cases prior to inclusion as study therapists. All sessions were audiotaped for treatment fidelity rating purposes. All CAPS interviews were tape-recorded and monitored for inter-rater reliability, supervised by J.D.F. Consensus conferences were also held to agree on comorbid diagnoses obtained by the Structured Clinical Interview for the DSM-IV (SCID) [35].
2.2. Biological outcomes
The primary biological measures were: (i) GR BCLI genotype, (ii) glucocorticoid sensitivity as assessed by lysozyme inhibition test in cultured peripheral blood mononuclear cells (PBMCs) stimulated by varying doses of dexamethasone (DEX), (iii) measures reflecting glucocorticoid levels (24 h urinary cortisol excretion) and circadian rhythm (bedtime salivary cortisol), and (iv) plasma cortisol, NPY and DHEA.
2.2.1. Blood measures and methods
Blood was drawn on two consecutive mornings at 8.00 into EDTA containing vacutainer tubes. PBMCs were purified as previously described [9]. Some cell pellets were cultured and used for determination of lysozyme IC50-DEX [36], and an aliquot was frozen for later DNA extraction [9]. Genotyping of the BCLI polymorphism used the allelic discrimination technique with a qPCR machine [37]. The BCLI is a single nucleotide polymorphism (rs41423247) located in the intron 2 of the NR3C1 gene. This C > G polymorphism was found to be in a Hardy–Weinberg equilibrium (MAF = 0.28), as expected. For the purpose of statistical analyses, genotype was divided into two groups to designate ‘carriers’ of the G-allele (both homozygous GG and heterozygous CG) and ‘non-carriers’ (homozygous wild-type).
Plasma for hormone analyses was stored at −80°C. Cortisol, NPY and DHEA were determined by radioimmunoassay (RIA) [9,38,39]. The intra- and inter-assay coefficients of variation were 2.7% and 10.2% for cortisol, 3.5% and 11.6% for NPY, and 3.6% and 6.1% for DHEA, respectively.
2.2.2. Urine and saliva samples
Urine samples were collected over a 24 h period and stored frozen until assayed for cortisol as previously described [9]. Creatinine concentrations were obtained to monitor completeness of collection. Participants also collected saliva into Salivette tubes (Starstedt, Numbrecht, Germany) from awakening until bedtime for cortisol determination [40]. The intra- and inter-assay coefficients of variation for the urine cortisol assays were 2.6 and 10.8, respectively, and for salivary cortisol were 2.4 and 10.2, respectively.
2.3. Data analyses
The primary aims were to identify biological indicators that predict versus change in association with symptom improvement. These aims were addressed by comparing pre- and post-treatment data. Data collected at follow-up reflect maintenance of effects, but given participant attrition, data from the follow-up are not included in analyses of pre- to post-treatment effects; however, they were used in correlational analyses to determine whether predictors of clinical outcome at post-treatment also predicted associations at follow-up.
Responder status was defined conservatively by the presence or the absence of a PTSD diagnosis at post-treatment as determined by an independent psychologist using the CAPS for DSM-IV. A variety of covariates were examined for their potential association with biological measures including age, gender, body mass index (BMI) and use of psychotropic medications. Significant covariates were used in analysis of covariance and partial correlations as warranted.
Neuroendocrine markers previously associated with PTSD or resilience in cross-sectional studies were measured before and after treatment. Three sets of planned analyses on all biological variables were conducted in order to distinguish predictors of symptom change from correlates of symptom state. Although multiple analyses were performed, the dependent variables were not independent constructs, but domains that are conceptually and biologically linked.
(1) The first set of analyses was performed on pre-treatment biological, demographic and clinical data grouped on the basis of response status at post-treatment time point (i.e. no longer meeting the diagnostic criteria for PTSD). Group differences in pre-treatment measures were considered predictors of recovery (i.e. treatment response status).
-
(2) The second set of analyses compared biological measures at pre- and post-treatment in repeated measures ANOVAs (or ANCOVAs, if appropriate) using the same dichotomous variable of response status as a between subjects factor. A biological variable was considered to be associated with treatment response if there was a significant group × time interaction. A variable demonstrating a main effect of time (treatment), without a group × time interaction was also analysed as a possible correlate of symptom change regardless of diagnostic status, or change in secondary, more general clinical measures.
In the event that an association was observed in either of the above analyses, follow-up correlational analyses were performed between the biological variable(s) and continuous measures of PTSD and associated symptom severity. This analysis was conducted in order to evaluate whether a biological change was also associated with extent of symptom improvement; difference scores (pre- minus post-treatment, and pre- minus follow-up) or symptom severity at post-treatment or follow-up were used in these correlational analyses.
(3) A third set of analyses examined cross-sectional relationships between biological and clinical indicators. It was hypothesized that biological measures associated with treatment response would correlate cross-sectionally with clinical severity at post-treatment. Given the restricted range of pre-treatment severity, these associations were tested only at post-treatment.
3. Results
3.1. Demographic, descriptive and clinical measures
Baseline comparisons of demographic and clinical characteristics between treatment completers who were assigned to MA or PE were made to evaluate whether randomization was successful. There were no significant differences in any of these variables, which included age, gender, ethnicity, psychotropic medication, combat era or exposure, psychiatric diagnosis and severity, and BMI, indicating a successful randomization to treatment type. There were no significant gender differences in either treatment assignment or treatment response. On average, veterans had a mean CAPS score of 80.8 ± 18.5 (mean ± s.d.) at pre-treatment, and a lifetime severity score of 96.0 ± 17.7, indicating that most people had severe PTSD at the initiation of treatment.
3.2. Demographic and clinical predictors of post-traumatic stress disorder symptom improvement
Table 1 shows that several baseline demographic and clinical variables predicted a positive clinical outcome (regardless of treatment condition). Younger veterans, those who were older when they experienced their first trauma, and those with higher self-reported social support (ISEL) were more likely to be responders. Although PTSD severity at pre-treatment was not a predictor of response status, lower ‘worst episode’ PTSD CAPS scores and greater global impairment as assessed by the SF-36 mental health subscale predicted treatment response. When considering the subset of persons receiving PE only (n = 25), the same predictors were observed (data not shown).
Table 1.
Baseline demographics of responders and non-responders to treatment.
| responders (n = 13) | non-responders (n = 24) | χ2 or t-test | p < 0.05 | |
|---|---|---|---|---|
| age | 42.7 ± 15.3 | 52.2 ± 12.1 | t35 = −2.07 | p = 0.046 |
| gender (female) | 1 (8%) | 3 (13%) | 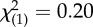 |
n.s. |
| education (years) | 13.2 ± 1.8 | 14.5 ± 2.3 | t35 = −1.78 | n.s. |
| full-time employed | 6 (46%) | 11 (46%) | 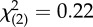 |
n.s. |
| ethnicity (hispanic) | 5 (39%) | 11 (46%) | 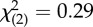 |
n.s. |
| psychiatric medications | 6 (46%) | 18 (75%) | 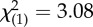 |
n.s. |
| body mass index (BMI) | 30.7 ± 4.4 | 28.8 ± 3.9 | t35 = 1.36 | n.s. |
| combat exposurea | 11.2 ± 3.2 | 9.6 ± 4.2 | t35 = 1.15 | n.s. |
| combat era (OIF/OEF) | 9 (69%) | 11 (46%) | 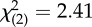 |
n.s. |
| age at first traumab | 22.7 ± 13.4 | 14.9 ± 7.5 | t35 = 2.27 | p = 0.029 |
| duration since combat trauma (years) | 14.7 ± 18.2 | 24.5 ± 18.5 | t35 = −1.55 | n.s. |
| social supportc | 79.5 ± 20.6 | 60.0 ± 15.8 | t35 = 3.21 | p = 0.003 |
| current clinician-rated PTSD severityd | 74.7 ± 18.6 | 84.2 ± 18.0 | t35 = −1.51 | n.s. |
| depressione | 19.9 ± 8.0 | 24.2 ± 10.4 | t35 = −1.30 | n.s. |
| anxietyf | 63.5 ± 17.6 | 71.5 ± 16.7 | t32 = −1.34 | n.s. |
| global impairmentg | 48.5 ± 15.6 | 37.8 ± 13.3 | t34 = 2.17 | p = 0.037 |
| other diagnosesh | ||||
| current major depressive disorder | 2 (15%) | 11 (46%) | 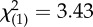 |
n.s. |
| past alcohol abuse/dependence | 3 (23%) | 11 (46%) | 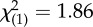 |
n.s. |
aDeployment Risk and Resilience Inventory, Combat Subscale (DRRI-I).
bTrauma History Questionnaire (THQ).
cInterpersonal Support Evaluation List (ISEL).
dClinician Administered PTSD Scale (CAPS).
eBeck Depression Inventory (BDI).
fState-Trait Anxiety Inventory (STAI).
gSF-36 Health Survey, Mental Health (SF-36).
hStructured Clinical Interview for the DSM-IV (SCID).
3.3. Treatment effects
There were no differences in drop-out rates associated with treatment condition: 12 out of 15 (80%) randomized to MA completed, and 25 out of 37 (68%) randomized to PE completed (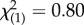 , p = 0.370). In the PE group, there were 11 responders and 14 non-responders; in the MA group, two responders and 10 non-responders. Chi-squared analysis does not show a significant difference in the proportion of responders and non-responders for the two treatment conditions (
, p = 0.370). In the PE group, there were 11 responders and 14 non-responders; in the MA group, two responders and 10 non-responders. Chi-squared analysis does not show a significant difference in the proportion of responders and non-responders for the two treatment conditions (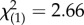 , p = 0.103).
, p = 0.103).
In order to determine whether there were any specific effects of treatment, a repeated measures ANOVA was conducted using treatment type as a between subjects factor. For the primary clinical outcome—PTSD symptom severity as measured by the CAPS total score—there was no significant treatment group by time interaction (F1,35 = 1.09, p = 0.303), signifying that post-treatment symptom severity was not a function of MA or PE. A significant main effect of time showed improvement from pre- to post-treatment for both groups (F1,35 = 16.52, p < 0.0005). An average reduction of 23 points on the CAPS (28.7%) was observed in those treated with PE, and those treated with MA showed a mean 14 point reduction (17.2%), a difference that failed to reach significance using χ2 analysis. Therefore, remaining analyses were conducted using treatment response or symptom severity as the primary outcome, regardless of treatment condition. When analyses were conducted additionally covarying for treatment modality, all results retained their significance. Additionally, when two-way ANCOVAs were performed evaluating treatment modality and responder status for each biological outcome, there were no treatment type by responder status interactions, indicating that type of treatment did not moderate the relationship between response status and the biological indicators. Finally, when the above analyses were repeated in the PE sample alone, results were unchanged other than in a manner that could be attributed to the reduction in sample size.
When data were subdivided by the conservative response status of no longer meeting criteria for PTSD, the group differences at post-treatment were extremely large (F1,35 = 38.48, p < 0.0005). Regarding the secondary outcomes, for depression there was a main effect of time (F1,37 = 5.45, p = 0.025) reflecting that depression scores were lower at post-treatment for the entire sample. There were no main effects or interactions with respect to anxiety and general mental health functioning. PTSD responders also showed significant improvement in all secondary measures at post-treatment (p < 0.001 for all measures).
3.4. Relationship between glucocorticoid receptor genotype (BCLI polymorphism), response status and glucocorticoid sensitivity
Chi-squared analysis demonstrated that responders were more likely to carry the GG or GC genotype than the CC genotype (71% versus 30%; 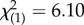 , p = 0.013; table 2). The functional significance of this finding is illustrated in figure 1, which shows a significant genotype by time interaction (F1,26 = 7.33, p = 0.012) with respect to glucocorticoid sensitivity (reflected by an increased lysozyme IC50-DEX), such that those with GG or GC genotypes showed a decrease in glucocorticoid sensitivity following treatment while those with the CC genotype did not. As will be described below, this relationship corresponds to the association of responder status with the change in lysozyme IC50-DEX over time.
, p = 0.013; table 2). The functional significance of this finding is illustrated in figure 1, which shows a significant genotype by time interaction (F1,26 = 7.33, p = 0.012) with respect to glucocorticoid sensitivity (reflected by an increased lysozyme IC50-DEX), such that those with GG or GC genotypes showed a decrease in glucocorticoid sensitivity following treatment while those with the CC genotype did not. As will be described below, this relationship corresponds to the association of responder status with the change in lysozyme IC50-DEX over time.
Table 2.
Pre-treatment biological characteristics of responders and non-responders.
| responders (n = 13) | non-responders (n = 24) | χ2 or ANOVA/ ANCOVAc | p < 0.05 | |
|---|---|---|---|---|
| genotype | ||||
| BCLI (GG or GC) | 10 (71%) | 7 (30%) | 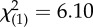 |
p = 0.013 |
| glucocorticoid sensitivity | ||||
| lysozyme IC-50DEX | 3.8 ± 2.2 | 3.9 ± 1.9 | F1,30 = 0.01 | n.s. |
| glucocorticoid circadian rhythm | ||||
| bedtime cortisol | 374.3 ± 268.4 | 254.7 ± 184.2 | F1,35 = 2.57 | n.s. |
| 24 h urinary cortisola | 56.1 ± 6.8 | 60.9 ± 30.3 | F1,34 = 0.32 | n.s. |
| resilience | ||||
| DHEA/cortisol ratiob | 0.7 ± 0.1 | 0.7 ± 0.1 | F1,33 = 0.00d | n.s. |
| NPYa,b | 75.8 ± 5.6 | 58.9 ± 3.9 | F1,32 = 5.69 | p = 0.023 |
Covariates: agender, bage.
cMean ± s.d. provided for ANOVAs, and estimated marginal mean ± s.e. for ANCOVAs.
dData were substantially skewed; statistic based on analysis following natural log transformation; unlogged mean ± s.e. displayed.
Figure 1.
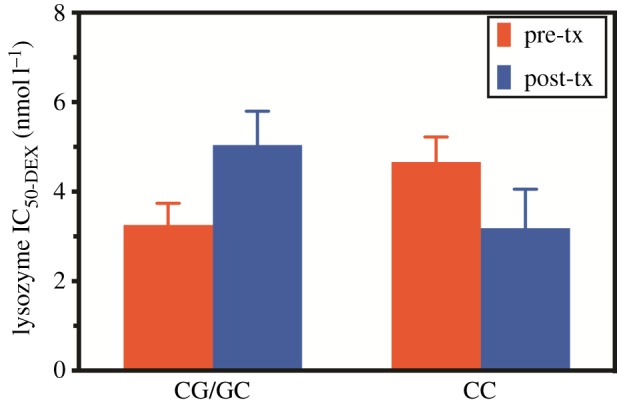
Glucocorticoid sensitivity at pre- and post-treatment depending on the GR BCLI polymorphism genotype (CG/GC or CC). Glucocorticoid sensitivity was measured with the lysozyme inhibition test in cultured peripheral blood mononuclear cells stimulated by varying doses of DEX. Pre-treatment measures are represented by blue (dark grey) bars and post-treatments measures by red (light grey) bars. Data were adjusted for age, gender and body mass index and are represented as mean ± s.e.m. Statistical significance was set at p < 0.05. (Online version in colour.)
3.5. Circadian and endocrine predictors of treatment response
Although no measure reflecting glucocorticoid circadian rhythm was significantly different based on response status at post-treatment, higher pre-treatment salivary cortisol at bedtime predicted greater change in PTSD symptom severity reflected by the change in total CAPS scores from pre- to post-treatment (r = 0.386, d.f. = 34, p = 0.020) and follow-up (r = 0.497, d.f. = 26, p = 0.007), controlling for gender. Pre-treatment 24 h urinary cortisol excretion predicted CAPS scores at post-treatment (r = 0.350, d.f. = 32, p = 0.042), self-reported PTSD symptoms on the PSS-SR at post-treatment (r = 0.454, d.f. = 31, p = 0.008) and the secondary outcome of self-reported depression on the BDI (r = 0.420, d.f. = 32, p = 0.013, all controlling for age, gender and BMI).
With respect to resilience-related measures, pre-treatment plasma NPY predicted treatment response status (table 2) (F1,32 = 5.69, p = 0.023, controlling for age and gender). Pre-treatment plasma NPY also predicted post-treatment self-reported global mental health (r = 0.403, d.f. = 31, p = 0.023) and anxiety on the STAI-S (r =−0.373, d.f. = 29, p = 0.039), and depression as measured by the BDI at both post-treatment and follow-up (r =−0.329, d.f. = 32, p = 0.058; r =−0.466, d.f. = 25, p = 0.014), all covaried for age and gender. The DHEA/cortisol ratio at pre-treatment was associated with post-treatment symptom severity as assessed by the PSS-SR, such that a lower pre-treatment ratio was correlated with a lower level of self-reported PTSD symptoms at post-treatment (r = 0.336, d.f. = 32, p = 0.052, controlling for age). Lower pre-treatment DHEA/cortisol also predicted lower post-treatment anxiety on the STAI-S (r = 0.390, d.f. = 30, p = 0.027, controlling for age).
3.6. Endocrine changes associated with treatment response
Figure 2 shows endocrine changes associated with treatment response. There was a significant group × time interaction for glucocorticoid sensitivity as assessed by the lysozyme IC50-DEX (F1,28 = 4.55, p = 0.042; figure 2a), indicating that, at post-treatment, responders demonstrated a decrease in glucocorticoid sensitivity, whereas non-responders tended to show a slight increase in glucocorticoid sensitivity. Changes in glucocorticoid sensitivity were associated with symptom improvement as evidenced by the significant correlation of pre- to post-treatment changes in lysozyme IC50-DEX with pre- to post-treatment changes in total CAPS scores (r =−0.371, n = 30, p = 0.044) as well pre-treatment to follow-up CAPS alterations (r =−0.519, n = 22, p = 0.013).
Figure 2.
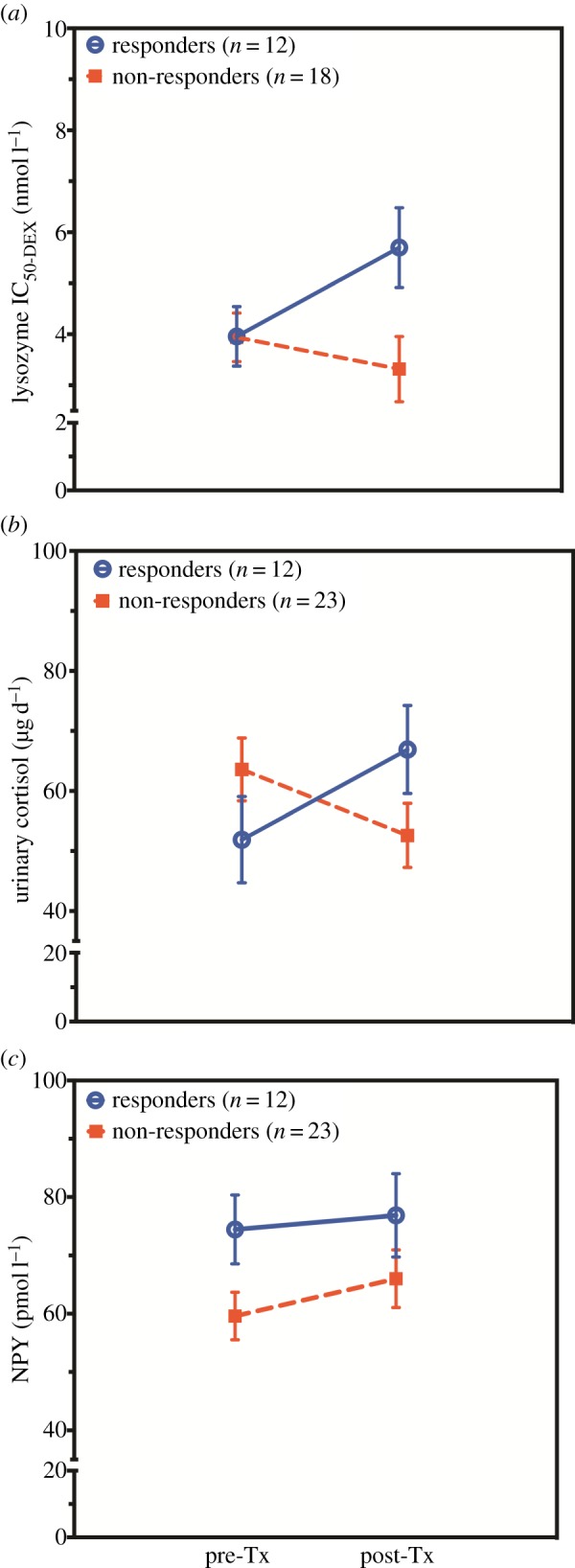
Biological predictors and treatment associated alterations from pre- to post-treatment for responders and non-responders: (a) peripheral blood mononuclear cells glucocorticoid sensitivity, (b) 24-h urinary cortisol excretion and (c) neuropeptide Y (NPY). Responders to treatment are represented by open blue circles and non-responders by filled red squares. All values are represented as mean ± s.e. NPY values were adjusted for age. Urinary cortisol values were adjusted for gender. Statistical significance was set at p < 0.05. (Online version in colour.)
There was a significant group by time interaction indicating that the change in 24 h urinary cortisol excretion from pre- to post-treatment was significantly different for responders than for non-responders (F1,33 = 5.76, p = 0.022, controlling for gender; figure 2b). Urinary cortisol levels increased for responders, and decreased for non-responders. In this case, the change in CAPS from lifetime (i.e. worst episode) to post-treatment was inversely correlated with change in urinary cortisol from pre- to post-treatment (r =−0.327, n = 36, p = 0.052), indicating that an increase in urinary cortisol was associated with a positive outcome. There were no effects of time or group × time interactions for measures reflecting salivary circadian rhythm.
Figure 2c demonstrates a different pattern in that plasma NPY increased over time for both responders and non-responders to treatment (F1,33 = 4.60, p = 0.040, covaried for age). There were no effects of time or group × time interactions observed for the DHEA/cortisol ratio.
3.7. Cross-sectional correlations at post-treatment
At post-treatment, glucocorticoid sensitivity reflected by the lysozyme IC50-DEX was correlated with self-reported measures of PTSD severity on the PSS-SR (r =−0.422, n = 29, p = 0.023), depression on the BDI (r =−0.361, n = 30, p = 0.050), and anxiety on the STAI-S (r =−0.500, n = 27, p = 0.006), indicating the utility of this measure as a reflection of state.
4. Discussion
A longitudinal approach in the context of a treatment study was used to distinguish biological predictors of recovery and biological correlates of PTSD and related symptoms. Biological measures that were found to be significant predictors of recovery, as defined by the absence of PTSD following treatment, were GR BCLI genotype and the resilience-related marker plasma NPY. Pre-treatment 24 h urinary cortisol excretion also predicted PTSD severity at post-treatment. Additionally, a measure of cortisol circadian rhythmicity, bedtime salivary cortisol, predicted the change in PTSD severity from pre- to post-treatment and follow-up.
Biological markers that changed from pre- to post-treatment in association with responder status were glucocorticoid sensitivity, as assessed by the lysozyme IC50-DEX, and 24 h urinary cortisol levels. Significant cross-sectional correlations at post-treatment between the lysozyme IC50-DEX and PTSD symptom severity, and PTSD-associated symptoms of depression and anxiety, confirmed the utility of this biomarker in association with state.
4.1. Effect of treatment on symptoms
The rationale for including MA as an alternative intervention was the concern that there might be an insufficient number of treatment non-responders to PE based on civilian efficacy studies. However, a significant advantage of PE over MA in reducing PTSD was not demonstrated for treatment completers. PE is considered a first line treatment for combat veterans with PTSD [41], though the effect size for this treatment in combat veterans is not as high as in civilians [42]. Having a biological predictor of positive response to PE, or any other psychotherapy, would be highly advantageous in a clinical environment [2].
4.2. Biological prediction of treatment response: implications of BCLI polymorphism
A positive treatment response was more common among G-carriers than in those homozygous for the wild-type C-allele. Bachmann et al. [26] previously demonstrated that the G allele was associated with higher GR sensitivity in Vietnam veterans. By contrast, van Zuiden et al. [43] recently reported that differences in BCLI genotype were not associated with the observation of an increased GR number at pre-deployment in veterans at increased risk for PTSD development following combat exposure. Thus, the BCLI genotype may associate with GR responsiveness as it is manifest in persons after they develop PTSD (i.e. as a prognostic indicator, in this case, of recovery in a group of veterans with PTSD), but may not be associated with the PTSD risk factor of increased GR number prior to trauma exposure, or even with the development of PTSD following exposure. Indeed, although allelic variations of this gene have been associated with GR responsiveness in a population-based cohort of more than 7000 healthy persons [25,44], they were not associated with PTSD risk per se, nor has this gene been identified in any genome-wide association studies in PTSD to date [45]. In this study, the BCLI genotype was not observed to associate with glucocorticoid sensitivity as measured by the lymphocyte lysozyme IC50-DEX at baseline within the narrow range of severe PTSD observed in this pre-treatment sample, but predicted the change in this measure in response to treatment.
4.3. Glucocorticoid sensitivity alterations associate with symptom improvement
The lysozyme IC50-DEX, an in vitro index of glucocorticoid sensitivity in PBMCs, and 24 h urinary cortisol excretion, a measure of adrenal glucocorticoid production, do not distinguish responders and non-responders at pre-treatment. However, both measures change over the course of treatment in association with responder status. As such, each may provide a clue to mechanisms of therapeutic change and may represent potential targets for treatment development.
Higher levels of bedtime salivary cortisol also predicted the change in PTSD severity from pre- to post-treatment and follow-up. Elevated levels of bedtime cortisol reflect a greater dynamic range of cortisol throughout the diurnal cycle, as previously observed in PTSD [46]. Cortisol circadian rhythmicity is thought to reflect a more upstream process that may in fact regulate glucocorticoid sensitivity in target tissues in a gene-specific fashion [47].
4.4. Resilience-related prognostic markers
NPY and DHEA/cortisol were associated with the prediction of treatment response to psychotherapy. The DHEA/cortisol ratio has been proposed as a resilience marker and associated with successful adaptation to stress [48]. DHEA is known to have neuroprotective and functionally enhancing properties in animal studies [49] and has been linked with resilience-related qualities such as self-efficacy, optimism, social support and well-being in a non-clinical sample [50]. We and others have reported increased DHEA and DHEA-S in association with recovery from PTSD in combat veterans [38,51], a finding that has also been observed in civilian trauma survivors with PTSD [14]. More recently, DHEA was demonstrated to have anabolic properties, whereas DHEA-S has been shown to have neuroprotective qualities in warfighters, a fact that has led to treatment trials with DHEA and DHEA-S supplementation during military training [48,52].
NPY is a peptide with behaviourally relevant effects on the hippocampus and is thought to function as an anxiolytic [53]. There are important functional interactions between NPY and corticotropin-releasing factor (CRF) such that NPY counteracts the anxiogenic effects of CRF in brain regions like the amygdala [53]. NPY also has counter-regulatory effects on catecholamines in many brain areas associated with anxiety, fear and depression [54,55]. Preliminary studies have demonstrated that persons under extreme stress with high NPY levels show better performance than those with low levels of NPY [56]. Similarly, patients with PTSD have reduced baseline plasma NPY levels and a blunted yohimbine-induced NPY increase in comparison to controls [57]. Plasma NPY levels were directly associated with recovery from PTSD, as they were significantly higher in combat veterans with past, but not current PTSD, compared with veterans with current PTSD or veterans who never developed PTSD [38]. More recently, NPY at post-treatment was predicted by pre-treatment GR 1F promoter methylation in a subset of this sample [9]. The association of NPY not only with PTSD reduction, but also with reduction in secondary outcome measures suggests that this peptide is a general marker of resilience rather than a specific indicator of reduced PTSD severity. In repeated measures ANCOVA, NPY was the only marker that was associated with treatment regardless of responder status, indicating that plasma NPY not only predicted change, but was itself changed even following more modest treatment gains.
4.5. State-related markers: glucocorticoid sensitivity
In this study, glucocorticoid sensitivity clearly emerged as a state-related marker for PTSD symptom severity at post-treatment using the lysozyme IC50-DEX. Notably, GR sensitivity as reflected by the lysozyme IC50-DEX was predicted by the BCLI genotype. Glucocorticoid sensitivity has been associated with PTSD risk, but was not associated with prediction of treatment response in this study.
4.6. Interaction between biological and psychological indices of post-traumatic stress disorder
In addition to the ability to detect potential clinically useful biomarkers, examinations of biological measures before and after treatment can also serve the purpose of enhancing our understanding of mechanisms underlying PTSD symptoms and recovery. We have previously suggested that enhanced glucocorticoid circadian rhythmicity in the context of lower cortisol and greater GR responsiveness reflects a greater capacity for engagement with the environment, and have contrasted the PTSD HPA-axis phenotype with that associated with major depression in which greater cortisol levels, decreased GR responsiveness and flattened glucocorticoid circadian rhythm result in a decreased capacity for environmental engagement [19]. It may be that in the absence of a therapeutic environment, the HPA-axis adaptations of PTSD lead to exaggerated or overgeneralized responses to traumatic triggers. However in the presence of positive or guided therapeutic influences these biological changes may catalyse greater therapeutic gains.
5. Summary
In sum, the results of this study suggest that G-carriers of the GR BCLI polymorphism, higher levels of plasma NPY, a lower DHEA/cortisol ratio, and lower levels of 24 h urinary cortisol excretion at pre-treatment predict recovery from PTSD or less severe PTSD symptom ratings at post-treatment. Higher bedtime salivary cortisol at pre-treatment also predicted greater symptom reduction in PTSD severity at post-treatment. Glucocorticoid sensitivity was reduced in concert with treatment-related PTSD symptom reduction. Other pre-treatment factors that emerged as predictors of a positive response included higher social support. The combination of these factors may serve to create a profile of a good treatment responder. The fact that large response differences did not emerge between people assigned to PE versus MA suggests that even minimal interventions can be effective in reducing symptoms of PTSD in veterans. Factors that changed in association with treatment response were related to glucocorticoid sensitivity, suggesting that they may serve as markers of treatment response. Finally, NPY emerged as the only marker that changed over time with symptom change, but not differentially in responders versus non-responders. This suggests that NPY is a general marker of state-related distress, including changes in PTSD and depressive symptoms.
While the range of conflicts and eras represented in the sample increase generalizability, the relatively small sample size is a limitation of this study, particularly regarding the genotype finding. Replication in larger studies with racially diverse samples of men and women is needed.
Funding statement
Funding for this study was provided by a grant from the Department of Defense to R.Y. (W81XWH-06-0032). The Department of Defense had no further role in the study design, in the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
References
- 1.Yehuda R, Neylan TC, Flory JD, McFarlane AC. 2013. The use of biomarkers in the military: from theory to practice. Psychoneuroendocrinology 38, 1912–1922. ( 10.1016/j.psyneuen.2013.06.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehrner A, Yehuda R. In press Biomarkers of PTSD: military applications and considerations. Eur. J. Psychotraumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yehuda R, Koenen KC, Galea S, Flory JD. 2011. The role of genes in defining a molecular biology of PTSD. Dis. Markers 30, 67–76. ( 10.3233/DMA-2011-0794) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glatt SJ, et al. 2013. Blood-based gene-expression predictors of PTSD risk and resilience among deployed marines: a pilot study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 313–326. ( 10.1002/ajmg.b.32167) [DOI] [PubMed] [Google Scholar]
- 5.Yehuda R, LeDoux J. 2007. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56, 19–32. ( 10.1016/j.neuron.2007.09.006) [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Li H, Benedek D, Li X, Ursano R. 2009. A strategy for the development of biomarker tests for PTSD. Med. Hypotheses 73, 404–409. ( 10.1016/j.mehy.2009.02.038) [DOI] [PubMed] [Google Scholar]
- 7.Yehuda R, Flory JD, Southwick S, Charney DS. 2006. Developing an agenda for translational studies of resilience and vulnerability following trauma exposure. Ann. NY Acad. Sci. 1071, 379–396. ( 10.1196/annals.1364.028) [DOI] [PubMed] [Google Scholar]
- 8.Heinrichs M, Wagner D, Schoch W, Soravia LM, Hellhammer DH, Ehlert U. 2005. Predicting posttraumatic stress symptoms from pretraumatic risk factors: a 2-year prospective follow-up study in firefighters. Am. J. Psychiatry 162, 2276–2286. ( 10.1176/appi.ajp.162.12.2276) [DOI] [PubMed] [Google Scholar]
- 9.Yehuda R, et al. 2013. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front. Psychiatry 4, 118 ( 10.3389/fpsyt.2013.00118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindauer RJ, Vlieger EJ, Jalink M, Olff M, Carlier IV, Majoie CB, Den Heeten GJ, Gersons BP. 2005. Effects of psychotherapy on hippocampal volume in out-patients with post-traumatic stress disorder: a MRI investigation. Psychol. Med. 35, 1421–1431. ( 10.1017/S0033291705005246) [DOI] [PubMed] [Google Scholar]
- 11.Levy-Gigi E, Szabo C, Kelemen O, Keri S. 2013. Association among clinical response, hippocampal volume, and FKBP5 gene expression in individuals with posttraumatic stress disorder receiving cognitive behavioral therapy. Biol. Psychiatry 74, 793–800. ( 10.1016/j.biopsych.2013.05.017) [DOI] [PubMed] [Google Scholar]
- 12.Bryant RA, Felmingham K, Kemp A, Das P, Hughes G, Peduto A, Williams L. 2008. Amygdala and ventral anterior cingulate activation predicts treatment response to cognitive behaviour therapy for post-traumatic stress disorder. Psychol. Med. 38, 555–561. ( 10.1017/S0033291707002231) [DOI] [PubMed] [Google Scholar]
- 13.Lindauer RT, van Meijel EP, Jalink M, Olff M, Carlier IV, Gersons BP. 2006. Heart rate responsivity to script-driven imagery in posttraumatic stress disorder: specificity of response and effects of psychotherapy. Psychosom. Med. 68, 33–40. ( 10.1097/01.psy.0000188566.35902.e7). [DOI] [PubMed] [Google Scholar]
- 14.Olff M, de Vries GJ, Guzelcan Y, Assies J, Gersons BP. 2007. Changes in cortisol and DHEA plasma levels after psychotherapy for PTSD. Psychoneuroendocrinology 32, 619–626. ( 10.1016/j.psyneuen.2007.04.001) [DOI] [PubMed] [Google Scholar]
- 15.Yehuda R, Bierer LM, Pratchett LC, Pelcovitz M. 2010. Using biological markers to inform a clinically meaningful treatment response. Ann. NY Acad. Sci. 1208, 158–163. ( 10.1111/j.1749-6632.2010.05698.x) [DOI] [PubMed] [Google Scholar]
- 16.Gerardi M, Rothbaum BO, Astin MC, Kelley M. 2010. Cortisol response following exposure treatment for PTSD in rape victims. J. Aggression, Maltreatment Trauma 19, 349–356. ( 10.1080/10926771003781297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mousavijazi M, Naderan A, Ebrahimpoor M, Sadeghipoor M. 2013. Association between psychological stress and stimulation of inflammatory responses in periodontal disease. J. Dent. 10, 103–111. [PMC free article] [PubMed] [Google Scholar]
- 18.Fredrickson BL, Grewen KM, Coffey KA, Algoe SB, Firestine AM, Arevalo JM, Ma J, Cole SW. 2013. A functional genomic perspective on human well-being. Proc. Natl Acad. Sci. USA 110, 13 684–13 689. ( 10.1073/pnas.1305419110). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yehuda R. 2002. Post-traumatic stress disorder. N. Engl. J. Med. 346, 108–114. ( 10.1056/NEJMra012941) [DOI] [PubMed] [Google Scholar]
- 20.Ehring T, Ehlers A, Cleare AJ, Glucksman E. 2008. Do acute psychological and psychobiological responses to trauma predict subsequent symptom severities of PTSD and depression? Psychiatry Res. 161, 67–75. ( 10.1016/j.psychres.2007.08.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Zuiden M, Geuze E, Willemen HL, Vermetten E, Maas M, Heijnen CJ, Kavelaars A. 2011. Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment. Am. J. Psychiatry 168, 89–96. ( 10.1176/appi.ajp.2010.10050706) [DOI] [PubMed] [Google Scholar]
- 22.Daskalakis NP, Lehrner A, Yehuda R. 2013. Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol. Metab. Clin. North Am. 42, 503–513. ( 10.1016/j.ecl.2013.05.004) [DOI] [PubMed] [Google Scholar]
- 23.Feder A, Nestler EJ, Charney DS. 2009. Psychobiology and molecular genetics of resilience. Nat. Rev. Neurosci. 10, 446–457. ( 10.1038/nrn2649nrn2649[pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foa EB, Hembree EA, Cahill SP, Rauch SA, Riggs DS, Feeny NC, Yadin E. 2005. Randomized trial of prolonged exposure for posttraumatic stress disorder with and without cognitive restructuring: outcome at academic and community clinics. J. Consult. Clin. Psychol. 73, 953–964. ( 10.1037/0022-006X.73.5.953) [DOI] [PubMed] [Google Scholar]
- 25.Derijk RH, de Kloet ER. 2008. Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. Eur. J. Pharmacol. 583, 303–311. ( 10.1016/j.ejphar.2007.11.072) [DOI] [PubMed] [Google Scholar]
- 26.Bachmann AW, Sedgley TL, Jackson RV, Gibson JN, Young RM, Torpy DJ. 2005. Glucocorticoid receptor polymorphisms and post-traumatic stress disorder. Psychoneuroendocrinology 30, 297–306. ( 10.1016/j.psyneuen.2004.08.006) [DOI] [PubMed] [Google Scholar]
- 27.Foa E, Hembree E, Rothbaum BO. 2007. Prolonged exposure therapy for PTSD: emotional processing of traumatic experiences therapist guide. Oxford, UK: Oxford University Press. [Google Scholar]
- 28.Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, Keane TM. 1995. The development of a Clinician-Administered PTSD Scale. J. Trauma. Stress 8, 75–90. ( 10.1002/jts.2490080106) [DOI] [PubMed] [Google Scholar]
- 29.Foa EB, Riggs DS, Dancu CV, Rothbaum BO. 1993. Reliability and validity of a brief instrument for assessing post-traumatic stress disorder. J. Trauma. Stress 6, 459–473. ( 10.1002/jts.2490060405) [DOI] [Google Scholar]
- 30.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. 1961. An inventory for measuring depression. Arch. Gen. Psychiatry 4, 561–571. ( 10.1001/archpsyc.1961.01710120031004) [DOI] [PubMed] [Google Scholar]
- 31.Spielberger CD. 1989. State-trait anxiety inventory: a comprehensive bibliography, 2nd edn Palo Alto, CA: Mind Garden. [Google Scholar]
- 32.Ware JE, Jr, Sherbourne CD. 1992. The MOS 36-item short-form health survey (SF-36): I. Conceptual framework and item selection. Med. Care 30, 473–483. ( 10.1097/00005650-199206000-00002) [DOI] [PubMed] [Google Scholar]
- 33.King LA, King DW, Vogt DS, Knight J, Samper RE. 2006. Deployment risk and resilience inventory: a collection of measures for studying deployment-related experiences of military personnel and veterans. Military Psychol. 18, 89–120. ( 10.1207/S15327876mp1802_1) [DOI] [Google Scholar]
- 34.Brookings JB, Bolton B. 1988. Confirmatory factor analysis of the interpersonal support evaluation list. Am. J. Community Psychol. 16, 137–147. ( 10.1007/BF00906076) [DOI] [PubMed] [Google Scholar]
- 35.First MB, Spitzer RL, Gibbon M, Williams JBW. 2002 Structured Clinical Interview for DSM-IV-TR Axis I Disorders, research version, patient edition (SCID-I/P). New York, NY: Biometrics Research, New York State Psychiatric Institute. [Google Scholar]
- 36.Yehuda R, Golier JA, Yang RK, Tischler L. 2004. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol. Psychiatry 55, 1110–1116. ( 10.1016/j.biopsych.2004.02.010) [DOI] [PubMed] [Google Scholar]
- 37.Lee LG, Connell CR, Bloch W. 1993. Allelic discrimination by nick-translation PCR with fluorgenic probes. Nucleic Acids Res. 21, 3761–3766. ( 10.1093/nar/21.16.3761) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yehuda R, Brand S, Yang RK. 2006. Plasma neuropeptide Y concentrations in combat exposed veterans: relationship to trauma exposure, recovery from PTSD, and coping. Biol. Psychiatry 59, 660–663. ( 10.1016/j.biopsych.2005.08.027) [DOI] [PubMed] [Google Scholar]
- 39.Yehuda R, Brand SR, Golier JA, Yang RK. 2006. Clinical correlates of DHEA associated with post-traumatic stress disorder. Acta Psychiatr. Scand. 114, 187–193. ( 10.1111/j.1600-0447.2006.00801.x) [DOI] [PubMed] [Google Scholar]
- 40.Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ. 1996. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol. Psychiatry 40, 79–88. ( 10.1016/0006-3223(95)00451-3) [DOI] [PubMed] [Google Scholar]
- 41.Rauch SA, Defever E, Favorite T, Duroe A, Garrity C, Martis B, Liberzon I. 2009. Prolonged exposure for PTSD in a veterans health administration PTSD clinic. J. Trauma Stress 22, 60–64. ( 10.1002/jts.20380) [DOI] [PubMed] [Google Scholar]
- 42.Bradley R, Greene J, Russ E, Dutra L, Westen D. 2005. A multidimensional meta-analysis of psychotherapy for PTSD. Am. J. Psychiatry 162, 214–227. ( 10.1176/appi.ajp.162.2.214) [DOI] [PubMed] [Google Scholar]
- 43.van Zuiden M, Geuze E, Willemen HL, Vermetten E, Maas M, Amarouchi K, Kavelaars A, Heijnen CJ. 2012. Glucocorticoid receptor pathway components predict posttraumatic stress disorder symptom development: a prospective study. Biol. Psychiatry 71, 309–316. ( 10.1016/j.biopsych.2011.10.026) [DOI] [PubMed] [Google Scholar]
- 44.Van Rossum EF, et al. 2003. Identification of the BclI polymorphism in the glucocorticoid receptor gene: association with sensitivity to glucocorticoids in vivo and body mass index. Clin. Endocrinol. 59, 585–592. ( 10.1046/j.1365-2265.2003.01888.x) [DOI] [PubMed] [Google Scholar]
- 45.Logue MW, et al. 2013. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol. Psychiatry 18, 937–942. ( 10.1038/mp.2012.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yehuda R, Teicher MH, Levengood RA, Trestman RL, Siever LJ. 1994. Circadian regulation of basal cortisol levels in posttraumatic stress disorder. Ann. NY Acad. Sci. 746, 378–380. ( 10.1111/j.1749-6632.1994.tb39260.x) [DOI] [PubMed] [Google Scholar]
- 47.Kino T. 2012. Circadian rhythms of glucocorticoid hormone actions in target tissues: potential clinical implications. Sci. Signal. 5, pt4 ( 10.1126/scisignal.2003333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH. 2009. Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front. Neuroendocrinol. 30, 65–91. ( 10.1016/j.yfrne.2008.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardounel A, Regelson W, Kalimi M. 1999. Dehydroepiandrosterone protects hippocampal neurons against neurotoxin-induced cell death: mechanism of action. Proc. Soc. Exp. Biol. Med. 222, 145–149. ( 10.1046/j.1525-1373.1999.d01-124.x) [DOI] [PubMed] [Google Scholar]
- 50.Petros N, Opacka-Juffry J, Huber JH. 2013. Psychometric and neurobiological assessment of resilience in a non-clinical sample of adults. Psychoneuroendocrinology 38, 2099–2108. ( 10.1016/j.psyneuen.2013.03.022) [DOI] [PubMed] [Google Scholar]
- 51.Spivak B, Maayan R, Kotler M, Mester R, Gil-Ad I, Shtaif B, Weizman A. 2000. Elevated circulatory level of GABA(A)—antagonistic neurosteroids in patients with combat-related post-traumatic stress disorder. Psychol. Med. 30, 1227–1231. ( 10.1017/S0033291799002731) [DOI] [PubMed] [Google Scholar]
- 52.Taylor MK, Padilla GA, Stanfill KE, Markham AE, Khosravi JY, Ward MD, Koehler MM. 2012. Effects of dehydroepiandrosterone supplementation during stressful military training: a randomized, controlled, double-blind field study. Stress 15, 85–96. ( 10.3109/10253890.2011.585189) [DOI] [PubMed] [Google Scholar]
- 53.Heilig M. 2004. The NPY system in stress, anxiety and depression. Neuropeptides 38, 213–224. ( 10.1016/j.npep.2004.05.002) [DOI] [PubMed] [Google Scholar]
- 54.Thorsell A, Carlsson K, Ekman R, Heilig M. 1999. Behavioral and endocrine adaptation, and up-regulation of NPY expression in rat amygdala following repeated restraint stress. Neuroreport 10, 3003–3007. ( 10.1097/00001756-199909290-00024) [DOI] [PubMed] [Google Scholar]
- 55.Guidi L, et al. 1999. Neuropeptide Y plasma levels and immunological changes during academic stress. Neuropsychobiology 40, 188–195. ( 10.1159/000026618) [DOI] [PubMed] [Google Scholar]
- 56.Morgan CA, III, Wang S, Southwick SM, Rasmusson A, Hazlett G, Hauger RL, Charney DS. 2000. Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol. Psychiatry 47, 902–909. ( 10.1016/S0006-3223(99)00239-5) [DOI] [PubMed] [Google Scholar]
- 57.Rasmusson AM, Hauger RL, Morgan CA, Bremner JD, Charney DS, Southwick SM. 2000. Low baseline and yohimbine-stimulated plasma neuropeptide Y (NPY) levels in combat-related PTSD. Biol. Psychiatry 47, 526–539. ( 10.1016/S0006-3223(99)00185-7) [DOI] [PubMed] [Google Scholar]