

Abstract
We have previously demonstrated the release of membranous structures by cells into their extracellular environment, which are termed exosomes, microvesicles or extracellular vesicles depending on specific characteristics, including size, composition and biogenesis pathway. With activation, injury, stress, transformation or infection, cells express proteins and RNAs associated with the cellular responses to these events. The exosomes released by these cells can exhibit an array of proteins, lipids and nucleic acids linked to these physiologic events. This review focuses on exosomes associated with traumatic brain injury, which may be both diagnostic and a causative factor in the progression of the injury. Based on current data, exosomes play essential roles as conveyers of intercellular communication and mediators of many of the pathological conditions associated with development, progression and therapeutic failures and cellular stress in a variety of pathologic conditions. These extracellular vesicles express components responsible for angiogenesis promotion, stromal remodelling, signal pathway activation through growth factor/receptor transfer, chemoresistance, immunologic activation and genetic exchange. These circulating exosomes not only represent a central mediator of the pro-inflammatory microenvironment linked with secondary brain injury, but their presence in the peripheral circulation may serve as a surrogate for biopsies, enabling real-time diagnosis and monitoring of neurodegenerative progression.
Keywords: exosomes, traumatic brain injury, neurodegeneration, inflammatory response, cytokines
1. Introduction to central nervous system injury
Brain injury can be the consequence of physical, chemical or biological insults and can result in disabling or potentially fatal consequences. There can be multiple outcomes resulting from different forms of brain injuries and these are difficult to predict. Owing to the inability to biopsy components of the central nervous system (CNS), injury-linked biomarkers are needed to define pathophysiological mechanisms and to predict neurological outcomes. To date, some of the brain-associated biomarkers assessed have included cytokines (such as interleukin (IL)-1β, IL-6, transforming growth factor-β (TGF-β)), S100B, glial proteins (such as glial fibrillary acidic protein and neurofilament light chain), and enzymes such as tissue transglutaminases), β-amyloid proteins and tau. The major clinical challenge to the accurate definition of prognosis is to identify the degree of CNS damage and the site of the lesion. A key goal is the development of assay markers that provide accurate prognosis in patients suffering from either acute or chronic CNS injuries, as well as definition of the extent and location of the primary injury, which is crucial for predicting mortality and morbidity. The review focuses on acute neurologic injuries.
Traumatic brain injury (TBI) is a significant public health issue with an estimated 1.6–3.8 million individuals experiencing TBI annually in the USA [1,2]. Of these, approximately 40% do not seek medical attention. For those seeking medical attention, the diagnosis of mild TBI is complicated by a lack of a consensus in defining mild TBI. Concussion and mild TBI are frequently used interchangeably. TBI commonly produces neurological morbidity and the neurologic consequences are seen even in mild TBI. Mild TBIs can lead to acute symptoms, including chronic traumatic encephalopathy, cognitive impairment, dementia, movement disorders and motor neuron dysfunction. Even within mild to moderate TBI, 51–54% exhibit long-term disability, along with 78% of those with severe TBI [3]. While the incidence of mild TBI is higher in the military population, a similar percentage do not seek medical attention. Between 2000 and 2011, Veterans Affairs medical records report approximately 7% of veterans from Iraq and Afghanistan were diagnosed with TBI [4,5]. Among military personnel who serve in Iraq or Afghanistan, 11–23% may have sustained at least mild TBI [6]. While many cases of TBI go unreported or unrecognized, US military records indicate 233 425 TBIs; among those 178 961 were listed as mild [6]. In Iraq and Afghanistan, mild TBIs have become more prominent due to the use of improvised explosive devices (IEDs), accounting for approximately 75% of all combat-related injuries to military personnel [7]. Approximately 50% of IED injuries result in mild TBIs, with the lifetime prevalence for mild TBI in US military personnel estimated to be 19.5%.
2. Diagnostic gap
Imaging modalities such as computed tomography (CT) and magnetic resonance imaging exhibit suboptimal sensitivities for detecting concussions (3–10% for CT and 10–57% for magnetic resonance imaging), since these are designed to detect major structural damage that may not be present in concussions [8]. For clinicians who evaluate and treat individuals with concussions, it is critical to definitively identify those who have sustained a concussion and those who are at risk for adverse outcomes. Most concussions present with vague symptoms. While signs and symptoms are difficult to quantify, approximately 1–2% of the population exhibit lifelong TBI-related disabilities [9]. These disabilities include memory impairment, diminished reasoning and impaired motor functions. In many cases, TBI, including concussions, can result in adverse outcomes, such as chronic traumatic encephalopathy, cognitive impairment, dementia, movement disorders and motor neuron dysfunction [10]. The inability to quantitatively assess individuals with concussions can also lead to the risk of ‘second-impact syndrome’ (SIS). SIS is a frequently fatal condition that occurs when an individual suffers a second concussion prior to resolution of the symptoms from the first concussion. For many sports, ‘return-to-play’ guidelines have been established to prevent SIS; however, the grading systems for defining symptoms and recovery are subjective. The most common scale is the Glasgow Coma Scale (GCS), which is used to assess the level of consciousness following a head trauma. However, the GCS was not intended to be used alone to define the severity of brain damage or to predict outcome. There are significant concerns regarding the poor inter-observer reliability and the lack of prognostic utility. Additionally, the failure of clinical trials is due in part to the difficulty of defining a quantitative severity scale to accurately stratify the array of TBI disease, and to the absence of biomarkers that reflect specific pathophysiological mechanisms. Currently, there remains a significant need for new diagnostic approaches to identify brain injury and to predict the risk of neurological deterioration.
3. Immunologic sequelae
In CNS injury, primary damage occurs as a direct result of trauma, leading to immediate physical, biochemical and cellular alterations. These alterations occur within seconds of the injury and are marked by systemic and local events. Diverse populations of cells and bioactive molecules from the nervous, immune and vascular systems are involved. The reactions of the immune system to acute injury are both cellular and molecular. At the injury site, the cumulative effect of the immune cells and regulatory proteins is inflammation [11]. Inflammation is a critical element in the secondary injury cascade that occurs immediately and can persist for weeks or months [12]. While the inflammatory response is essential for the clearance of cellular debris, the ‘over-activation’ of the inflammatory response can damage healthy tissue and exacerbate the primary injury. Microglia represent the innate immune cells of the CNS and these cells have a range of functions, which can vary dependent on time and the microenvironment [13]. Inflammation, resulting from the primary injury, attracts neutrophils, monocytes, microglia and T-lymphocytes [14]. Neutrophils release cytokines, proteases and free radicals, which activate other inflammatory and glial cells to perpetuate the inflammatory cascade, leading to neuron injury or death. Shortly after the primary injury, monocytes infiltrate and differentiate into macrophages, where activated microglia and macrophages secrete various pro-inflammatory cytokines, free radicals and growth factors [15,16].
The activated immune cells secrete pro-inflammatory cytokines, including IL-1β, IL-6 and tumour necrosis factor-α (TNF-α), all of which increase the extent of inflammation. Following CNS injury, there is a marked increase in the blood–brain barrier permeability due to both direct mechanical disruption by the primary injury and the effects on endothelial cells by numerous inflammatory mediators and other compounds that are upregulated. While in normal rat brain, IL-1β and its mRNA are present, both are increased in certain disease states, including ischaemic brain injury and TBI [17]. Two inflammatory cytokines (TNF-α and IL-1β) are upregulated and are known to increase vascular permeability. Increased production of IL-1β may contribute to injury processes by causing microvascular injury, which disrupts the blood–brain barrier and leads to oedema after brain injury [18]. Furthermore, IL-1β stimulates the proliferation and hypertrophy of astrocytes and thus may be causally involved in the formation of the astroglial scar that develops in the injured CNS. Studies with an antagonist of the IL-1β receptor suggest that neuronal damage produced by ischaemia is mediated in large part by IL-1β [19].
Currently, there is no clear consensus on the role of endogenous TNF-α in CNS acute injury. TNF-α is a key inflammatory cytokine that is upregulated in neurons, glia and endothelial cells and its production at the injury site appears to be involved in secondary tissue [20]. Investigators have shown the presence of TNF-α protein and mRNA at the sites of traumatic lesions within minutes and the TNF-α concentrations peak in the cerebrospinal fluid (CSF) within 24 h [21,22]. Six hours after TBI, TNF-α expression is higher in the CSF than in serum; TNF-α expression did not correlate with outcome [23]. The differential patterns of localization of TNF-α receptors in neuronal and glial cells, their state of activation and the downstream effectors are all thought to play an important role in determining whether TNF-α will exert a beneficial or harmful effect on the CNS. Additionally, TNF-α contributes to the tissue injury induced by neutrophils by directly activating them, as well as by increasing the expression of such molecules as E-selectin, which cause the activated neutrophils to adhere to the surface of the endothelial cells [24]. It has also been shown that the inhibition of neutrophil adhesion to the endothelial cell surface markedly reduces the severity of the CNS injury induced by compressive trauma.
4. Biomarkers
For clinicians who evaluate and treat individuals with mild TBI, it is critical to definitively identify those who have sustained a concussion and those who are at further risk for adverse outcomes. Most mild TBIs present as vague symptoms and imaging characteristics [25]. It is also difficult to monitor therapeutic responses; this failure is due in part to the difficulty of defining a quantitative severity scale to accurately stratify the array of TBI disease, and to the absence of biomarkers that reflect specific pathophysiological mechanisms.
Currently, functional neurologic measurements are used to stratify injury severity and predict neurologic outcome; however, in acutely injured patients these metrics are often difficult to determine. Biomarkers have potential utility as diagnostic, prognostic and therapeutic adjuncts in the setting of TBI. Circulating biomarkers have been investigated for the definitive diagnosis and monitoring of treatment for TBI. Such circulating biomarkers could also serve to monitor disease progression, by assessing tissue injury and predicting risk of neurological deterioration. They could be used to help determine which patients should receive which treatments. Two approaches are being used, namely, assessing markers of structural damage, and quantifying mediators of the cellular, biochemical or molecular cascades in secondary injury and repair. Circulating biomarkers have been proposed to be promising for the definitive diagnosis and monitoring of treatment in CNS injury. Defining TBI-linked biomarkers has numerous advantages, such as diagnosing the disease, identifying processes that are difficult to image (such as diffuse axonal injury), predicting outcome by identifying patients at risk for long-term neurocognitive consequences, defining injury-specific molecular and pathological alterations for developing therapeutic targets and monitoring responses to acute interventions [26,27]. These circulating biomarkers could also serve to monitor disease progression, by assessing tissue injury and predicting risk of neurological deterioration. However, circulating biomarkers are problematic and exhibit several critical issues. Free protein and nucleic acid biomarkers are extremely unstable in the circulation, thus a high steady-state must be reached for detection, which is generally not observed except in severe TBI. Minor changes over time (essential for monitoring) are difficult to quantify by current technologies. Furthermore, these current used biomarkers are sensitive to sample handling and require strict procedures for collecting, handling and processing. Thus, differentiating the degree of severity and predicting the extent of neurologic recovery remains enigmatic.
5. Exosomes
To circumvent these issues, we have initiated the use of exosome-associated biomarkers. Our work has demonstrated that these are extremely stable within the circulation, in the order of days (versus minutes for traditional soluble markers). In addition to serving as biomarkers of injury, our data demonstrate that exosomes may mediate in vitro events associated with TBI. Thus, their characterization may provide insight into the ‘driver’ mechanisms of neurodegenerative disease.
In 1979, our group discovered and published the initial observation of circulating exosomes and proposed their diagnostic potential [28]. The release of 50–200 nm-sized membranous vesicles into biological fluids by viable cells has since been demonstrated in multiple cell types and systems over the past 30 years. In vivo, these nano-sized vesicles released by tumour cells accumulate in biologic fluids, including blood, urine, ascites and pleural fluids [29]. These released vesicles have been identified by various terms, including high molecular weight complexes, membrane fragments, exosomes, microvesicles, microparticles and extracellular vesicles. The terms, ‘microvesicles’ and ‘microparticles’ appear to be misnomers as these are nanometres in size and should have been termed ‘nanovesicles’. Additional terms have been created to differentiate vesicles from specific cell types, such as ‘oncosomes’, ‘texosomes’ and ‘tumoursomes’ for tumour-derived vesicles, ‘prostasomes’ for prostate cell-derived vesicles and ‘dexosomes’ for dendritic cell-derived vesicles. The use of these various terms has only served to confuse the field. While, more recently, restrictive definitions have been applied to these cell-derived vesicles, significant overlap (in terms of size, markers, cargoes and function) exists between structures identified as exosomes and microvesicles. Within the circulation, it may not be possible to differentiate 50–100 nm ‘exosomes’ from 50 to 200 nm ‘microvesicles’. With recent technological developments, we are beginning to define the extremely complex compositions of these vesicles, which are likely to provide insights into their classifications, cellular and molecular origins and biologic functions.
In many studies, uncharacterized cell-derived vesicles (in terms of markers or size) are termed ‘microvesicles’, while numerous studies define ‘exosomes’ solely based on density and the presence of the cell surface markers tetraspanins. These overlaps in vesicle properties and terms suggest that these distinctions may not be clear-cut. While many of the definitions are still used, their flaws are now recognized. The apparent size and shape of exosomes are artefacts of fixation and drying associated with electron microscopy. Principal markers of exosomes are tetraspanins, which as plasma membrane associated components are present on most vesicles, regardless of their origin. The importance of the endocytic pathway of vesicle formation has also been questioned as knock-out studies with Rab proteins only diminished vesicle release by approximately 30% (based on exosomal protein) [30]. The ‘exosome’ term was coined in 1981 for any ‘exfoliated membrane vesicles with 5′-nucleotidase activity’ [31]. This term, ‘exosome’, originated from the discovery of neoplastic cell line-derived exfoliated vesicles, which mirrored the 5′-nucleotidase activity of the parent cells [31]. For these reasons, this review uses the term ‘exosomes’ to include all 50–200 nm released vesicles.
Over the past three decades, shed vesicles have been characterized for multiple human cell types; they are not exact replicates of the plasma membrane or other membranous compartments of the originating tumour cells, but they represent ‘micromaps’ with enhanced expression of tissue-specific antigens as well as other macromolecules, including major histocompatibility antigens [29]. Exosomes can be viewed as cytoplasm enclosed in a lipid layer with the external domains of transmembrane proteins exposed to the extracellular environment in their normal cellular orientation. Electron microscopic studies have demonstrated the fusion of the limiting membrane of multivesicular bodies (MVB) with the plasma membrane as well as release of intraluminal vesicles in different cell types of haematopoietic origin, such as Epstein–Barr virus-transformed B cells [32], mastocytes [33], dendritic cells [34], platelets [35], macrophages [36] and cells of non-haematopoietic origin such as neurons and epithelial cells [37]. We have developed genomic, proteomic and metabolomic technologies to define stable, disease-specific exosomal markers for detection, disease characterization and predicting prognosis [38]. These temporal changes in exosomal profiles have been demonstrated to accurately predict disease recurrence and overall patient survival [39]. The proteomic and genomic profiles of circulating exosomes provide a real-time monitor of therapeutic response, serving as a companion diagnostic. By correlating these circulating markers with the molecular characteristics and real-time clinical parameters, we have established the use of circulating exosomes as a ‘liquid biopsy’. In 2008, our group published the initial demonstration of circulating exosomal RNA for diagnosis [40]. Many studies have examined the diagnostic utility of profiling total circulating microRNA in specific pathologies. While there is a significant exosome contribution from immune cells, platelets and vascular endothelial cells, little is known regarding these normal vesicular RNA contents or how they respond to exogenous ‘stressors’. Proliferating and injured cells, both in vivo and in vitro, have been demonstrated to release membranous structures, defined as microvesicles or exosomes, consisting of an array of macromolecules derived from the originating cells, including proteins, lipids and nucleic acids. In the peripheral circulation of brain cancer patients, we have demonstrated the presence brain-derived exosomes (figure 1), based on the combined presence of β-amyloid, tau and S100β.
Figure 1.

Quantification of brain-derived exosomes in the circulation of a patient with brain cancer. Following chromatographic isolation, circulating exosomes were quantified using a NanoSight NS300. The x-axis presents the size range of observed vesicles, the y-axis the concentration of vesicles and the z-axis the relative intensity. Panel (a) shows the circulating total exosome population, defined by the NS300 in light scatter mode. Panel (b) identifies those vesicles expressing a marker of brain-derived exosomes (β-amyloid), determined using the NS300 in fluorescent mode.
While described over three decades, only recently have the roles of these vesicular components in intercellular communication become elucidated. Significant evidence has demonstrated that exosomes can exert a broad array of detrimental effects on the immune system, ranging from apoptosis of activated cytotoxic T cells to impairment of monocyte differentiation into dendritic cells, to induction of myeloid-suppressive cells and T regulatory cells. Through the expression of molecules involved in angiogenesis promotion, stromal remodelling, signalling pathway activation through growth factor/receptor transfer and genetic intercellular exchange, exosomes represent a central mediator of a tissue's microenvironment. TBI is associated with induction of inflammation, oxidative stress, oedema and hypoxia, which can occur over hours to weeks, leading to brain tissue damage. Following initial injury, inflammatory mediators play a critical role in mediating leucocyte adhesion and cellular stress responses. There is a dramatic elevation of pro-inflammatory cytokines within minutes of injury, in particular TNF-α, IL-1β, IL-6, IL-8, IL-10 and TGF-β. These cytokines are primarily produced by infiltrating lymphoid and mononuclear cells. Our work has demonstrated that these same pro-inflammatory cytokines are induced by specific exosomes (figure 2) from mononuclear cells. We have demonstrated that injury-derived exosomes mediate the production of pro-inflammatory cytokines, including IL-1β [41]. Following TBI, elevations in the IL-1 cytokine family are observed, including IL-1β, IL-1α, IL-1 receptor antagonist and IL-18. Of these, IL-1β is the most prominently enhanced cytokine following TBI [42]. After TBI, IL-1β is released primarily by microglia and acts as a pro-inflammatory pyrogen, increasing expression of other cytokines, proteases and matrix metalloproteinases (MMPs). IL-1β is a potent mitogen for astroglia and has been shown to be elevated within 10 days after injury [43]. Studies have demonstrated that IL-1β increases within the initial 24 h in the circulation and CSF [42]. These IL-1β levels may exhibit prognostic value, as CSF IL-1β levels have been shown to be greatest in severe CNS injuries with elevated intracranial pressures and unfavourable outcomes [44]. By contrast, IL-1ra, the receptor antagonist that diminishes IL-1 responses, has been shown to be greatest in patients with favourable outcomes [45].
Figure 2.
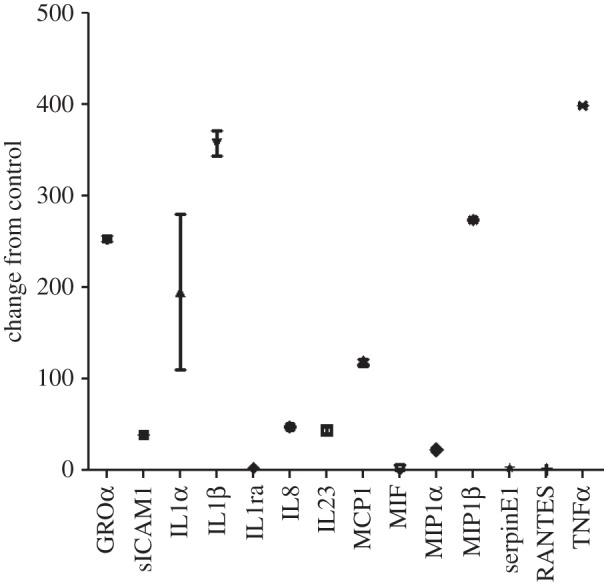
Induction of cytokines in human macrophages following exposure to 100 µg ml−1 exosomes. Human macrophages were incubated in the presence of 100 µg ml−1 exosomes for 20 h and the levels of specific cytokines were identified in the media by the Cytokine Profiler Array A. Exosomes were chromatographically isolated from pooled normal human serum (control) or from tumour cell conditioned media. Cytokine expression in tumour exosome-treated macrophages was expressed relative to the expression observed with addition of the ‘control’ exosomes.
6. Functions of exosomes
While some early studies implicated exosomes as ‘garbage bags’ of the cells [46,47], some vesicles released from tumour cells have gained increasing recognition as ‘vehicles’ for intercellular communication. Intercellular communication has been thought to be limited to cell-to-cell adhesion conduits (gap junctions) or secreted signals, such as hormones, neurotransmitters and cytokines, released from cells and acting in an autocrine or paracrine manner. These exosomes interact with the plasma membrane of a recipient cell by ligand/receptor binding, fusion or internalization (or a combination of these). If the exosomes fuse with the target cell, they can transfer their cargo to that recipient cell. Owing to the presence of cell-type-specific adhesion molecules, exosomes can interact with specific cells and deliver their ‘cargoes’, including bioactive lipids, cytokines, growth factors, receptors and genetic material. In this manner, exosomes represent a pathway for intercellular transfer of information, similar to that observed with direct cell–cell contact, but that can function at distance. Exosomes provide stable conformational conditions for their protein content, conserve bioactivity of their proteins, improve bio-distribution and support an efficient interaction with target cells.
The complexity of extracellular vesicle-associated bioactive macromolecules supports a critical role in generating the tumour microenvironment [48,49]. Exosomes can transfer specific proteins to target cells for the delivery of signalling pathways [50,51]. The presence of tumour-derived exosomes can increase MMP secretion and VEGF expression in target cells through the expression of pro-angiogenic molecules, such as members of the tetraspanin family, thereby promoting neo-angiogenesis even at secondary metastatic sites [52]. The released MMPs can digest the extracellular matrices where they arise. This degradation is enhanced when MMPs are co-released with exosome-associated extracellular matrix metalloproteinase inducer (EMMPRIN) [53].
Studies have shown that cancer ascites-derived exosomes carry extracellular matrix-remodelling enzymes, such as MMP-2 and MMP-9 [54,55], and urokinase plasminogen activator [56], leading to an increase in extracellular matrix degradation. The expression of matrix-remodelling enzymes increases the tumour's invasive phenotype and promotes metastasis. The presence of pro-angiogenic factors supports neovascularization of the developing tumour. A commonly identified cellular component of the tumour microenvironment is the monocyte/macrophage. Within the microenvironment, tumour-associated macrophages have been shown to assist in tumour progression by expressing cytokine/chemokine profiles that promote angiogenesis, stimulate tumour growth, and elicit immunosuppression by suppressing Th1 responses [57,58]. The tumour microenvironment is characterized by pro-inflammatory profiles, including IL-1β. This profile is generally produced by infiltrating macrophages following interactions with tumours or their components. While we proposed that exosomes could ‘educate’ macrophages to produce pro-inflammatory cytokines following internalization, we recently demonstrated that the induction of IL-1β was observed even when internalization of vesicles was blocked [59]. Arginine, glycine and aspartate containing peptides (RGD peptides), which are used to block fibronectin binding to macrophage α5β1 integrin, were observed to abrogate vesicle induction of IL-1β production and downstream phosphorylation of Akt and c-Jun [60]. This approach reveals the importance of receptor/ligand interactions in vesicle communication.
When released exosomes fuse with their target cells, they can transfer specific membrane components, including receptors and ligands, which can express an activated phenotype. This transfer of receptors from exosomes to target cells was demonstrated by the observation that bystander B cells acquire antigen receptors from activated B cells by membrane transfer [61]. This transfer allows the amplified expansion of antigen-binding B cells with the ability to present specific antigens to CD4 T cells. Exosomes can transfer the adhesion molecule CD41 from platelets to endothelial cells or to tumour cells, conferring pro-adhesive properties to the target cell [62]. Exosome-mediated transfer of Fas ligand from tumour cells induces apoptosis of activated T cells enabling tumour immune escape [63]. Exosomes can also be protective for tumour cells by removing molecules such as Fas or the membrane attack complex from their membranes.
The horizontal transfer of macromolecules and their functional consequences has been demonstrated in human gliomas [64,65]. In this model, only a fraction of the cells exhibiting a transformed phenotype expressed the truncated epidermal growth factor receptor, EGFRvIII, associated with dysregulated tumour growth [64]. Al-Nedawi et al. [65] demonstrated transfer of the oncogenic EGFRvIII from human glioma cancer cells expressing the receptor to glioma cells without the EGFRvIII via the fusion of exosomes. After transfer, the glioma cells originally lacking the receptor were transformed to express EGFRvIII-regulated genes, including VEGF, Bcl-xL and p27 [64]. Subsequent studies demonstrated that the oncogenic EGFRvIII from human squamous cell carcinoma cells was transferred via exosomes to tumour-associated endothelial cells to activate MAPK and Akt cell signalling pathways and promote endothelial VEGF expression [66].
Epigenetic changes have been frequently demonstrated in various tumours, resulting in regulation of gene transcription, altered proliferation, differentiation and therapeutic resistance [67]. Genetic information can be transferred through two proposed mechanisms: vertical gene transfer, which is gene exchange from parent to the next generation, and horizontal gene transfer, induced through, for example, bacteriophages or viruses. Since exosomes have been implicated as a potent source of macromolecule transfer to neighbouring and distant cells, viruses and other pathogens appear to exploit this system. Exosomes are postulated to contribute to the spread of infective agents, such as human immunodeficiency virus type 1 [68]. In macrophages receiving chemokine receptors, this can induce an increased risk of HIV infection together with resistance to apoptosis. The transfer of the chemokine (CXC motif) receptor 4 and the chemokine (CC motif) receptor 5 chemokine co-receptors for human immunodeficiency virus type I by released exosomes can enhance the entry of the virus into cell types other than the lympho-haemopoietic lineage [69]. In addition to transferring receptors, exosomes can transfer viruses, contained within exosomes, by the ‘Trojan exosome hypothesis’ involving direct delivery [70].
Cell-derived exosomes represent another mechanism of horizontal gene transfer. Genomic instability may be mediated by horizontal transfer of tumour-derived materials via exosomes. Horizontal transfer of macromolecules, including RNA, proteins and lipids, via exosomes has been shown in multiple tumour systems, including gliomas, monocytes, mast cells and T cells [71]. Tumour-derived exosomes have been shown to be capable of transferring surface components (proteins and lipids) and RNAs to monocytes. Janowska-Wieczorek et al. [72] demonstrated that exosomes derived from murine embryonic stem cells (ESCs) could induce epigenetic reprogramming of target cells. ESC-derived exosomes were shown to improve survival of haematopoietic stem/progenitor cells, to induce upregulation of early pluripotent and early haematopoietic markers, and to induce phosphorylation of mitogen-activated protein kinase p42/44 and Akt. ESC-derived exosomes were shown to express mRNAs for several pluripotent transcription factors that can be delivered to target cells and translated to their corresponding proteins [73]. As RNase treatment inhibited their exosome-mediated biological effect, the involvement of mRNA in the observed biological effects was suggested. Yuan et al. [74] have shown that in addition to mRNA, exosomes can transfer microRNA to target cells. They demonstrated that exosomes derived from ESCs contain abundant microRNA and that they can transfer a subset of microRNAs to mouse embryonic fibroblasts in vitro. Since microRNAs are regulators of protein translation, this observation raised the possibility that stem cells can alter the expression of genes in neighbouring cells by transferring exosomal microRNAs. When shed vesicles fuse with their target cells, the portion of cytosol segregated within their lumen is discharged to and integrates with the cytosol of the target cell. Because this transfer can also include transmission of specific mRNAs, it can ultimately contribute to the epigenetic and proteomic properties of target cells.
One of the key mechanisms proposed for immune regulation by exosomes is the macromolecular transfer of small non-coding RNAs. These small non-coding RNAs, including microRNA, have been recognized in the regulation of cellular processes in maintenance of health and development of disease [75]. Specific microRNAs have been associated with the development of neurological disorders [76,77], contributing to the onset and progression of complications linked with TBI [78]. MicroRNAs associated with exosomes derived from injured brain tissue appear to represent surrogates and should have utility as stable, clinically accessible biomarkers to improve TBI detection and serve as sensitive measures for therapeutic outcomes.
7. Cargoes of exosomes
Exosomes contain proteins, non-coding RNAs and mRNAs, and the exosomal lipid bilayer appears to protect these materials from degradation. While protein and RNA cargoes of exosomes vary depending on the originating cell, there are conserved proteins among exosomes from different cellular origins [79]. The protein composition of exosomes has been extensively analysed by various techniques including Western blotting, fluorescence-activated cell sorting, immuno-electron microscopy and mass spectrometry. All exosomes exhibit cytoskeleton proteins (such as ezrin and actins), proteins associated with the MVB biogenesis (such as alix and TSG101), membrane transport and fusion proteins (e.g. annexins and Rab proteins) and tetraspanins (e.g. CD9, CD63 and CD81). A catalogue of proteins, RNAs and lipids associated with exosomes can be found at www.microvesicles.org. Currently, Vesiclepedia (formerly ExoCarta) lists entries (many redundant) for 43 731 proteins, 20 196 mRNAs, 2400 microRNAs and 342 lipids associated with exosomes.
We have analysed the cancer patient-derived exosomal proteome using ion trap mass spectrometry and identified 232 unique proteins. These proteins were classified as percentage of the identified total proteins into molecular chaperones (8.5%), vesicle fusion (8.5%), cytoskeletal proteins and proteins involved in the assembly/disassembly of the cytoskeletal networks (17.6%), anionic and cationic ion transport channels (3.7%), proteins involved in lipid (6.9%), carbohydrate (3.2%) and amino acid (2.1%) metabolisms, and proteins involved in DNA replication (6.9%), mRNA splicing (5.3%), transcription/translation (5.3%), post-transcriptional protein modification (13.8%) and signal transduction (2.7%). Our studies demonstrated that cytosolic proteins were highly represented, and we observed a diverse array of cytoskeletal constituents (actin, α-actinin-1, cofilin, filamin-A, -B, -C, tubulins, gelsolin, profilin-1, spectrin, symplekin, talin, vinculin, myosins). We identified that transmembrane proteins were also abundant, including multiple integrins (β1, α3, αv), intercellular adhesion molecule 1 (ICAM-1) and mucin-4. A variety of channels were observed, such as the voltage-dependent anion-selective channel protein 2 and 3, chloride intracellular channel protein 1, sodium/potassium-transporting ATPase subunit β-3, long sodium/potassium-transporting ATPase subunit α-1 and transitional endoplasmic reticulum ATPase. In line with their endocytic origin, exosomal proteins belonging to the ESCRT complexes, which are important protein complexes involved in ubiquitin-dependent exosome biogenesis, have also been observed [80]. These ESCRT-associated proteins include vacuolar protein sorting-associated protein 35 (VPS-35), Alix, ubiquitin-like modifier-activating enzyme and ubiquitin carboxyl-terminal hydrolases. We demonstrated that proteins involved in membrane trafficking and fusion processes were enriched (annexin A2, A5, A6, clathrin heavy chain 1/2, coatomer subunit β, Rab1b, Rab2a and Rab7a). A group of markers of endosomes and lysosomes were also detected (cathepsin-C, -D, EH domain-containing protein 1 and β-hexoaminidase), and several chaperones were identified (HSP60, HSP70, HSP90, HSC70, GRP75, HSP110, GRP78, GRP94, HSP47, HSP27, HSP9 alpha members; T-complex protein 1, endoplasmin and protein disulfide-isomerase A3, A4, A6) [79,81,82].
8. Exosome-associated RNA
The current hypothesis for the stability of circulating RNAs is that they are released from cells in membranous vesicles. Recent data confirm that extracellular RNA can exist in several forms: protein-associated RNA (including Argonaute 2-bound RNA), high-density lipoprotein-bound RNA and vesicle-associated RNA. This review focuses on RNA associated with exosomes. These exosomes are generated constitutively by most, but not all, cell types and contain both mRNAs and non-coding RNA. The ability of exosomes to transfer genetic information may facilitate cancer spread by delivering genetic material and oncogenic proteins. RNA profiles of exosomes differ from that of cellular RNA, since vesicles contain primarily small RNA, as mRNA (potentially debris and full length) and microRNA [40,71].
The presence of circulating RNAs has been extensively investigated, despite the presence of RNases, which should degrade any free RNA. The majority of the circulating RNAs have been defined as microRNAs based on the molecular weight [83]. Studies also demonstrated that microRNAs not only have high stability in body fluids, but also survive in the unfavourable physiological conditions such as freeze–thawing, extreme variations in pH and long time at room temperature [84–86]. Addition of detergents, such as Triton X100 or SDS, to serum or plasma allows the miRNAs to be easily degraded by RNases [86]. The results indicate there are at least two approaches responsible for the stability of extracellular microRNAs: packaging in membrane-encapsulated vesicles and protection by RNA-binding proteins. The stability of extracellular microRNAs has been hypothesized to be due to the formation of the RNA-vesicle. During RNA-induced silencing complex (RISC) disassembly in the cytoplasm, some microRNAs are found to be sorted into MVBs, which are commonly considered to form exosomes by fusion with the plasma membrane [87]. Both exosome and microvesicle can easily translocate across the cell membrane, which enables microRNAs to enter recipient cells easily and mediate cell-to-cell communication.
Our studies have indicated that many of the RNAs enriched in the exosomes may not be abundant, or even detectable, in the originating cell or the RNA might be highly expressed within the cell and low or absent within exosomes, indicating sorting of specific RNAs into exosomes. These released microRNAs can be classified into three categories based on the ratio between the amount of microRNA released from the cells and the amount retained in the cell [88]. The first group is selectively released microRNAs, which are characterized by being primarily released from tumour cells with relatively low concentrations remaining in the cell. By contrast, normal cells do not release appreciable quantities of these microRNAs [88]. An additional group of released microRNAs are those released in levels equal to those appearing within the cell, termed ‘neutrally’ released microRNAs. These neutrally released microRNAs include miR16 and miR21, where the abundance in exosomes reflects that in the tumour cells. The selective release of specific microRNAs differs depending on the cell type and appears to be influenced by malignant transformation. Breast and ovarian tumour cells have been demonstrated to release greater than 99% of the miR451 and miR1246 produced by the cells [88–90]. These selectively released microRNAs have been linked to the malignant phenotype. miR451 has been identified as a tumour suppressor, defining proliferation and cell polarity. miR451 has also been shown to induce chemosensitivity. miR1246 induces p53-dependent apoptosis triggered by DNA damage [91]. The changes in the release of cancer-related microRNAs may suggest a role for selective microRNA export in malignant transformation, and it may provide a cancer signature within the exported, circulating microRNA population. While the mechanism of this selective sorting is unclear, some researchers have postulated that this selectivity relates to microRNA/RISC components. Exosomes contain components of the microRNA/RISC, such as Argonaute 2, together with several RNA-binding proteins known to regulate RNA traffic between the nucleus and the cytoplasm. It can therefore be hypothesized that, during vesicle biogenesis, these RNA binding proteins regulate the accumulation of selected RNAs within exosomes. Studies on the transfer of reporter mRNAs and their translation into proteins, demonstrated both in vitro and in vivo, suggest that the mRNA delivered by exosomes is functional [92,93].
Most investigations on small RNAs in exosomes have been limited to microRNAs; however, next-generation sequencing of small RNAs in exosomes is expanding the populations identified. While intracellular microRNAs have been defined in many biological processes, identification of extracellular vesicle-associated microRNAs represents a non-invasive approach to investigate disease-specific microRNA and may provide a method for disease diagnosis [85]. To detect, analyse and quantitate the RNA signatures of exosomes derived from biologic fluids, several approaches have been used, including microarrays, quantitative real-time PCR and next-generation sequencing. The development of high detection sensitivity in next-generation sequencing technologies has expanded the identification of the exosomal transcriptome, beyond microRNA. While most studies have focused on exosomal microRNAs, we now recognize the presence of numerous other small RNAs within these circulating exosomes, as well as fragments of larger RNAs. These exosomal small non-coding RNAs are less than 200 nucleotides in length (generally 20–30 nt). There are several primary populations of small non-coding RNAs, including microRNAs and Piwi-interacting RNAs (piRNAs) [94]. Small non-coding RNAs have been shown to be key regulators in development, apoptosis, stem cell self-renewal, differentiation and cell integrity maintenance. piRNAs are generated from intergenic elements, including transposable elements, through Dicer-independent pathways. These piRNAs function through the Piwi-Argonaute sub-family (AGO3, Aubergine and Piwi), leading to silencing of transposable elements. A link between piRNAs and cancer has been demonstrated in gastric cancers where two aberrantly expressed piRNAs, piRNA-651 and piRNA-823, were found in gastric tumour tissue versus paired normal tissue [95,96].
9. Exosomes as diagnostic markers
Circulating biomarkers have been proposed to be promising for the definitive diagnosis and monitoring of therapeutic responses. Defining circulating biomarkers has numerous advantages as diagnostic biomarkers. Their presence can serve to identify processes that are difficult to image. The RNA cargoes of these exosomes may predict outcome by identifying patients at risk for therapeutic failure, defining molecular and pathological alterations for developing therapeutic targets and monitoring responses to interventions [97]. Such biomarkers could also serve to monitor disease progression and predict risk of recurrence. Circulating biomarkers are problematic and exhibit several critical issues, since free protein and nucleic acid biomarkers are extremely unstable in the circulation. Thus, to detect these a high steady state must be reached for detection, which is generally not observed except in late-stage disease, and minor changes over time (essential for monitoring) are difficult to quantify, in addition to these biomarkers being sensitive to sample handling. The use of exosome-associated biomarkers appears to be capable of circumventing these issues.
Exosomes provide stable, disease-specific markers for detection, disease characterization and predicting prognosis [98]. Temporal changes in exosomal RNA profiles have been demonstrated to accurately predict disease recurrence and overall patient survival [99]. The proteomic and genomic profiles of circulating exosomes provide a real-time monitor of therapeutic response, serving as a companion diagnostic. By correlating these circulating markers with the molecular characteristics and real-time clinical parameters, our group as well as others has established the use of circulating exosomes as a ‘liquid biopsy’. Since our original description of exosomal microRNA, many studies have examined the diagnostic utility of profiling total circulating microRNA in specific pathologies. The release of exosomal RNAs provides features with utility for diagnostic biomarkers, as they can be detected at early stages, are present in routinely obtained biologic fluids (blood, CSF, urine and saliva), are derived from specific tissues and can be easily and accurately quantified.
10. Conclusion
While the use of exosomes as biomarkers in the clinical setting is in the development phase, the findings presented within this review demonstrate their significant diagnostic potential in TBI. In the 6 years since our initial demonstration of the diagnostic utility of RNA profiles from exosomes, new and sensitive techniques have been developed, including deep sequencing and technologies based on microarrays and real-time PCR, enabling the monitoring of pathology-derived exosomes. The potential of using circulating exosomal proteins and RNAs as biomarkers extends beyond the diagnostic arena and provides biomarkers linked with patient stratification, therapy selection, monitoring of therapeutic responses and pathologic disruptions. Early definitive identification of TBI will be critical for significant impact in improving patient outcomes, particularly in patients without symptoms as well as to stratify a heterogeneous patient population. Further, it appears that exosomes not only are diagnostic of TBI, but also may be an essential causative agent in cases of its progression to long-term neurodegenerative conditions. Biomarkers can be used to determine the potential to develop progressive disease, measure its progress or predict prognosis. Identifying informative biomarkers is an exceptionally valuable tool for evaluating clinical trial outcomes and for assisting physicians in choosing and personalizing treatment options.
References
- 1.Faul M, Xu L, Wald MM, Coronado VG. 2010. Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002–2006. Atlanta, GA, USA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. [Google Scholar]
- 2.Coronado VG, Xu L, Basavaraju SV, Mcguire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD. 2011. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill. Summ. 60, 1–32. [PubMed] [Google Scholar]
- 3.W.H. Organization. 2006. Neurological disorders: public health challenges. Geneva, Switzerland: WHO Press. [Google Scholar]
- 4.Taylor BC, Hagel EM, Carlson KF, Cifu DX, Cutting A, Bidelspach DE, Sayer NA. 2012. Prevalence and costs of co-occurring traumatic brain injury with and without psychiatric disturbance and pain among Afghanistan and Iraq War Veteran V.A. users. Med. Care 50, 342–346. ( 10.1097/MLR.0b013e318245a558) [DOI] [PubMed] [Google Scholar]
- 5.Schwab KA, Ivins B, Cramer G, Johnson W, Sluss-Tiller M, Kiley K, Lux W, Warden D. 2007. Screening for traumatic brain injury in troops returning from deployment in Afghanistan and Iraq: initial investigation of the usefulness of a short screening tool for traumatic brain injury. J. Head Trauma Rehabil. 22, 377–389. ( 10.1097/01.HTR.0000300233.98242.87) [DOI] [PubMed] [Google Scholar]
- 6.MacDonald CL, et al. 2011. Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 364, 2091–2100. ( 10.1056/NEJMoa1008069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svetlov SI, Larner SF, Kirk DR, Atkinson J, Hayes RL, Wang KKW. 2009. Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J. Neurotrauma 26, 913–921. ( 10.1089/neu.2008.0609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maas AI, Stocchetti N, Bullock R. 2008. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7, 728–741. ( 10.1016/S1474-4422(08)70164-9) [DOI] [PubMed] [Google Scholar]
- 9.Kochanek PM, Bramlett H, Dietrich WD, Dixon CE, Hayes RL, Povlishock J, Tortella FC, Wang KK. 2011. A novel multicenter preclinical drug screening and biomarker consortium for experimental traumatic brain injury: operation brain trauma therapy. J. Trauma 71, S15–S24. ( 10.1097/TA.0b013e31822117fe) [DOI] [PubMed] [Google Scholar]
- 10.Thornhill S, et al. 2000. Disability in young people and adults one year after head injury: prospective cohort study. Br. Med. J. 320, 1631–1635. ( 10.1136/bmj.320.7250.1631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popovich PG, Longbrake EE. 2008. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 9, 481–493. ( 10.1038/nrn2398) [DOI] [PubMed] [Google Scholar]
- 12.Fehlings MG, Nguyen DH. 2010. Immunoglobulin G: a potential treatment to attenuate neuroinflammation following spinal cord injury. J. Clin. Immunol. 1, S109–S112. ( 10.1007/s10875-010-9404-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz M, Butovsky O, Bruck W, Hanisch UK. 2006. Microglial phenotype: is the commitment reversible? Trends Neurosci. 29, 68–74. ( 10.1016/j.tins.2005.12.005) [DOI] [PubMed] [Google Scholar]
- 14.Bareyre FM, Schwab ME. 2003. Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci. 26, 555–563. ( 10.1016/j.tins.2003.08.004) [DOI] [PubMed] [Google Scholar]
- 15.Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. 1999. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur. J. Neurosci. 11, 3648–3658. ( 10.1046/j.1460-9568.1999.00792.x) [DOI] [PubMed] [Google Scholar]
- 16.Anderson AJ. 2002. Mechanisms and pathways of inflammatory responses in CNS trauma: spinal cord injury. J. Spinal Cord Med. 25, 70–79. [DOI] [PubMed] [Google Scholar]
- 17.Gercel-Taylor C, Tullis RH, Atay S, Kesimer M, Taylor DD. 2012. Nanoparticle analysis of circulating cell-derived vesicles in ovarian cancer patients. Anal. Biochem. 428, 44–53. ( 10.1016/j.ab.2012.06.004) [DOI] [PubMed] [Google Scholar]
- 18.Esteller M. 2011. Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. ( 10.1038/nrg3074) [DOI] [PubMed] [Google Scholar]
- 19.Sibley CR, Wood MJ. 2011. The miRNA pathway in neurological and skeletal muscle disease: implications for pathogenesis and therapy. J. Mol. Med. (Berl). 89, 1065–1077. ( 10.1007/s00109-011-0781-z) [DOI] [PubMed] [Google Scholar]
- 20.Rasouli J, Lekhraj R, White NM, Flamm ES, Pilla AA, Strauch B, Casper D. 2012. Attenuation of interleukin-1beta by pulsed electromagnetic fields after traumatic brain injury. Neurosci. Lett. 519, 4–8. ( 10.1016/j.neulet.2012.03.089) [DOI] [PubMed] [Google Scholar]
- 21.Adamczak S, Dale G, DeRivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. 2012. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as markers of functional outcome. J. Neurosurg. 117, 1119–1125. ( 10.3171/2012.9.JNS12815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frugier T, Morganti-Kossmann MC, O'Reilly D, McLean CA. 2010. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 27, 497–507. ( 10.1089/neu.2009.1120) [DOI] [PubMed] [Google Scholar]
- 23.Unterberg AW, Stover J, Kress B, Kiening KL. 2004. Edema and brain trauma. Neuroscience 129, 1021–1029. ( 10.1016/j.neuroscience.2004.06.046) [DOI] [PubMed] [Google Scholar]
- 24.Woodcock T, Morganti-Kossmann MC. 2013. The role of markers of inflammation in traumatic brain injury. Front Neurol. 4, 18 ( 10.3389/fneur.2013.00018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu GW, Wang F, Liu WG. 2009. Classification and prediction of outcome in traumatic brain injury based on computed tomographic imaging. J. Int. Med. Res. 37, 983–995. ( 10.1177/147323000903700402) [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Rasmussen IA, Lagopoulos J, Haberg A. 2007. Diffuse axonal injury in severe traumatic brain injury visualized using high-resolution diffusion tensor imaging. J. Neurotrauma 24, 753–765. ( 10.1089/neu.2006.0208) [DOI] [PubMed] [Google Scholar]
- 27.Smith DH, David F, Meaney DF. 2000. Axonal damage in traumatic brain injury. Neuroscientist 6, 483–495. ( 10.1177/107385840000600611) [DOI] [Google Scholar]
- 28.Taylor DD, Doellgast GJ. 1979. Quantitation of peroxidase-antibody binding to membrane fragments using column chromatography. Anal. Biochem. 98, 53–59. ( 10.1016/0003-2697(79)90704-8) [DOI] [PubMed] [Google Scholar]
- 29.Taylor DD, Gercel-Taylor C. 2011. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Sem. Immunopathol. 33, 441–454. ( 10.1007/s00281-010-0234-8) [DOI] [PubMed] [Google Scholar]
- 30.Peinado H, et al. 2012. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891. ( 10.1038/nm.2753) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trams EG, Lauter CJ, Salem C, Jr, Heine U. 1981. Exfoliation of membrane ecto-enzymes in the form of microvesicles. Biochim. Biophys. Acta 645, 63–70. ( 10.1016/0005-2736(81)90512-5) [DOI] [PubMed] [Google Scholar]
- 32.Zumaquero E, et al. 2010. Exosomes from human lymphoblastoid B cells express enzymatically active CD38 that is associated with signaling complexes containing CD81, Hsc-70, and Lyn. Exp. Cell Res. 316, 2692–2706. ( 10.1016/j.yexcr.2010.05.032) [DOI] [PubMed] [Google Scholar]
- 33.Admyre C, Telemo E, Almqvist N, Lotvall J, Lahesmaa R, Scheynius A, Gabrielsson S. 2008. Exosomes—nanovesicles with possible roles in allergy inflammation. Allergy 63, 404–408. ( 10.1111/j.1398-9995.2007.01600.x) [DOI] [PubMed] [Google Scholar]
- 34.Montecalvo A, et al. 2008. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J. Immunol. 180, 3081–3090. ( 10.4049/jimmunol.180.5.3081) [DOI] [PubMed] [Google Scholar]
- 35.Rak J. 2010. Microparticles in cancer. Semin. Thromb. Hemost. 36, 888–906. ( 10.1055/s-0030-1267043) [DOI] [PubMed] [Google Scholar]
- 36.Anand PK. 2010. Exosomal membrane molecules are potent immune response modulators. Commun. Integr. Biol. 3, 405–408. ( 10.4161/cib.3.5.12474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kesimer M, Scull M, Brighton B, DeMaria G, Burns K, O'Neal W, Pickles RJ, Sheehan JK. 2009. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J. 23, 1858–1868. ( 10.1096/fj.08-119131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor DD, Gercel-Taylor C. 2013. The origin, function and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front. Genet. 4, 0142 ( 10.3389/fgene.2013.00142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graner MW, et al. 2013. Circulating exosomes as new biomarkers for brain disease and injury. In Proc. SPIE 8723, Sensing Technologies for Global Health, Military Medicine, and Environmental Monitoring III, 87230R Baltimore, MD, USA ( 10.1117/12.2027435) [DOI] [Google Scholar]
- 40.Taylor DD, Gercel-Taylor C. 2008. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 110, 13–21. ( 10.1016/j.ygyno.2008.04.033) [DOI] [PubMed] [Google Scholar]
- 41.Atay S, Gercel-Taylor C, Suttles J, Mor G, Taylor DD. 2011. Trophoblast derived exosomes mediate monocyte recruitment and differentiation. Am. J. Reprod. Immunol. 65, 65–77. ( 10.1111/j.1600-0897.2010.00880.x) [DOI] [PubMed] [Google Scholar]
- 42.Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT. 2006. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J. Trauma 60, 152–157. ( 10.1097/01.ta.0000196345.81169.a1) [DOI] [PubMed] [Google Scholar]
- 43.Kuhlow CJ, Krady JK, Basu A, Levison SW. 2003. Astrocytic ceruloplasmin expression, which is induced by IL-1β and by traumatic brain injury, increases in the absence of the IL-1 type 1 receptor. Glia 44, 76–84. ( 10.1002/glia.10273) [DOI] [PubMed] [Google Scholar]
- 44.Kochanek PM, Berger RP, Bayr H, Wagner AK, Jenkins LW, Clark RS. 2008. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr. Opin. Crit. Care 14, 135–141. ( 10.1097/MCC.0b013e3282f57564) [DOI] [PubMed] [Google Scholar]
- 45.Youn YA, Kim SJ, Sung IK, Chung SY, Kim YH, Lee IG. 2012. Serial examination of serum IL-8, IL-10 and IL-1Ra levels is significant in neonatal seizures induced by hypoxic–ischaemic encephalopathy. Scand. J. Immunol. 76, 286–293. ( 10.1111/j.1365-3083.2012.02710.x) [DOI] [PubMed] [Google Scholar]
- 46.Pan BT, Blostein R, Johnstone RM. 1983. Loss of the transferrin receptor during the maturation of sheep reticulocytes in vitro: an immunological approach. Biochem. J. 210, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. 1987. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 262, 9412–9420. [PubMed] [Google Scholar]
- 48.Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M. 2008. CD44 and EpCAM: cancer-initiating cell markers. Curr. Mol. Med. 8, 784–804. ( 10.2174/156652408786733667) [DOI] [PubMed] [Google Scholar]
- 49.Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK. 2010. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol. Cell Proteomics 9, 1085–1099. ( 10.1074/mcp.M900381-MCP200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong BS, et al. 2009. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 10, 556 ( 10.1186/1471-2164-10-556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiang X, et al. 2009. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 124, 2621–2633. ( 10.1002/ijc.24249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M, Lochnit G, Preissner KT, Zöller M. 2010. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 70, 1668–1678. ( 10.1158/0008-5472.CAN-09-2470) [DOI] [PubMed] [Google Scholar]
- 53.Keller S, König AK, Marmé F, Runz S, Wolterink S, Koensgen D, Mustea A, Sehouli J, Altevogt P. 2009. Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett. 278, 73–81. ( 10.1016/j.canlet.2008.12.028) [DOI] [PubMed] [Google Scholar]
- 54.Dolo V, D'Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A, Vittorelli ML, Canevari S, Pavan A. 1999. Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin. Exp. Metastasis 17, 131–140. ( 10.1023/A:1006500406240) [DOI] [PubMed] [Google Scholar]
- 55.Dolo V, Ginestra A, Cassara D, Ghersi G, Nagase H, Vittorelli ML. 1999. Shed membrane vesicles and selective localization of gelatinases and MMP-9/TIMP-1 complexes. Ann. N Y Acad. Sci. 878, 497–499. ( 10.1111/j.1749-6632.1999.tb07707.x) [DOI] [PubMed] [Google Scholar]
- 56.Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA. 2004. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 64, 7045–7049. ( 10.1158/0008-5472.CAN-04-1800) [DOI] [PubMed] [Google Scholar]
- 57.Lewis CE, Pollard JW. 2006. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612. ( 10.1158/0008-5472.CAN-05-4005) [DOI] [PubMed] [Google Scholar]
- 58.Whiteside TL. 2013. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour derived exosomes). Biochem. Soc. Trans. 41, 245–251. ( 10.1042/BST20120265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Atay S, Gercel-Taylor C, Kesimer M, Taylor DD. 2011. Morphologic and proteomic characterization of exosomes released by cultured extravillous trophoblast cells. Exp. Cell Res. 317, 1192–1202. ( 10.1016/j.yexcr.2011.01.014) [DOI] [PubMed] [Google Scholar]
- 60.Atay S, Roberson C, Gercel-Taylor C, Taylor DD. 2013. Ovarian cancer patient-derived exosomal fibronectin induces pro-inflammatory IL-1β production by macrophages. Exosomes and Microvesicles 1, 1 ( 10.5772/56180) [DOI] [Google Scholar]
- 61.McLellan AD. 2009. Exosome release by primary B cells. Crit. Rev. Immunol. 29, 203–217. ( 10.1615/CritRevImmunol.v29.i3.20) [DOI] [PubMed] [Google Scholar]
- 62.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. 2006. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20, 1487–1495. ( 10.1038/sj.leu.2404296) [DOI] [PubMed] [Google Scholar]
- 63.Ichim TE, et al. 2008. Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J. Transl. Med. 6, 37 ( 10.1186/1479-5876-6-37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. 2008. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 10, 619–624. ( 10.1038/ncb1725) [DOI] [PubMed] [Google Scholar]
- 65.Al-Nedawi K, Meehan B, Rak J. 2009. Microvesicles: messengers and mediators of tumor progression. Cell Cycle 8, 2014–2018. ( 10.4161/cc.8.13.8988) [DOI] [PubMed] [Google Scholar]
- 66.Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. 2009. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl Acad. Sci. USA 106, 3794–3799. ( 10.1073/pnas.0804543106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. 2010. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 78, 838–848. ( 10.1038/ki.2010.278) [DOI] [PubMed] [Google Scholar]
- 68.Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM. 2010. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 11, 110–122. ( 10.1111/j.1600-0854.2009.01006.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Izquierdo-Useros N, et al. 2009. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 113, 2732–2741. ( 10.1182/blood-2008-05-158642) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Izquierdo-Useros N, Naranjo-Gómez M, Erkizia I, Puertas MC, Borràs FE, Blanco J, Martinez-Picado J. 2010. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathog. 6, e1000740 ( 10.1371/journal.ppat.1000740) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Skog J, et al. 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. ( 10.1038/ncb1800) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, Ratajczak MZ. 2005. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 113, 752–760. ( 10.1002/ijc.20657) [DOI] [PubMed] [Google Scholar]
- 73.Genneback N, Hellman U, Malm L, Larsson G, Ronquist G, Waldenstrom A, Morner S. 2013. Growth factor stimulation of cardiomyocytes induces changes in the transcriptional contents of secreted exosomes. J. Extracellular Ves. 2, 20167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuan XL, et al. 2010. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin. Immunol. 134, 277–288. ( 10.1016/j.clim.2009.10.005) [DOI] [PubMed] [Google Scholar]
- 75.Cohen SM. 2010. microRNA in CNS development and neurodegradation. In Macro roles for microRNAs in the life and death of neurons (eds de Stooper B, Christen Y.), pp. 69–77. Berlin, Germany: Springer. [Google Scholar]
- 76.Abe M, Bonini NM. 2013. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 23, 30–36. ( 10.1016/j.tcb.2012.08.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gascon E, Gao FB. 2012. Cause or effect: misregulation of microRNA pathways in neurodegeneration. Front. Neurosci. 6, 48 ( 10.3389/fnins.2012.00048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Redell JB, Moore AN, Ward NH, Hergenroeder GW, Dash PK. 2010. Human traumatic brain injury alters plasma microRNA levels. J. Neurotrauma 27, 2147–2156. ( 10.1089/neu.2010.1481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mathivanan S, Simpson RJ. 2009. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics 9, 4997–5000. ( 10.1002/pmic.200900351) [DOI] [PubMed] [Google Scholar]
- 80.Doring T, Gotthardt K, Stieler J, Prange R. 2010. γ2-Adaptin is functioning in the late endosomal sorting pathway and interacts with ESCRT-I and -III subunits. Biochim. Biophys. Acta 1803, 1252–1264. ( 10.1016/j.bbamcr.2010.08.001) [DOI] [PubMed] [Google Scholar]
- 81.Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, Bigner DD. 2009. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23, 1541–1557. ( 10.1096/fj.08-122184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simpson RJ, et al. 2012. Vesiclepedia: a compendium for extracellular vesicles that enables continuous annotation. PLoS Biol. 10, e1001450 ( 10.1371/journal.pbio.1001450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mitchell PS, et al. 2008. Circulating microRNAs as stable blood-based markers for cancer detection . Proc. Natl Acad. Sci. USA 105, 10 513–10 518. ( 10.1073/pnas.0804549105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X, Liang H, Zhang J, Zen K, Zhang CY. 2012. Horizontal transfer of microRNAs: molecular mechanisms and clinical applications . Protein Cell 3, 28–37. ( 10.1007/s13238-012-2003-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duggagupta R, Jiang R, Gollub J, Getts RC, Jones KW. 2011. Impact of cellular miRNAs on circulating miRNA biomarker signatures. PLoS ONE 6, e20769 ( 10.1371/journal.pone.0020769) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y, et al. 2010. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 39, 33–144. ( 10.1016/j.molcel.2010.06.010) [DOI] [PubMed] [Google Scholar]
- 87.Simons M, Raposo G. 2009. Exosomes: vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 21, 575–581. ( 10.1016/j.ceb.2009.03.007) [DOI] [PubMed] [Google Scholar]
- 88.Pigati L, Yaddanapudi SC, Iyengar R, Kim DJ, Hearn SA, Danforth D, Hastings ML, Duelli DM. 2010. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 5, e13515 ( 10.1371/journal.pone.0013515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rabinowits G, Gercel-Taylor C, Day JM, Taylor DD, Kloecker GH. 2009. Exosomal microRNA: a diagnostic marker for lung cancer. Clin. Lung Cancer 10, 42–46. ( 10.3816/CLC.2009.n.006) [DOI] [PubMed] [Google Scholar]
- 90.Mittelbrunn M, Gutiérrez-Vázquez C, Villarroya-Beltri C, González S, Sánchez-Cabo F, González MÁ, Bernad A, Sánchez-Madrid F. 2011. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2, 282 ( 10.1038/ncomms1285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Liao JM, Zeng SX, Lu H. 2011. p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep. 12, 811–817. ( 10.1038/embor.2011.98) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL, Wood MJ. 2012. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 7, 2112–2126. ( 10.1038/nprot.2012.131) [DOI] [PubMed] [Google Scholar]
- 93.Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G. 2013. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 44, 11–19. ( 10.1007/s12020-012-9839-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang X, et al. 2013. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 14, 319 ( 10.1186/1471-2164-14-319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng J, Deng H, Xiao B, Zhou H, Zhou F, Shen Z, Guo J. 2012. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 315, 12–17. ( 10.1016/j.canlet.2011.10.004) [DOI] [PubMed] [Google Scholar]
- 96.Cheng J, Guo JM, Xiao BX, Miao Y, Jiang Z, Zhou H, Li QN. 2011. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta 412, 1621–1625. ( 10.1016/j.cca.2011.05.015) [DOI] [PubMed] [Google Scholar]
- 97.Kim JW, Galanzha EI, Zaharoff DA, Griffin RJ, Zharov VP. 2013. Nanotheranostics of circulating tumor cells, infections and other pathological features in vivo. Mol. Pharm. 10, 813–830. ( 10.1021/mp300577s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liang B, et al. 2013. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteomics 80C, 171–182. ( 10.1016/j.jprot.2012.12.029) [DOI] [PubMed] [Google Scholar]
- 99.Takeshita N, et al. 2013. Serum microRNA expression profile: miR-1246 as a novel diagnostic and prognostic biomarker for oesophageal squamous cell carcinoma. Br. J. Cancer 108, 644–652. ( 10.1038/bjc.2013.8) [DOI] [PMC free article] [PubMed] [Google Scholar]