

Abstract
Antibodies, most commonly IgGs, have been widely used as targeting ligands in research and therapeutic applications due to their wide array of targets, high specificity and proven efficacy. Many of these applications require antibodies to be conjugated onto surfaces (e.g. nanoparticles and microplates); however, most conventional bioconjugation techniques exhibit low crosslinking efficiencies, reduced functionality due to non-site-specific labeling and random surface orientation, and/or require protein engineering (e.g. cysteine handles), which can be technically challenging. To overcome these limitations, we have recombinantly expressed Protein Z, which binds the Fc region of IgG, with an UV active non-natural amino acid benzoylphenyalanine (BPA) within its binding domain. Upon exposure to long wavelength UV light, the BPA is activated and forms a covalent link between the Protein Z and the bound Fc region of IgG. This technology was combined with expressed protein ligation (EPL), which allowed for the introduction of a fluorophore and click chemistry-compatible azide group onto the C-terminus of Protein Z during the recombinant protein purification step. This enabled crosslinked-Protein Z-IgG complexes to be efficiently and site-specifically attached to aza-dibenzycyclooctyne-modified nanoparticles, via copper-free click chemistry.
Keywords: Antibody, conjugation, site-specific, click chemistry, nanoparticle
1. Introduction
Personalized medicine promises advancements in healthcare by emphasizing early diagnosis of disease, more accurate disease classification and the use of targeted rather than systemic treatments. These goals are increasingly within reach thanks to the emergence of targeted nanoplatforms to detect, classify and treat diseases. A central element of many targeted therapies is the use of antibodies. These protein molecules have a wide array of potential targets, high binding affinities (e.g. nM range), mature production techniques and proven clinical efficacies. All of these features make antibodies the most important class of targeting ligand.[1–3] In order to use antibodies as targeting ligands, they frequently need to be conjugated to nanoparticles carrying the therapeutic or diagnostic payloads. While ideal conjugation methods should possess several features, including biocompatibility, high efficiency, site-specificity, and broad applicability, most existing conjugation methods unfortunately do not meet these requirements.
Some of the most widely used conjugation techniques are based on carbodiimide (e.g. EDC) and/or N-hydroxysuccinimide (NHS) chemistries.[4] These techniques link carboxylated surfaces with primary amines that are randomly located throughout the antibody, hence they can potentially create products with poorly oriented antibodies that have sterically hindered antigen binding domains.[5–8] The products are also often heterogeneous with respect to the density of antibody on the surface, thereby complicating purification and lowering the therapeutic indices.[8–11] Perhaps the most significant limitation of these methods is that the conjugation efficiencies are generally poor, with only 10–20% of antibodies being conjugated in most cases.[1, 12, 13] Since antibodies are often produced in complex biological media such as ascites fluid or stored with carrier proteins such as BSA, they first must be purified from the other amine-containing proteins before being conjugated using EDC/NHS-based methods. This introduces additional burdens and lowers the final product yield.
Thiol groups or click-chemistry groups (e.g. azides and alkynes) can be introduced onto antibodies, using chemicals such as N-succinimidyl S-acetylthioacetate (SATA)) or NHS-azide, to create a unique handle for bioconjugation; however, these approaches suffer from many of the same shortcomings as EDC/NHS. Moreover, the indiscriminate labeling of antibodies with click- and thiol-reactive groups has the potential to render the antibodies nonfunctional, especially since tens of reactive groups must often been introduced onto each antibody to maximize surface conjugation.[1] Thiolated antibodies have the additional shortcoming of being prone to undesirable disulfide formation, aggregation, and precipitation.
While some existing methods have been developed for conjugating antibodies in a site-specific manner, they often are not broadly applicable. For example, a popular method utilizes carbohydrates that are only found in some post-translationally glycosylated Immunoglobulin G’s (IgGs).[4, 14] Other site-specific conjugation methods require cysteine residues to be engineered into recombinant IgGs, a costly and lengthy endeavor.[15–18] Yet another approach was recently developed by Alves et. al., where an UV-active nucleotide analogue was used to covalently crosslink a conserved Nucleotide Binding Site (NBS) found in nearly all IgGs.[19] However, as this NBS is situated within the Fab region, conjugates bound via the NBS have the potential to sterically hinder antigen binding. Additionally, this method requires crosslinking with short wavelength UV light (250nm), which has the potential to damage proteins. Despite their shortcomings however, these conjugation methods nonetheless have been illustrative in showing that when site-specific modifications are possible, the resulting antibody conjugates often possess much higher activity.[6, 7, 19–23]
Recently, several groups have used natural IgG binding domains, such as Protein A [24–26] or Protein G, [21] to site-specifically attach antibodies to various substrates. In particular, the 58 amino acid Protein Z, which is derived from the B domain of Protein A, provides a convenient scaffold for orienting IgG as it is recombinantly produced and only binds to the Fc region.[27–29] Unfortunately, Protein A, G, and Z only associate with IgG non-covalently and as a result these complexes are not biostable. To address this limitation, Konrad et. al. [30] and Yu et. al. [31] have achieved covalent conjugations by incorporating photoreactive crosslinkers into the Protein Z binding domain and then using UV light to site-specifically crosslink Protein Z onto IgGs at the Fc region. Benzophenone-based crosslinkers, such as the amino acid analog benzoyl-phenylalanine (BPA) as was used by Konrad et al., are ideal for this application since they are not easily photo-bleached and can be activated by the non-harmful, long wavelength UV light (365nm) for crosslinking.[32, 33]
One downside of these methods however, stems from the difficulty associated with incorporating the crosslinker into Protein Z, which requires either using peptide synthesis as was done by Konrad et al. or post-translational modification of recombinant Protein Z as was done by Yu et al. Peptide synthesis and purification can be difficult for peptides as large as Protein Z, particularly when incorporating additional functionalities, such as photocrosslinkers and other moieties for nanoparticle conjugation (e.g. haptens or click moieties). Likewise, post-translational modification of recombinant Protein Z can also be problematic since additional reaction and purification steps are required to introduce the photocrossslinker. If more than one chemical moiety is needed, as is the case for photocrosslinking and nanoparticle conjugation, this approach may not be tractable. Therefore, neither method is ideal for large-scale production and downstream applications.
One approach that overcomes the shortcomings of peptide synthesis and post-translational modification involves producing photoreactive Protein Z in an entirely recombinant manner. This can be achieved by using an engineered E. coli that is capable of incorporating unnatural amino acids, such as BPA, into proteins during translation.[34] The engineered E. coli are simply transformed with a plasmid encoding both an amber suppressor tRNA and an engineered aminoacyl-tRNA synthetase that can charge the suppressor tRNA with BPA. These two components allow the E. coli to incorporate BPA into the peptide chain during translation when amber codons (UAG) are encountered. This technique has been used previously to introduce BPA into endogenous proteins in order to study protein-protein interactions in vivo and assist in drug discovery.[32, 35–37] Here, this system was used for the efficient production of recombinant Protein Z containing BPA moieties.
In addition to introducing a photoreactive moiety into the binding site of recombinant Protein Z, additional functionalities are also needed for IgG-Protein Z complexes to be subsequently attached to nanoparticles. One option is to incorporate a biotin tag onto the recombinant protein in situ using a biotinylation peptide sequence;[38, 39] however, while the biotin-streptavidin interaction is well suited for in vitro applications, the presence of endogenous biotins and the immunogenicity of streptavidins preclude their use for in vivo applications.[40–42]
Azide-alkyne based click reactions, on the other hand, offer a favorable option for downstream bioconjugations. These reactions form covalent bonds, are highly efficient, and also are bioorthogonal as they do not react with endogenous molecules. The recently developed strain-promoted alkyne azide cycloaddition (SPAAC), also known as copper-free click reaction, have further improved the versatility, simplicity, and biocompatibility of click reactions.[43, 44] While it can be challenging to site-specifically incorporate azido moieties into proteins, our group has previously developed an intein-mediated Expressed Protein Ligation (EPL) technique that allows azide- and fluorescently-labeled peptides to be efficiently and site-specifically ligated to the carboxy-terminus of recombinant proteins during the affinity purification process.[45, 46]
This system was applied here to create a “tri-functional” Protein Z domain. Specifically, EPL was used to incorporate a short peptide, containing a fluorophore for imaging and a terminal azide for bioconjugations, onto a recombinantly expressed photoreactive Protein Z. Herein, we show that this protein can not only be site-specifically photo-crosslinked to various IgGs (purified or in complex biological fluids), but that these Protein Z-IgG complexes can subsequently be site-specifically and efficiently attached to superparamagnetic iron oxide (SPIO) nanoparticles.
2. Results
2.1. In vivo incorporation of BPA during protein expression
The coding sequence for wild-type Protein Z was cloned into an EPL-compatible plasmid pTXB1 (New England Biolabs), generating a construct that encodes Protein Z fused to a self-cleaving intein domain followed by a Chitin Binding Domain (CBD) (Figure 1A: Ligation). To allow for incorporation of the unnatural amino acid, BPA, into the fusion protein during translation, site-directed mutagenesis was performed to introduce an amber codon (i.e. UAG) into the IgG binding site of Protein Z. The BPA replaced a phenylalanine in the thirteenth position (F13). This site was selected due to the structural similarities between BPA and phenylalanine (BPA is a derivative of phenylalanine), F13’s postulated role in IgG binding and the outward orientation of its side chain, which can minimize the possibility of intramolecular crosslinking.[28, 47] Additionally, in order to compare the performance of F13BPA Protein Z with that of the F5BPA variant previously synthesized by others, a second construct was prepared with phenylalanine at the fifth position mutated to BPA.[30]
Figure 1. Schematic describing the production and surface conjugation of Protein Z-IgG complexes.

(A) A fusion protein containing an unnatural amino acid benzoylphenylalanine (BPA) in the Protein Z domain is expressed in frame with an intein and a chitin binding domain. During the affinity purification process, the intein is used to drive expressed protein ligation between the Protein Z and an azido fluorescent peptide (AzFP) containing an N-terminal cysteine, a “clickable” azide group and a 5-FAM fluorophore. (B) After the protein Z-AzFP conjugate is mixed with IgG, long-wavelength UV irradiation is used to create a site-specific covalent bond between the BPA and Fc region of IgG. (C) The crosslinked IgG, now containing both a fluorophore and an azide moiety, can be used for site-specific conjugation with any substrate containing a free alkyne via click chemistry. A strained alkyne, aza-dibenzocyclooctyne (ADIBO), capable of copper-free click reactions is shown.
Host E. Coli were co-transformed with the pTXB1 plasmids encoding either the photoreactive protein Z or wild-type protein Z and the pEVOL-pBpF plasmid[34], which carries the tRNA/aminoacyl transferase pair. Analysis of the expressed proteins by Coomassie-stained SDS-PAGE revealed that while wild-type fusion protein could be expressed in the absence of BPA, the amber mutant protein required BPA for expression (Figure 2). This is expected since, in the absence of BPA, the amber stop codon is not suppressed and translation is terminated early. This also confirms that there is no “leaky” background incorporation of other amino acids in response to the amber codon, as was seen when some other proteins containing UAAs (i.e. ochre codons) were expressed using similar approaches.[34] Additionally, the expression levels for the BPA-containing mutant fusion proteins (F13BPA and F5BPA) were comparable to that of the wild type Protein Z. Representative protein expression data of the F13BPA mutant fusion protein is shown in Figure 2. Mass spectrometry of the purified F13BPA mutant fusion protein confirmed the identity of Protein Z with a single substitution of BPA in place of phenylalanine (Figure S1).
Figure 2. SDS-PAGE with Coomassie staining confirming the in vivo incorporation of BPA into expressed Protein Z.
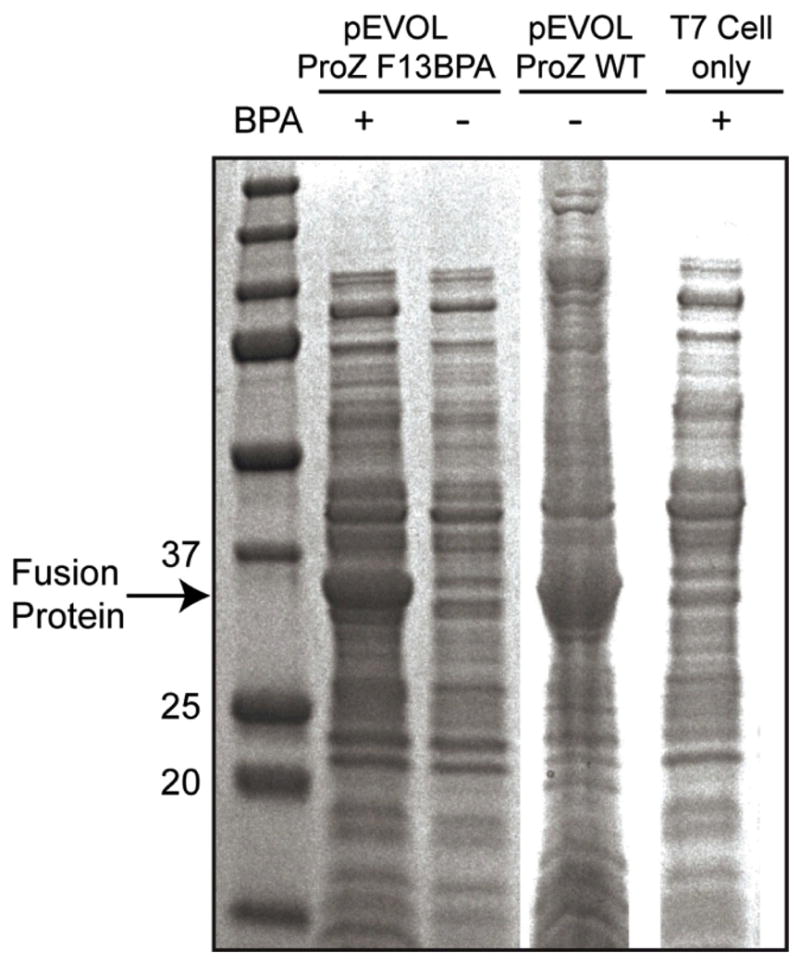
T7 competent E. coli were co-transformed with the pEVOL-pBpf plasmid containing the amber suppressor tRNA/aminoacyl transferase pair and the pTXB1 plasmid, which codes for Protein Z with an amber codon mutation (ProZ F13BPA). Following induction of protein expression, cell lysates, with or without BPA in the media, were evaluated by SDS-PAGE stained with Coomassie (lanes 1 and 2, respectively). Analogous studies were performed with E. coli that express wild-type Protein Z (lane 3) and unmodified T7 competent cells (lane 4).
2.2. Protein purification and Expressed Protein Ligation
Following protein expression, a ten amino acid peptide (AzFP) with a fluorophore (5-FAM) and an azide moiety was ligated onto the C-terminus of Protein Z via intein-mediated Expressed Protein Ligation (EPL). This enables the crosslinked Protein Z-IgG complexes to be subsequently visualized and attached to alkyne-functionalized substrates via click chemistry. EPL is a versatile technique that allows multiple functional groups to be introduced simultaneously during standard recombinant protein purification procedures, without requiring any additional steps (Figure 1A, Ligation). The chitin column-bound fusion protein is simply incubated with sodium 2-sulfanylethanesulfonate (MESNA) along with AzFP, causing its ligation to the C-terminus of the Protein Z. Analogous procedures were also performed with cysteine in place of AzFP to create a Protein Z-cysteine ligation product, which was used as an unmodified size standard, while the AzFP-ligated Protein Z (abbrev. Pz-AzFP) was used for conjugation with alkyne-functionalized substrates.
Both the AzFP and cysteine forms of Protein Z were analyzed on a tris-tricine SDS-PAGE gel. Protein bands were visualized by Coomassie-staining and fluorescence (Figure 3A). The lane containing the AzFP-ligated Protein Z clearly showed a band migrating at the approximate molecular weight of 8kD, by both Coomassie staining and fluorescence (Figure 3A, right lane). In contrast, the cysteine-ligated Protein Z was only observed by Coomassie staining, at the expected approximate molecular weight of 6.6kD, but not by fluorescence (Figure, 3A, left lane).
Figure 3. SDS-PAGE and HPLC analysis of Protein Z following ligation with cysteine or an azido fluorescent peptide.

(A) Tricine SDS-PAGE analysis of Protein Z following intein-mediated expressed protein ligation with either a cysteine (Pz-Cys) or an azido-fluorescent peptide (Pz-AzFP). Formation of the conjugate was evaluated via a white light image of the gel (left) and further confirmed by fluorescent imaging of the gel (right), which showed that only the Protein Z-AzFP conjugate was fluorescent. (B) Pz-Cys and Pz-AzFP ligation products were also analyzed by HPLC.
The two Protein Z variants were further analyzed using reverse-phase HPLC (RP-HPLC). The resulting spectra clearly showed the two variants to be distinct species, with the Protein Z – AzFP variant eluting in more organic conditions due to the addition of the AzFP peptide (Figure 3B).
2.3. UV-induced Protein Z crosslinking onto IgG
To crosslink photoreactive Protein Z onto IgGs, the F13BPA variant was first incubated with the humanized IgG1 monoclonal antibody rituximab and exposed to long wavelength UV light (365nm) (Figure 1B: Crosslinking). The extent of crosslinking was assessed using both reducing and non-reducing SDS-PAGE gels. In the reducing gel, the formation of an additional band above the IgG heavy chain was clearly observed after the crosslinking procedure (Figure 4A). This additional band corresponds to those IgG heavy chains that have been crosslinked with a Protein Z. No analogous band was observed above the IgG light chain, indicating the Fc-specific nature of Protein Z binding and crosslinking.
Figure 4. Evaluation of UV crosslinked Protein Z-rituximab conjugates via SDS-PAGE.

Samples containing photoreactive Protein Z and/or rituximab (Ritux) were UV or mock irradiated and analyzed via SDS-PAGE under reducing and non-reducing conditions. (A) The reducing gel shows that the exposure of samples containing photoreactive Protein Z and rituximab to UV irradiation results in an additional band above the heavy chain (lane 5), corresponding to Protein Z crosslinked heavy chains. No additional bands were observed above the light chain. (B) The non-reducing gel clearly shows the appearance of two additional bands when rituximab is crosslinked with Protein Z (lane 5), corresponding to 1 or 2 crosslinked Protein Z per IgG. The formation of the additional bands is dependent on both the presence of rituximab and Protein Z, as well as on exposure to UV.
While the reducing SDS-PAGE showed that approximately 50% of the heavy chains were crosslinked, which would be consistent with one Protein Z per IgG, it has previously been reported that each IgG could bind up to two Protein Zs.[31, 47] Therefore analysis was also performed using a non-reducing SDS-PAGE gel, which did in fact show two additional bands in the crosslinked rituximab sample, corresponding to whole rituximab crosslinked with either one or two Protein Zs (Figure 4B). Image analysis of the gel showed that up to 80% of rituximab was crosslinked with at least one Protein Z.
In addition to the F13BPA variant, the F5BPA variant of Protein Z, as was described previously by Konrad et. al, was also crosslinked onto rituximab under the same conditions.[30] Compared to the F13BPA variant, which showed 50% crosslinking as assessed by reducing gel, the F5 variant showed a significantly lower crosslinking efficiency of only 20% (Figure S2). Additionally, the F5BPA variant was observed to form two additional bands upon crosslinking. It is speculated that the BPA may be forming crosslinks to two different sites within the IgG heavy chain, thereby generating differentially unfolded proteins with different migration shifts.
2.4. Crosslinking monoclonal antibodies in ascites solution
In order to highlight Protein Z’s specific binding towards IgG, crosslinking was also performed with monoclonal antibodies in ascites fluid. AzFP-ligated Protein Z was directly incubated with ascites fluid containing anti-BSA antibodies. As a control, Protein Z was also incubated with purified rituximab. Both samples were then exposed to UV light and examined by reducing SDS-PAGE gel. In the gel, an additional fluorescent band was observed in both crosslinked samples at a slightly higher apparent molecular weight than the IgG heavy chain, indicative of Protein Z-crosslinking (Figure 5). No other fluorescent band was observed in the UV exposed ascites fluid sample. Together, these findings suggest that even in a complex biological solution such as the ascites fluid, Protein Z’s capacity to crosslink IgG is not only preserved but also remains Fc-specific.
Figure 5. Crosslinking of photoreactive Protein Z and IgG in ascites fluid.
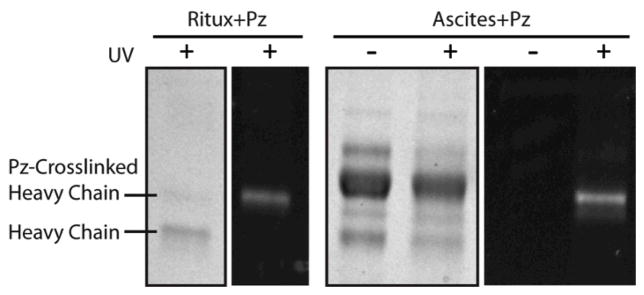
Photoreactive F13BPA Protein Z-AzFP conjugates that were incubated with rituximab or monoclonal anti-BSA IgG in ascites fluid were either mock or UV irradiated and analyzed via a reducing SDS-PAGE gel. Gels were imaged under white light and via fluorescence.
2.5. B cell labeling using rituximab targeted SPIO nanoparticles
To underscore the versatility of Protein Z-crosslinked antibodies and their utility in nanotechnology applications, photoreactive Protein Z-AzFP was first crosslinked to rituximab and then site-specifically conjugated to ADIBO-functionalized SPIO nanoparticles (Figure 1C: Conjugation). These rituximab-conjugated nanoparticles were analyzed by dynamic light scattering (DLS), which showed an increase in the hydrodynamic diameter following conjugation, from 51 nm for unlabeled nanoparticles to 76 nm for rituximab-conjugated-nanoparticles (Figure S3). The correct conjugation was further confirmed via reducing SDS-PAGE gel. Since nanoparticles are too large to enter the gel matrix, the particles and their covalently conjugated Protein Z- heavy chain complex should not be able to migrate into the gel. This was indeed seen in Figure 6A (fluorescent image), where the fluorescent Protein Z is seen to be stuck in bottom of the well containing rituximab-conjugated SPIO. Due to the reduction of disulfide bonds, the non-conjugated half of the IgG heavy chains and all of the light chains should be able to migrate normally into the gel. This effect is also seen in Figure 7A (Coomassie image), where compared to the lane containing rituximab only sample, the lane containing rituximab-conjugated SPIO showed a selective reduction in the amount of heavy chains relative to light chains. It is also worth noting that since some rituximab molecules have both heavy chains crosslinked with a Protein Z, in cases where only one of the two Protein Z-linked heavy chains is immobilized on the SPIO, the non-immobilized Protein Z-heavy chain complex can still migrate into the gel. This is indicated by the fluorescent band that appears above the IgG heavy chain, in the lane containing rituximab-conjugated SPIO (Figure 7A fluorescent image).
Figure 6. Evaluation of B cell-targeted SPIO nanoparticles with site-specifically conjugated rituximab.

(A) Photoreactive F13BPA Protein Z-AzFP was crosslinked to rituximab and conjugated onto ADIBO-functionalized SPIO (SPIO-Ritux). The SPIO-Ritux and rituximab alone were analyzed via a reducing SDS-PAGE gel. (B) Rituximab-conjugated SPIOs were used to label B cells in the presence and absence of excess free rituximab and the results analyzed using flow cytometry. (C) B cells were incubated with unmodified SPIO or SPIO-ritux in the presence or absence of excess free rituximab and T2*-weighted MR images of the respective cell pellets were acquired.
To assay the binding capacity of rituximab-conjugated nanoparticles, the nanoparticles were used to label B-cells in vitro. Since each Protein Z carries a 5-FAM fluorophore, successful labeling of B cells can be established by flow cytometry (Figure 6B) and fluorescence microscopy (Figure S4). Upon treating B cells with rituximab-coated SPIO particles, there was a clear increase in the fluorescent signal. When free rituximab was used as a competitive inhibitor, the labeling was blocked, indicating the specificity of targeting. Receptor-specific cell targeting was also confirmed using T2-weighted magnetic resonance (MR) imaging (Figure 6C). B cell suspensions were imaged in a microplate following incubation with either rituximab-conjugated SPIO, in the presence or absence of excess free rituximab, or with non-functionalized SPIO. Consistent with the flow cytometry studies, B cells incubated with rituximab-conjugated SPIO exhibited much higher MR contrast (indicated by hypointense pixels), compared with B cells incubated with either non-targeted SPIO or targeted SPIO in the presence of a competitive inhibitor.
3. Discussion
Given the tremendously diverse applications requiring antibody conjugates and the variability, non-uniformity, and/or low efficiency of existing conjugation methods, there is a clear need for an enabling technology that is simple, site-specific and broadly applicable. Here, we have shown that an engineered bacterial expression system can efficiently produce a recombinant “tri-functional” Protein Z-based IgG binding domain containing: 1. A photoactive moiety that can be used to crosslink IgG. 2. an azido moiety at the C-terminus that can be used to attach conjugates. 3. a fluorophore for imaging. This construct allows for site-specific antibody bioconjugations and is broadly applicable, due to Protein A’s natural affinity towards a variety of IgG Fc regions.
BPA was selected as the photoreactive crosslinker because it has several favorable properties. Specifically, BPA’s benzophenone group can be activated by long wavelength UV light (365nm), which is not harmful to antibodies or other proteins. Additionally, even after being UV excited to its triplet state, benzophenone can relax back to its unreactive ground state if there are no abstractable hydrogen atoms in close proximity.[32, 33] This allows the photoreactive Protein Z to be produced and handled in ambient light conditions with low risk of photobleaching.
By using the Expressed Protein Ligation (EPL) technique, we were also able to easily introduce a fluorophore and an azide moiety onto the Protein Z during the protein purification step. The 5-FAM fluorophore provides a convenient means to visualize and track conjugated antibodies while the azido group allows further derivitization with a broad array of conjugates. Azide is a particularly attractive choice because it is a click chemistry moiety that is biocompatible, highly reactive and yet also highly specific.[43, 48] Pending user preference, other application-specific functions can also be easily incorporated into our Protein Z since the short EPL peptide that carries the functional groups can be easily synthesized. We have already successfully incorporated a terminal alkyne in place of the azido group and an Atto 740 near infrared dye in place of the 5-FAM fluorophore.[45, 49]
Lastly, since the reported Protein Z is produced recombinantly, it can be produced efficiently and in high yields. This makes this conjugation method readily accessible to a large number of researchers and scalable for commercial applications, especially given the ease of use and widespread availability of the E coli. expression system. Notably, for potential clinical applications, immunogenecity of Protein Z does not appear to be a significant problem.[25, 50] A variant of Protein Z known as an Affibody® has been reported to induce little to no detectable activation of the immune system.[51] In the cases where an immune response was measurable, it did not have an adverse effect on the binding capacity of the Affibody® molecule. Interactions of Protein A with EGFR have been reported, but immunoprecipitation studies with Protein A show no significant binding, and Protein Z shows minimal ability to induce sTNFR1.[52]
To demonstrate the applicability of our conjugation method, we utilized it to create targeted nanoparticles. While nanoparticles that are currently in clinical use (i.e. Doxil) mostly rely on the Enhanced Permeability and Retention effect (EPR) to accumulate in disease tissues, it is increasingly clear that in order to achieve more sensitive diagnosis, safer therapeutic index and more exquisite control of pharmacokinetics, targeting is required.[53] In our experience, a major hurdle in preparing targeted nanoparticles is the difficulty of conjugating targeting ligands efficiently and economically. Moreover, nanoparticle avidity is likely to be particularly sensitive to poorly oriented antibodies generated by traditional conjugation methods, due to their limited surface area. Therefore, a versatile, efficient, site-specific conjugation method has the potential to significantly benefit and simplify the preparation of various targeted nanoparticle formulations. Given the many choices of potential biological targets, the array of antibody clones that bind to each antigen, and the high cost of each antibody, a conjugation method that spares the Fab region and obviates IgG prepreufication will also improve targeting capacity and allow for rapid prototyping and scale-up of targeted nanoparticles.
Despite the promising initial results obtained with the photoreactive Protein Z construct developed here, there remain a number of potential areas for improvement. For example, while the 80% crosslinking efficiency of the F13BPA variant is already adequate for most applications, it may be possible to obtain even faster and more complete crosslinking by incorporating multiple BPAs into the Protein Z. Additionally, since Protein Z only binds weakly to the mouse IgG1 antibody subtype (mIgG1), it is not optimized for conjugating many commonly used research antibodies. However, Yu et. al. recently reported a F5I mutation for Protein Z that can substantially increase its affinity for mIgG1.[31] Therefore, incorporating this mutation may be one option for increasing the repertoire of antibodies that can be efficiently conjugated with this approach.
Lastly, besides the nanoparticle-based application already tested here, there are a number of additional areas that may readily benefit from this Protein Z based conjugation method. For example, Protein Z can be used to functionalize microplates with higher antibody densities in a site-specific manner or control the amount and location of drug attachment in antibody-drug conjugates, thereby improving their therapeutic index.[9–11] Additionally, the photo-dependent nature of the conjugation can be used for photo-immobilizing IgGs onto surfaces in defined patterns. Such photo-patterned immunoreactive surfaces herald interesting diagnostic and research potentials.[54–56]
4. Conclusion
We have successfully demonstrated that a recombinantly expressed, photoactive Protein Z domain can be used to site-specifically and covalently conjugate IgGs onto nanoparticles. Our conjugation method is favorable for several reasons. 1. It is broadly applicable to a wide range of native full-length antibodies. 2. It conjugates IgGs at their Fc domains, hence avoiding steric hindrance or destruction of the antigen binding domain. 3. The mild yet versatile nature of our technique suggests various potential applications beyond those described here.
5. Experimental Section
Materials
An azido fluorescent peptide (AzFP) with the sequence NH2-CDPEK(5-FAM)DSGK(N3)S-CONH 2 was custom synthesized by Anaspec (Fremont, CA). The K(5-FAM) represents a lysine with a 5-carboxyfluorescein covalently attached to its side chain amino group and the K(N3) represents a lysine with an azido group attached to its side chain-amino group. The ADIBO-NHS ester was synthesized as previously described.[44] The SPIO coating material, dextran T10, was purchased from Pharmacosmos (Denmark). Mouse Anti-BSA monoclonal antibody in ascites (B2901) was acquired from Sigma Aldrich (St. Louis, MO), and clinical grade anti-CD20 antibody, rituximab, was obtained from Genentech (South San Francisco, CA). Human lymphoma B cells (Burkitt GA-10) were obtained from the American Type Culture Collection (Manassas, VA). The 70 mm volume coil used for radiofrequency transmission and reception was purchased from Insight Neuroimaging Systems, LLC (Worcester, MA). All other reagents were purchased from Thermo Fisher Scientific (Waltham, MA) unless otherwise noted.
Cloning of Protein Z Recombinant Protein into pTXB1 Vector
Two complimentary oligonucleotides encoding the Protein Z amino acid sequence [27] and flanked at both ends by 15 base sequences homologous to the desired restriction sites of the destination vector were ordered from Integrated DNA Technologies (Coralville, IA). To improve subsequent cleavage from the affinity column, an additional 9 base pairs encoding a “MRM” amino acid sequence were included in the oligonucleotides at the C-terminal end of the Protein Z sequence. The full amino acid sequence for the Protein Z can be found in Supporting Information.
Oligonucleotides were incubated together at a final concentration of 5 μM and hybridized at room temperature for 30 min. The resulting Protein Z sequence was gel purified and directly ligated with gel-purified NdeI- SapI double digested pTXB1 vector (New England Biolabs, Inc) via the CloneEZ kit (Genscript). Insertion of the Protein Z sequence was verified by DNA sequencing using the T7 promoter as the sequencing primer. Site-directed mutagenesis of selected codons into amber codon (TAG) was done using Quikchange Mutagenesis Kit (Agilent).
Expression and Purification of Protein Z Recombinant Protein
The pTXB1 and pEVOL-pBpF (Prof. Peter Schultz, Addgene.org) plasmids were cotransformed into the T7 Expression Crystal Competent Cells (New England Biolabs). Bacterial cell cultures were initially grown overnight in an air shaker (225 rpm) at 37° C in 3 mL of lysogeny broth (LB) media. Cultures were scaled up to 50 mL of LB media and grown overnight under the same conditions, and then inoculated into 1 L LB media containing 50 mg/L of ampicillin and 25 mg/L of chloramphenicol.
At optical density (OD) 600 nm = 0.6, Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM to induce T7 RNA polymerase-based expression. For BPA incorporation, L-benzoylphenylalanine (Bachem, King of Prussia, PA) was added into the culture for a final concentration of 300 μM and the culture was left to grow for 30 min. Next, IPTG was added to a final concentration of 0.5 mM and arabinose to a final concentration of 0.1% to begin induction.
Cultures were allowed to express for 2 h at 37°C. Bacterial cultures were centrifugally pelleted at 10,000 g for 5 min, resuspended in 5 mL of column buffer (20 mM Na-HEPES, 0.5 M NaCl, 1 mM EDTA, pH 8.5) containing 0.75 g/L lysozyme and 50 mM phenylmethylsulfonyl fluoride. Cells were lysed by pulse sonication on ice. Cells were centrifuged at 15,000 g for 30 min at 4°C. Supernatant was collected and stored at −20°C. For the following purification steps, all procedures were run at 4°C. The supernatant (1 mL) was incubated for 10 min in a 10 mL Poly-Prep chromatography column (Bio-Rad, Hercules, CA) packed with 1 mL of chitin beads (New England Biolabs, Inc). Supernatant was allowed to pass through the column and chitin beads were washed with 50 mL of column buffer at a flow rate of approximately 2 mL/min.
Expressed Protein Ligation
MESNA (3 mL, 50 mM pH 7.4) was quickly passed through the column in order to evenly distribute the MESNA throughout the chitin beads, and flow was stopped. To make AzFP-ligated Protein Z, the AzFP (0.1 mM) was added into the column and incubated overnight at room temperature. For protein Z with a free C-terminus or cysteine-ligated protein Z, dithiothreitol (50mM) or free cysteine (50 mM), respectively, was added instead of AzFP into the column and incubated overnight at room temperature. The next day the column was eluted using 4 mL buffer (0.1 M Tris-HCl, pH 8.5). The EPL product and excess AzFP, DTT or cysteine were separated on a Superdex 30 chromatography column. Purification and concentration of the final product can also be performed using a 3 kD molecular weight cut-off (MWCO) filter.
16% Tricine-SDS-PAGE gels were used to visualize the separation between the cysteine and AzFP ligated Protein Zs. Gels were run in accordance with previously published methods for the visualization of small proteins.[57]
Protein Z can also be purified further with RP-HPLC (Varian Prostar). A C8 300 μm 5 μm column (Varian) was used. Protein Zs were eluted at 1 mL/min using a mixture of water and acetonitrile, both containing 0.1% TFA. The solvent gradient used was: 95%-75% water over the first 10 min, then 75%-69% over the next 60 min. Absorbance was monitored at 215 nm. The collected fractions were then dried using vacuum centrifuge concentrator (Labconco) and reconstituted in 0.1 M Tris-HCl buffer, pH 8.5.
Matrix-assisted laser desorption/ionization (MALDI) –mass spectrometry analysis of the HPLC purified product was carried out using a Voyager System 6030 (Applied Biosystems), using ubiquitin as the internal calibration.
Photocrosslinking
Stock rituximab (10 mg/mL) was diluted to 1 mg/mL in 0.1 M Tris buffer and mixed with HPLC purified Protein Z at the final concentrations of 100 μg/mL rixtuximab and 1 mg/mL Protein Z or Protein Z-AzFP. Incubation was done at 25°C for 1 hour with shaking. Afterwards, the sample was then placed in an ice bath under a 365 nm UV lamp (UVP UV L56, Upland, CA) for 3 hours.
Uncrosslinked Protein Z was then dissociated by buffer exchanging with pH 3.5 buffer (0.2 M Glycine). Both dissociated and excess Protein Z were then filtered out using an Amicon 50 kDa MWCO filter. The buffer was then exchanged back to 0.1 M Tris-HCl, pH 8.5 using the filter.
In the case of anti-BSA monoclonal antibody, the ascites fluid was incubated with HPLC-purified Protein Z-AzFP (1 mg/mL) at the volume-to-volume ratio of 1:10, and then UV crosslinked as above. The crosslinked solution was then washed 3 times using 0.2 M Glycine buffer and filtered with a 50 kDa MWCO filter as above and finally buffer exchanged back into 0.1 M Tris-HCl, pH 8.5 for a final dilution of 1:10 of the original ascites.
SPIO Nanoparticle Synthesis and Amination
Dextran-coated SPIO nanoparticles (NPs) were prepared using the co-precipitation method.[58] Specifically, 1.5 g FeCl2 and 4 g FeCl3 were each dissolved in 12.5 mL of degassed dH2O and added to 25 g dextran T10 in 50 mL diH2O. Keeping this mixing solution at 4°C, 15 mL ammonium hydroxide was slowly added to this mixture. The resulting black solution was then heated to 90°C for 1 h and cooled overnight. Purification of SPIO NPs was accomplished by ultracentrifugation of the mixture at 20k relative centrifugal force (RCF) for 30 min. Pellets were discarded, and the supernatant was subjected to diafiltration against greater than 20 volumes of 0.02 M citrate, 0.15 M sodium chloride buffer using a 100 kDa MWCO filter (GE Healthcare). The purified particles were then crosslinked by reacting the particles (10 mg Fe mL −1) with 25% (v/v) 10 M NaOH and 33% epichlorohydrin. After mixing for 24 h, the particles were briefly dialyzed and then functionalized with amines by adding 25% ammonium hydroxide. This reaction was allowed to continue for another 24 h followed by diafiltration as described above.
ADIBO Modification of SPIO Nanoparticles
Surface amines on dextran-coated SPIO NPs (5 mg/mL) were reacted with the amine-reactive ADIBO-NHS, diluted 10 times from stock (138mM) in dimethyl sulfoxide (DMSO), in 0.1 M sodium phosphate buffer, pH 9. The linker was added at 100 times molar excess to the aminated SPIO nanoparticles. All nanoparticles solutions were mixed overnight at room temperature. ADIBO-SPIO NPs were purified via PD-10 chromatography columns (GE Healthcare, Piscataway, NJ).
Copper-free click conjugation of ADIBO-SPIO NPs was performed by mixing the SPIO NP (2 mg/mL) with 30 μM of filtered Pz-rituximab in sodium carbonate buffer (pH 8). Reactions were mixed overnight at room temperature and then purified on MACS μ columns (Miltenyi Biotec, Bergisch Gladbach, Germany) and equilibrated with PBS.
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano from Malvern Instruments. (Malvern Instruments, Malvern, UK) The scattering angle was held constant at 90°.
Microscopy and Flow Cytometric Analysis
SPIO NPs were incubated with 100 μL of 4 × 106 cells/mL Burkitt GA-10 lymphoma B cells for 30 minutes at 37°C, 5% CO2, in a 96-well plate. SPIO NPs were added at final concentrations of 10 μg Fe/mL. In the antibody inhibition experiments, 100 μg/mL (final concentrations) of free anti-CD20 was added prior to the addition of the antibody-conjugated NPs. The free, unbound particles were purified from the cells through three PBS washes at 1,000 RCF for 5 minutes each. Cells were finally resuspended in 300 μL of PBS and placed in a 96-well plate.
Microscopy images were acquired with an Olympus IX 81 inverted fluorescence microscope using a LUC PLAN 40x objective (numerical aperture 0.6; Olympus) and an SOLA light engine excitation source (Lumencor, Beaverton, OR). Fluorescent images were acquired using a back-illuminated electron multiplying charge-coupled device camera (Andor Technology PLC, Belfast, Northern Ireland).
Flow cytometry of the washed cell samples was performed using a Guava Easycyte Plus system (Guava Technologies, Hayward, CA). Flow cytometry data were analyzed using FlowJo (TreeStar Inc., San Francisco, CA).
Cell Pellet MR Imaging
GA-10 cells were combined to form a single cell pellet for each nanoparticle formulation. The samples were transferred to a 384-well plate, and the cells were pelleted to the bottom of each well with brief, low-speed centrifugation. The plate was then imaged on a 9.4 T magnet interfaced to a Varian INOVA console using a 70 mm inner diameter volume coil for radio-frequency transmission and reception. T2*-weighted gradient echo (GEMS) MR images were collected using parameters as follows: repetition time (TR) = 200 ms, echo time (TE) = 5 ms, flip angle = 20°, slice thickness = 0.5 mm, field of view (FOV) = 4 cm × 4 cm, number of acquisitions = 8, resolution = 256 × 256.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institute of Health (NIH) NIBIB/R01-EB012065 (AT), NCI/R01-CA157766 (AT), and NCI/R01-CA175480 (ZC).
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
James Zhe Hui, Department of Bioengineering, University of Pennsylvania, 210 S. 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA.
Ajlan Al Zaki, Department of Bioengineering, University of Pennsylvania, 210 S. 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA.
Prof. Zhiliang Cheng, Department of Bioengineering, University of Pennsylvania, 210 S. 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA
Prof. Vladimir Popik, Department of Chemistry, University of Georgia, Athens, GA 30602, USA
Prof. Hongtao Zhang, Department of Pathology and Lab Medicine, University of Pennsylvania, PA 19104, USA
Prof. Eline T. Luning Prak, Department of Pathology and Lab Medicine, University of Pennsylvania, PA 19104, USA
Prof. Andrew Tsourkas, Email: atsourk@seas.upenn.edu, Department of Bioengineering, University of Pennsylvania, 210 S. 33rd Street, 240 Skirkanich Hall, Philadelphia, PA 19104, USA
References
- 1.Thorek DL, Elias DR, Tsourkas A. Mol Imaging. 2009;8 (4):221. [PubMed] [Google Scholar]
- 2.Carter PJ. Nat Rev Immunol. 2006;6 (5):343. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 3.Allen TM. Nature reviews Cancer. 2002;2 (10):750. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 4.Hermanson GT. Bioconjugate techniques. Academic Press; San Diego: 1996. p. xxv. [Google Scholar]
- 5.Porstmann T, Kiessig ST. J Immunol Methods. 1992;150(1–2):5. doi: 10.1016/0022-1759(92)90061-w. [DOI] [PubMed] [Google Scholar]
- 6.Kausaite-Minkstimiene A, Ramanaviciene A, Kirlyte J, Ramanavicius A. Anal Chem. 2010;82 (15):6401. doi: 10.1021/ac100468k. [DOI] [PubMed] [Google Scholar]
- 7.Tajima N, Takai M, Ishihara K. Anal Chem. 2011;83 (6):1969. doi: 10.1021/ac1026786. [DOI] [PubMed] [Google Scholar]
- 8.Vashist SK. Anal Biochem. 2012;421 (1):336. doi: 10.1016/j.ab.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 9.Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, Smider VV, Schultz PG. Proc Natl Acad Sci U S A. 2012;109 (40):16101. doi: 10.1073/pnas.1211023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. Nat Biotechnol. 2008;26 (8):925. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 11.Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, Ho WH, Farias S, Casas MG, Abdiche Y, Zhou D, Chandrasekaran R, Samain C, Loo C, Rossi A, Rickert M, Krimm S, Wong T, Chin SM, Yu J, Dilley J, Chaparro-Riggers J, Filzen GF, O’Donnell CJ, Wang F, Myers JS, Pons J, Shelton DL, Rajpal A. Chem Biol. 2013;20 (2):161. doi: 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. J Control Release. 2007;120(1–2):18. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Natarajan A, Gruettner C, Ivkov R, DeNardo GL, Mirick G, Yuan A, Foreman A, DeNardo SJ. Bioconjug Chem. 2008;19 (6):1211. doi: 10.1021/bc800015n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeglis BM, Davis CB, Aggeler R, Kang HC, Chen A, Agnew BJ, Lewis JS. Bioconjug Chem. 2013;24 (6):1057. doi: 10.1021/bc400122c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyons A, King DJ, Owens RJ, Yarranton GT, Millican A, Whittle NR, Adair JR. Protein Eng. 1990;3 (8):703. doi: 10.1093/protein/3.8.703. [DOI] [PubMed] [Google Scholar]
- 16.McDonagh CF, Turcott E, Westendorf L, Webster JB, Alley SC, Kim K, Andreyka J, Stone I, Hamblett KJ, Francisco JA, Carter P. Protein Eng Des Sel. 2006;19 (7):299. doi: 10.1093/protein/gzl013. [DOI] [PubMed] [Google Scholar]
- 17.Stimmel JB, Merrill BM, Kuyper LF, Moxham CP, Hutchins JT, Fling ME, Kull FC., Jr J Biol Chem. 2000;275 (39):30445. doi: 10.1074/jbc.M001672200. [DOI] [PubMed] [Google Scholar]
- 18.Voynov V, Chennamsetty N, Kayser V, Wallny HJ, Helk B, Trout BL. Bioconjug Chem. 2010;21 (2):385. doi: 10.1021/bc900509s. [DOI] [PubMed] [Google Scholar]
- 19.Alves NJ, Kiziltepe T, Bilgicer B. Langmuir. 2012;28 (25):9640. doi: 10.1021/la301887s. [DOI] [PubMed] [Google Scholar]
- 20.Cho IH, Paek EH, Lee H, Kang JY, Kim TS, Paek SH. Anal Biochem. 2007;365 (1):14. doi: 10.1016/j.ab.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Choi HK, Lee SY, Lim MW, Chang JH. Biosens Bioelectron. 2011;28 (1):146. doi: 10.1016/j.bios.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Peluso P, Wilson DS, Do D, Tran H, Venkatasubbaiah M, Quincy D, Heidecker B, Poindexter K, Tolani N, Phelan M, Witte K, Jung LS, Wagner P, Nock S. Anal Biochem. 2003;312 (2):113. doi: 10.1016/s0003-2697(02)00442-6. [DOI] [PubMed] [Google Scholar]
- 23.Cho IH, Seo SM, Jeon JW, Paek SH. J Biomed Biotechnol. 2009;2009:104094. doi: 10.1155/2009/104094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo JS, Lee S, Poulter CD. J Am Chem Soc. 2013;135(24):8973. doi: 10.1021/ja402447g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiandra L, Mazzucchelli S, De Palma C, Colombo M, Allevi R, Sommaruga S, Clementi E, Bellini M, Prosperi D, Corsi F. ACS Nano. 2013;7 (7):6092. doi: 10.1021/nn4018922. [DOI] [PubMed] [Google Scholar]
- 26.Mazzucchelli S, Colombo M, De Palma C, Salvade A, Verderio P, Coghi MD, Clementi E, Tortora P, Corsi F, Prosperi D. ACS Nano. 2010;4 (10):5693. doi: 10.1021/nn101307r. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlen M. Protein Eng. 1987;1 (2):107. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 28.Cedergren L, Andersson R, Jansson B, Uhlen M, Nilsson B. Protein Eng. 1993;6 (4):441. doi: 10.1093/protein/6.4.441. [DOI] [PubMed] [Google Scholar]
- 29.Cai Z, Fu T, Nagai Y, Lam L, Yee M, Zhu Z, Zhang H. Cancer Res. 2013;73 (8):2619. doi: 10.1158/0008-5472.CAN-12-3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konrad A, Karlstrom AE, Hober S. Bioconjug Chem. 2011;22 (12):2395. doi: 10.1021/bc200052h. [DOI] [PubMed] [Google Scholar]
- 31.Yu F, Jarver P, Nygren PA. PLoS One. 2013;8 (2):e56597. doi: 10.1371/journal.pone.0056597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorman G, Prestwich GD. Biochemistry. 1994;33 (19):5661. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 33.Kauer JC, Erickson-Viitanen S, Wolfe HR, Jr, DeGrado WF. J Biol Chem. 1986;261 (23):10695. [PubMed] [Google Scholar]
- 34.Young TS, Ahmad I, Yin JA, Schultz PG. J Mol Biol. 2010;395 (2):361. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 35.England PM. Biochemistry. 2004;43 (37):11623. doi: 10.1021/bi048862q. [DOI] [PubMed] [Google Scholar]
- 36.Kawamura A, Hindi S, Mihai DM, James L, Aminova O. Bioorg Med Chem. 2008;16(19):8824. doi: 10.1016/j.bmc.2008.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wittelsberger A, Mierke DF, Rosenblatt M. Chem Biol Drug Des. 2008;71 (4):380. doi: 10.1111/j.1747-0285.2008.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schatz PJ. Biotechnology (N Y) 1993;11(10):1138. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 39.Jung Y, Lee JM, Kim J-w, Yoon J, Cho H, Chung BH. Anal Chem. 2009;81 (3):936. doi: 10.1021/ac8014565. [DOI] [PubMed] [Google Scholar]
- 40.Breitz HB, Weiden PL, Beaumier PL, Axworthy DB, Seiler C, Su FM, Graves S, Bryan K, Reno JM. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2000;41(1):131. [PubMed] [Google Scholar]
- 41.Goldenberg DM, Sharkey RM, Paganelli G, Barbet J, Chatal JF. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24 (5):823. doi: 10.1200/JCO.2005.03.8471. [DOI] [PubMed] [Google Scholar]
- 42.Paganelli G, Grana C, Chinol M, Cremonesi M, De Cicco C, De Braud F, Robertson C, Zurrida S, Casadio C, Zoboli S, Siccardi AG, Veronesi U. European journal of nuclear medicine. 1999;26 (4):348. doi: 10.1007/s002590050397. [DOI] [PubMed] [Google Scholar]
- 43.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, Lo A, Bertozzi CR. Proc Natl Acad Sci U S A. 2010;107 (5):1821. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuzmin A, Poloukhtine A, Wolfert MA, Popik VV. Bioconjug Chem. 2010;21 (11):2076. doi: 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- 45.Elias DR, Cheng Z, Tsourkas A. Small. 2010;6 (21):2460. doi: 10.1002/smll.201001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci U S A. 1998;95 (12):6705. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deisenhofer J. Biochemistry. 1981;20 (9):2361. [PubMed] [Google Scholar]
- 48.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, Kwon IC, Jeong SY, Kim K. Angew Chem Int Ed Engl. 2012;51 (47):11836. doi: 10.1002/anie.201206703. [DOI] [PubMed] [Google Scholar]
- 49.Cheng Z, Elias DR, Kamat NP, Johnston ED, Poloukhtine A, Popik V, Hammer DA, Tsourkas A. Bioconjug Chem. 2011;22 (10):2021. doi: 10.1021/bc200214g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dima S, Medesan C, Mota G, Moraru I, Sjoquist J, Ghetie V. Eur J Immunol. 1983;13(8):605. doi: 10.1002/eji.1830130802. [DOI] [PubMed] [Google Scholar]
- 51. [accessed March 18, 2014];Affibody Annual Report. http://www.affibody.com/upload/Other/AnnualReport2003.pdf.
- 52.Gomez MI, O’Seaghdha M, Magargee M, Foster TJ, Prince AS. J Biol Chem. 2006;281 (29):20190. doi: 10.1074/jbc.M601956200. [DOI] [PubMed] [Google Scholar]
- 53.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338 (6109):903. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ekblad T, Liedberg B. Curr Opin Colloid Interface Sci. 2010;15 (6):499. [Google Scholar]
- 55.Feyssa B, Liedert C, Kivimaki L, Johansson LS, Jantunen H, Hakalahti L. PLoS One. 2013;8 (7):e68918. doi: 10.1371/journal.pone.0068918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin TA, Herman CT, Limpoco FT, Michael MC, Potts GK, Bailey RC. ACS Appl Mater Interfaces. 2011;3 (9):3762. doi: 10.1021/am2009597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schagger H. Nat Protoc. 2006;1 (1):16. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 58.Thorek DL, Tsourkas A. Biomaterials. 2008;29 (26):3583. doi: 10.1016/j.biomaterials.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.