

Abstract
For four decades, liposomes composed of both naturally occurring and synthetic lipids have been investigated as delivery vehicles for low molecular weight and macromolecular drugs. These studies paved the way for the clinical and commercial success of a number of liposomal drugs, each of which required a tailored formulation; one liposome size does not fit all drugs! Instead, the physicochemical properties of the liposome must be matched to the pharmacology of the drug. An extensive biophysical literature demonstrates that varying lipid composition can influence the size, membrane stability, in vivo interactions, and drug release properties of a liposome. In this review we focus on recently described synthetic lipid headgroups, linkers and hydrophobic domains that can provide control over the intermolecular forces, phase preference, and macroscopic behavior of liposomes. These synthetic lipids further our understanding of lipid biophysics, promote targeted drug delivery, and improve liposome stability. We further highlight the immune reactivity of novel synthetic headgroups as a key design consideration. For instance it was originally thought that synthetic PEGylated lipids were immunologically inert; however, it’s been observed that under certain conditions PEGylated lipids induce humoral immunity. Such immune activation may be a limitation to the use of other engineered lipid headgroups for drug delivery. In addition to the potential immunogenicity of engineered lipids, future investigations on liposome drugs in vivo should pay particular attention to the location and dynamics of payload release.
Keywords: Biophysics, Immunology, Liposomes, Payload Release, Targeting
1. Introduction
Lipid vesicles, or liposomes, are micro- or nano-structures formed from a bilayer of lipid surrounding an aqueous core. In the past 30 years they have been widely used to modify the pharmacokinetics, biodistribution, and cellular trafficking of drugs, nucleic acids, and proteins. Liposomal therapeutics have had preclinical and commercial success with more than 46,000 publications, 850 patents, and 13 clinically approved liposome agents with greater than $750 million in revenue in 2011. These drugs continue to advance through the clinic, and the results from a number of pivotal phase III trials, including those from Merrimack Pharmaceuticals (NCT01494506) and Celator Pharmaceuticals (NCT01696084), will be available in the next 18 months.
Liposomes can be tailored to deliver a range of cargo using a diverse toolbox of lipids with well-characterized biophysical behavior. Lipids in this toolbox can be naturally occurring or rationally designed using a variety of hydrophilic headgroups, linkers, and hydrophobic moieties. Selecting the appropriate combination of lipids and the method of assembly provides control over liposome macrostructures, biophysical characteristics, and subsequent in vivo behavior.
At the most fundamental level, the properties of a liposome depend upon the subtle physicochemical interactions among the various lipid species in its composition. A wealth of research has focused on the design, synthesis and characterization of naturally occurring and synthetic lipids. Individual lipids can be combined to form a myriad of superstructures including bilayers, and bilayer properties can be tuned to modulate drug release and membrane stability (Figure 1A,B). In a simplified bilayer model acyl chain length dictates bilayer thickness and phase transition temperature (Tm), acyl chain saturation controls bilayer fluidity, and headgroup interactions impact inter- and intra-lipid molecular forces (Figure 1B). Liposome behavior can be adjusted by incorporating synthetic lipids such as lipid prodrugs, fusogenic lipids and functionalizable lipids into the bilayer (Figure 1C). As a result, there have been 50 years of synthetic efforts to develop novel lipids with properties that improve delivery while maintaining low cytotoxicity and immunogenicity. A number of databases classify lipids by structure [1], organize information related lipid Tm and phase preferences into phase diagrams [2], or detail methods for liposome characterization (cyberlipid.org; lipidmaps.org). This abundance of information provides accessible resources to guide the development of lipids for drug delivery.
Figure 1. Modulating liposome behavior.
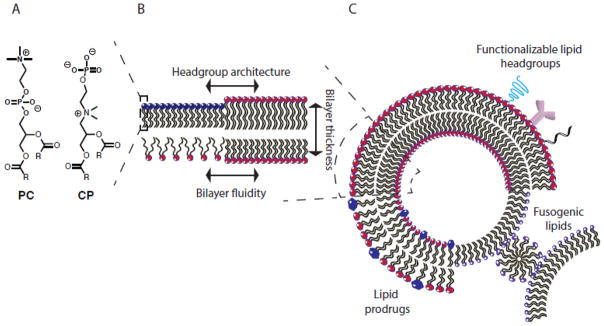
Liposome behavior can be controlled across a number of length scales: (A) engineering of individual lipids (B) modification of bilayer biophysics and (C) inclusion of lipids that direct macroscopic liposome behavior and interactions.
As a starting point, nature has provided a variety of lipids that have evolved to satisfy diverse structural and functional purposes. Phospholipids with neutral, zwitterionic, or anionic headgroups, such as: phosphatidylcholine (PC), sphingomyelin, and phosphatidylethanolamine (PE), are the primary components of cell membranes and are essential for membrane stability and intracellular trafficking. Glycerides are neutral lipids that serve as energy sources and signaling molecules in mammalian cells. Naturally occurring anionic lipids, including phosphatidylglycerol, phosphatidylinositol, cardiolipin, phosphatidic acid, and phosphatidylserine are also found in mammalian cell membranes, and play a critical role in cellular signaling, lipid-protein interactions, and membrane trafficking [3–7]. These naturally occurring lipids are components of FDA approved therapeutics such as Doxil®, AmBisome®, and DepoCyt® [8]. Half a century of characterization of the physicochemical properties of these lipids allows the lipid engineer to build from a wealth of structure-function relationships to design systems with control over stability and payload release.
1.1 Synthetic lipids for drug delivery
There are three key steps in liposomal drug delivery that can be improved with synthetic lipids: 1) extended circulation of the liposome after intravenous administration, 2) directed lipid headgroup interactions and cell targeting and 3) controlled payload release (Figure 2, 3). Synthetic lipids can be formulated in liposomes alongside naturally occurring lipids to serve these structural or functional roles.
Figure 2. Overview of lipid engineering.

Potential modifications to a single lipid are highlighted along with their potential functional consequences.
Figure 3. Three key steps in liposome drug delivery.
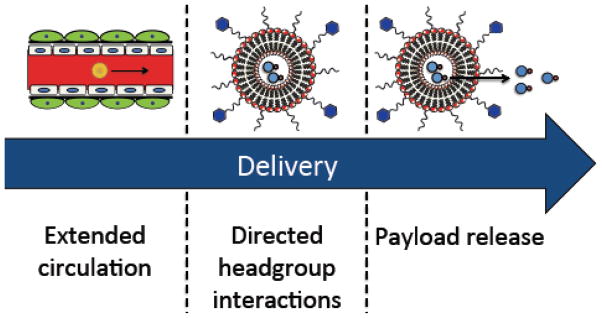
After intravenous administration, liposomes circulate in the bloodstream and accumulate at the disease site. Directing liposome headgroup interactions allows for targeting to the appropriate cellular or tissue compartment. Finally, membrane thickness, fluidity and interfacial charge orientation control payload release.
After administration, liposomes circulate in the bloodstream and accumulate in tumors by the enhanced permeability and retention (EPR) effect. Increasing the circulation half-life of the liposomes allows a higher fraction of the dose to transit to the tumor and increases the probability that liposomes will extravasate into the tumor parenchyma. Functionalizing lipid headgroups with polymers, proteins, or peptides, can extend liposome circulation time by reducing liposome adhesion to mononuclear phagocyte system (MPS) cells and preventing destabilizing interactions with serum proteins (Figure 2).
The next step in delivery involves delivering payload to specific cell types by attachment of targeting ligands. While cell targeting is not a requisite liposome characteristic, a number of next-generation delivery systems look to take advantage of targeting particular cell types. To that end, engineering lipids with chemistries that allow for facile attachment of proteins, sugars, or other targeting moieties are of particular interest. In addition to targeting, lipids with specific and programmable interactions in the headgroup can direct macrostructure formation and membrane biophysics. Such synthetic lipids can be used to probe the intra- and inter-molecular forces governing superstructure formation.
In the final delivery step, liposomes must release their cargo in the appropriate tissue or cellular compartment over the desired timescale. There are a number of ways to control contents release from liposomes: 1) change the liposome formulation, 2) make the cargo more hydrophilic such that it loads into the aqueous compartment, and 3) use remote loading to cause the cargo to accumulate inside the liposome [9]. These techniques are the subject of a number of excellent reviews and will not be discussed in this article [10]. An additional method to control liposome contents release relies on including triggerable lipids that are sensitive to environmental stimuli (pH, shear stress, oxidative environment, etc.) in the liposome formulation. Such lipids allow for burst delivery in the appropriate cellular compartment and have been of particular interest in siRNA delivery, where engineered pH-dependent fusogenic lipids allow for delivery to the cytosol. These lipids have also been extensively reviewed [11,12]. Herein we focus on a number of alternatives to these triggerable systems to control liposome functionalization and payload release, including lipid prodrugs, lipids with inverted headgroup architectures, and lipids with covalently attached hydrophobic structural or drug moieties (Figure 2).
While naturally occurring lipids are the workhorses of liposomal systems, clinical advances would not have been possible without the development of a number of synthetic lipids. In this review we focus on synthetic lipids developed over the past 10 years to probe certain aspects of liposome behavior or to improve liposome in vivo stability and therapeutic activity. We concentrate on lipids for drug delivery and highlight selected successes and failures of such lipids in animal studies or in the clinic.
2. Lipids that extend circulation
2.1 Polymer headgroup lipids
Conventional liposomes (CL) are cleared from circulation by the phagocytic cells of the MPS and preferentially accumulate in the spleen and liver [13]. Sterically stabilized liposomes (SL) are modified on the surface with a hydrophilic polymer that extends from the surface in a brush or mushroom configuration. This steric barrier around the liposome decreases protein adsorption to the lipid membrane, conceals surface charge, reduces liposome adhesion to cell surfaces, and, as a consequence of these factors, extends circulation time. This prolonged circulation improves the exposure of rapidly eliminated drugs by extending payload release and drug exposure and improving biodistribution to the target site. This is especially true for passive targeting strategies that depend on the EPR effect [14], in which liposomes preferentially escape the poorly organized tumor vasculature and accumulate near the tumor blood vessels. Steric stabilization further reduces the fraction of the drug that distributes to the liver, spleen, and bone marrow.
The first SLs incorporated glycolipids such as GM1 ganglioside, cerebroside sulfate, or phosphatidylinositol [15,16]. A significant breakthrough in the design of SLs came in the late 1980’s with the attachment of poly(ethyleneglycol) (PEG) to the liposome surface. PEG increased circulation half-life of proteins and other biomaterials, and it was discovered that similar effects were achievable by including PEGylated lipids into liposome formulations [17,18]. Doxil®, a liposomal doxorubicin formulation incorporating PEG-2000-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PEG2000-DSPE) is the only FDA approved PEGylated liposomal therapeutic [19,20]; its development was recently reviewed [21]. By forming a steric barrier around the liposome, PEG-modified lipids embedded in a lipid bilayer decrease interactions with serum opsonins, cellular ligands/receptors and other pre-existing serum factors [22] while reducing adhesion to other membrane surfaces [23].
In the early work on PEGylated proteins it was assumed that PEG was immunologically inert [17]. However, it is now recognized that PEG can induce an immune response under certain conditions. Importantly, PEGylated drugs and liposomes can initiate the accelerated blood clearance (ABC) effect [24]. In this effect, PEGylated materials induce an anti-PEG IgM response upon the first injection [26]. Subsequent injections are then labeled for removal and are rapidly cleared from circulation and accumulate largely in the liver and spleen [24,25]. This effect reduces the widespread utility of PEG.
Other synthetic polymer-modified lipids also increase circulation half-life by providing a hydrophilic steric coat, and may serve as alternatives to PEG (Table 1, Figure 4). HPMA (poly[N-(2-hydroxypropyl) methacrylamide]) [27–29], PVP (poly(vinylpyrrolidone)) [30,31], PMOX (poly(2- methyl-2- oxazoline)) [32,33], PAcM (poly(N-acryloyl morpholine)) [34,35], PAA (poly(acrylamide)) [36], PG (poly(glycerol)) [37,38], PVA (poly(vinylalcohol)) [39–41], pNIPAM (poly(n-isopropylacrylamide)) [42], and pAAs(poly(amino acids)) [43–46] all increase the circulation half-life of liposomes in vivo. However, inconsistent experimental procedures make it difficult to directly compare these polymers. While only PMOX coated liposomes demonstrated similar circulation times to PEG [33], a number of favorable properties could make these alternative polymers a better choice for certain drug delivery applications [44]. In particular, polymers that avoid the ABC effect and have a lower viscosity than PEG may be advantageous for the therapeutic delivery [44]. Future studies should focus on developing, characterizing, and comparing these polymers.
Table 1.
Summary of published data on polymer modified long circulating liposomes.
| Polymer | Mw | Lipid anchor | Liposome characteristics | Model | Circulation half-life | Ref. |
|---|---|---|---|---|---|---|
| PVP | 6–15 kDa | PE/Palmityl | 3 and 7 mol % polymer 155–190 nm | Mice | 0.7–2.2 h | [34,36] |
| PMOX | 2–5 kDa | DSPE | 5 mol % polymer 90–112 nm | Mice/Rats | 17.8 h | [33,47] |
| PAcM | 6 kDa | PE | 3 and 7 mol % polymer 155–180 nm | Mice | 1.5–2.8 h | [34] |
| HPMA | 3–4 kDa | Oleic acid | 0.3 and 3 mol % polymer 150–200 nm | Mice | 0.8–2.5 h | [48] |
| PVA | 6–20 kDa | (C16H33-S-) | 0.33–8.5 mol % polymer 100–300 nm | Rats | NA | [39–41] |
| PG | 0.2–3 kDa | DPPG/DSPE | 2–15 mol % polymer 100–150 nm | Mice/Rats | 8.8–22.1 h | [37,38] |
| pNIPAM-MAA | 29 kDa | DODA | 0.3 w/w | Rats | 7.7–9.8 h | [42] |
| pAAs | 3–4 kDa | PE/SA/DOD/DODA | 1–15 mol % polymer 105–370 nm | Rats | 2.8 h | [43,44] [45,46] |
| PEG | 2 kDa | DSPE | 5–7 mol % 100 nm | Mice | >20 h | [49] |
Figure 4.
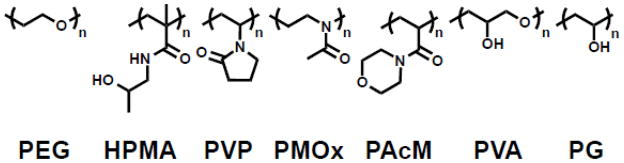
Polymer headgroup lipids for extended circulation of liposomes.
2.2 Alternatives to polymer headgroups for extending circulation
Polymer coatings for liposomes are limited by their high intrinsic viscosity and their induction of the IgM-mediated ABC effect. They can also hinder liposome uptake into diseased cells [50] and fail to stop absorption of serum proteins that promote clearance such as IgG [51]. Approaches that avoid these limitations have been promising in preclinical studies. Masking nanoparticles with markers of “self” is also emerging as an alternative approach to extending liposome circulation. Red blood cell surface proteins have been investigated as nanoparticle coatings that allow for evasion of the RES. Hu and colleagues coated PLGA nanoparticles by extruding them along with disrupted erythrocyte membranes [52]. These nanoparticles, coated in an erythrocyte membrane, had extended circulation compared to nanoparticles coated with PEG or unmodified nanoparticles in mice. In a more defined approach, Rodriguez and colleagues computationally designed a 21 amino acid peptide mimetic of CD47, a marker of self on erythrocytes that impedes phagocytosis by signaling through phagocyte receptor signal regulatory protein-α (SIRP-α) [51]. Nanoparticles opsonized with IgG were coated with peptide or with a PEG brush. While PEG had no mitigating impact on macrophage uptake of the opsonized particles in vitro and in vivo, the peptide prolonged nanoparticle circulation four fold. Further, nanoparticles coated with hCD47 showed a longer circulation time than those coated with PEG. To our knowledge, this peptide has yet to be tested in a liposomal system. A number of additional factors, including shape, surface chemistry, and mechanical properties are under investigation to improve the in vivo properties of nanoparticles [53]. Going forward, it will be important to perform controlled studies comparing these alternatives to PEG and other polymers for extended circulation in vivo. Such studies should place a heavy emphasis on understanding the immunogenicity of these systems.
3. Immunological considerations in lipid headgroup design
Humoral immune responses to natural and synthetic lipids occur under a myriad of conditions in various animals [55], and immune responses to drug delivery systems have been recently reviewed by Jiskoot and colleagues [54]. B cells secrete antibodies when the B cell receptor both possesses an affinity for a specific epitope and is activated (Figure 5). Recognition of antigen by a resting or naïve B cell occurs at the cell surface by membrane bound IgM or IgD. Subsequent B cell activation and antibody induction can occur through different mechanisms involving either a T cell dependent (T-D) response or T cell independent (T-I) response. The pathway for antibody generation during a T-D response requires protein to be processed and loaded in the major histocompatibility complex class II (MHC II) by antigen presenting cells (APCs) for T cell activation and release of cytokines for B cell activation. Activated B cells then undergo class switching to generate excreted forms of IgM, IgG, IgE or IgA depending on the cytokine stimulus from the T cell. T-D responses arising from liposomal protein and peptide antigens in the context of vaccine design have been reviewed [55] and are not the focus of this discussion. Rather, we will examine antibody responses to various non-protein molecules presented on liposomes to identify potential issues to consider in the design of novel lipids for liposomal drug delivery.
Figure 5. Pathways to B cell activation.
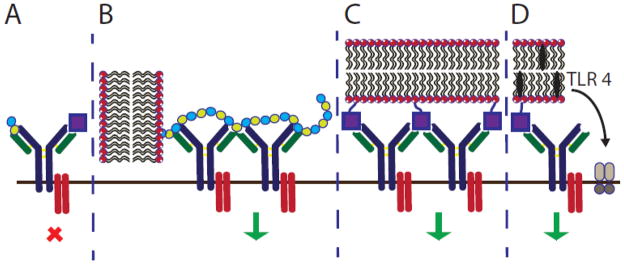
(A) Short polymers or monomeric haptens fail to activate B cells. (B) Long polymers, such as PEG, or (C) multivalent haptens on liposomes can activate B cells by cross-linking B cell receptors. (D) Inclusion of TLR4 agonists alongside haptens can also activate B cells.
In the absence of protein and consequent T cell activation, B cells orchestrate a humoral immune response in the T-I pathway to generate IgM and IgG antibodies. Both antibody isotypes are responsible for complement activation, which may result in adverse response to non-protein antigens displayed on liposomes. There are two types of T-I responses: T-I type 1 responses depend on toll-like receptor (TLR) agonists to activate B cells, and T-I type 2 responses depend on multivalent ligand interactions to activate B cell receptors for activation.
In general, most naturally occurring lipids do not exhibit a T-I response. Exceptions to this generalization include the glycosphingolipids (e.g. ceramide) and acidic phospholipids (e.g. cardiolipin), which are observed in certain autoimmune disorders, such as anti-phospholipid syndrome. This syndrome results in severe blood clots leading to stroke, heart attack and miscarriage [56]. Some synthetic headgroup modified lipids have been shown to induce immune responses including polymer and hapten conjugates. As discussed, polymer modifications, including PEG [25], induces an IgM response leading to the advanced blood clearance by the MPS. This IgM response is thought to be caused by B cell activation through receptor ligation by the polymer repeat units (Figure 5B) [57]. While the ABC effect has only been observed to date with PEG, this phenomenon must be considered when developing other liposome-polymer conjugates.
Haptens do not naturally induce immune responses unless conjugated to a carrier such as a protein or liposome (Figure 5A,C). While the immune response is generated against the hapten presented on the carrier, the response persists toward the unconjugated hapten. The first immune response from a synthetic hapten-lipid conjugate was reported by Uemura and colleagues in 1974 with the attachment of 2,4-dinitrophenol or fluorescein isothiocyanate (FITC) to the headgroup of PE lipids [58]. Since then, a number of haptenated liposomes have been investigated [59,60] and a number of biophysical studies have identified the role of membrane fluidity [61], linker length between the lipid and hapten [62], and hapten concentration (Table 2, left) [63] on immunogenicity. While antibody responses to PC lipids are rare, immune responses were observed when the PC headgroup was appended to PE lipids using a six-carbon spacer [64]. Importantly, memory IgM was also observed in response to haptenated liposomes, suggesting that multiple injections would also lead to an advanced blood clearance of the haptenated formulations [65]. Several haptenated liposomes have been evaluated, but the Super Hapten Database reports more than 7000 known haptens [66]. Given the vast number of known haptens and their chemical diversity, we believe that most synthetic lipid headgroups will induce an immune response.
Table 2.
(Left) Selected examples of hapten conjugated lipids that induce immune responses in the absence of adjuvants. (Right) Selected examples of hapten conjugated lipids that induce immune responses in the presence of adjuvants. In all structures, the R group represents a non-specific lipid moiety.
| −Adjuvant | +Adjuvant | ||
|---|---|---|---|
PEG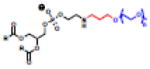
|
[25,75–80] | MPLA, MPLA
|
[67] |
Theophylline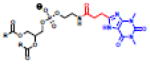
|
[59] | ||
TEMPO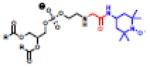
|
[61] | phosphatidylcholine MPLA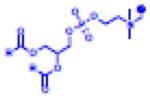
|
[67] |
Atrazine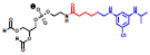
|
[63] | Sphingomyelin, MPLA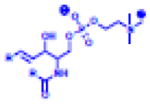
|
[67] |
Azobenzone arsonate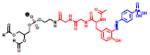
|
[65] | Squalene, MPLA |
[68] |
Dinitrobenzone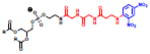
|
[65] | Biotin, CpG |
[60] |
Trinitrophenyl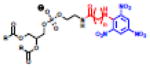
|
[62] | FITC, CFA/IFA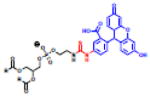
|
[58] |
Phosphocholine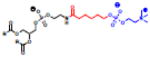
|
[64] | Dinitrobenzone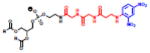
|
[58] |
Immunomodulatory molecules may also be incorporated in liposomes to induce specific immune responses. Adjuvants cause a T-I type 1 response through activation of the TLR signaling pathway. Monophosphoryl lipid A (MPLA), a component of endotoxin, has been the most widely studied adjuvant incorporated in liposomes and provides B cell activation as an agonist for TLR 4 [67,68]. Immune responses to haptens in the presence of other adjuvants, including nucleic acid CpG motifs [60] and Freund’s Adjuvant [58], have also been investigated (Figure 5D; Table 2, right). Incorporation of MPLA in liposomes induces antibody responses toward the liposome in the form of MPLA specific antibodies as well as anti-phospholipid antibodies including anti-phosphocholine, anti-sphingomyelin [67] and anti-squalene antibodies [68]. Antibody responses toward the liposome may destabilize the membrane through complement dependent damage [69]. Lipid specific antibodies directed toward DMPG, MPLA, cholesterol and phosphatidyl inositol-4-phosphate (PIP) were induced in human subjects after immunization with a candidate vaccine to Plasmodium falciparum [70]. Although antibodies toward liposomal lipids were observed, the authors suggest that these formulations appear to have acceptable safety records and that such vaccines would be well tolerated [70]. Longitudinal studies, however, must be performed to determine the long-term effects of such antibodies in serum. Other lipid adjuvants including the lipid core peptide (LCP) system [71] or Pam3Cys [72] have also been incorporated into liposomal formulations to induce peptide specific immune responses. In each of these reports, however, the adjuvant is included in the formulations to achieve an immune response toward a specific antigen. These data indicate that it is critical to prepare endotoxin free liposomal formulations to avoid the immunogenicity of drug delivery systems [73].
3.1 Inducing immune tolerance with liposomes
Liposome encapsulated cytotoxic agents have been widely used in drug delivery applications [74], but their activity has also been exploited to induce immune tolerance. Depleting a specific population of B cells using targeted cytotoxic liposomes leads to immune tolerance against a specific epitope. Liposomes encapsulating cytotoxic agents can be targeted to specific B cell receptors. Upon interaction with the B cell membrane, the liposome is endocytosed and the payload is released, thereby ablating the specific B cell population. A number of examples of this approach exist in the literature, and Doxil® is a particularly relevant example. While PEGylated liposomes induce an ABC effect, this effect is not seen in Doxil®, as B cells with a receptor that recognizes PEG are deleted by the cytotoxic activity of doxorubicin. Deletion of these B cells leads to immune tolerance to PEG and prevents the generation of IgM and the ABC effect [75–80]. This tolerance effect has also been observed with protein agents. Liposomes displaying bovine serum albumin (BSA) and containing methotrexate were found to depress the anti-BSA plaque-forming cells in mice [81,82]. In a similar fashion, tolerance to ovalbumin was achieved in the presence of doxorubicin [83,84], which was reexamined again 16 years later and published as a novel strategy [85]. The exciting concept of tolerance induction using cytotoxic agents is a field of research that is largely unexplored, but may be exploited for autoimmune disorders and allergic immune responses.
In summary, it is critical to be aware of the potential immune responses that may be induced by synthetically engineered lipids and novel formulations. The potential for immunoreactivity using synthetic lipids and lipid conjugates and the clearance of liposomal formulations through immune dependent mechanisms, including B cell responses, serum antibodies and complement activation, suggest that immune responses to novel lipids must be evaluated. The effect of immunoreactivity and clearance may also have implications on the pharmacokinetics, biodistribution, safety, and efficacy of liposomal formulations.
4. Lipids for directed headgroup interactions
Lipid headgroups can act as points for liposome functionalization for cargo, polymer coatings, or targeting ligands. These functionalization points often are covalent coupling moieties that allow for the attachment of ligands that alter pharmacokinetics or biodistribution, such as cell or tissue targeting groups, membrane-active peptides, or polymer coatings. In addition, headgroups contribute to lipid-lipid interactions, and influence the biophysical characteristics and macromolecular behavior of a liposome. Modifications to the headgroup allow for the control of specific molecular interactions such as H-bonding, pi-stacking, and electrostatics.
4.1 Lipids with a nucleic acid headgroup
Nucleolipids are lipids with a nucleic acid headgroup, and are unique because they can interact with nucleic acids via hydrogen bonding, pi stacking, and electrostatics. They are an excellent model system to study the hierarchy of molecular forces between lipid headgroups and how those forces contribute to superstructure formation. Further, these headgroups are biologically relevant, as they complex with single-stranded nucleic acids via base pairing and are useful in nucleic acid and drug delivery [88] and as lipid prodrugs [6]. Advancements in nucleolipid structures and their applications in transfection have recently been reviewed [86,89,90]. Herein we focus on engineered lipids with a monomeric nucleoside or nucleotide headgroup with a particular emphasis on understanding the forces governing headgroup interactions with nucleic acids.
The deoxyribose sugar is the scaffold for nucleolipid construction (Figure 6A). In general, a nucleobase (A, T, C, G or U) is conjugated to the 1′ position by a beta-glycosidic linkage. This base allows for selective interactions with other nucleolipids, single-stranded nucleic acids, or drugs via electrostatics, hydrogen bonding, and pi-stacking. The 2′ and 3′ positions of the ribose are generally functionalized to hydrophobic domains via an ester or ether linker. Finally, anionic, cationic, zwitterionic and non-ionic groups have been conjugated to the 5′ position in order to alter the behavior and distinct self-assembly properties of these lipids [89].
Figure 6. Nucleolipid headgroup structures.

Predicted forces governing interactions with nucleic acids are shown.
A few studies isolate the role of H-bonding and electrostatics in nucleolipid headgroup interactions with nucleic acids by engineering lipids that lack the functional groups required for such interactions. Ceballos and colleagues synthesized nucleolipids with 3-nitropyrrole, 5-nitroindole or 4-nitroimidazole headgroups, universal bases known to form complementary base pairs with all four natural bases [91,92]. The headgroups are further functionalized with a quaternized amine at the 5′ position and two oleyl chains to allow for lipid self-assembly (Figure 6B–D). These bases lack hydrogen-bonding capabilities, and are limited to interactions via pi-stacking interactions and electrostatics. These forces were sufficient to complex siRNA and knock down glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein levels in a number of cell lines. However, the hierarchies between pi-stacking and electrostatic interactions remain unclear in these lipids. To our knowledge, this is the first example of protein knockdown using nucleolipids transfecting siRNA. Interestingly, the authors increased the nucleolipid affinity for siRNA by altering the stereochemistry of the base at the 1′ position. This increase in affinity correlates with an increase in transfection efficiency, and this relationship was corroborated by density functional theory modeling, a quantum mechanical method to investigate the electronic structure of a molecule [92]. Defining such quantitative structure-function relationships is useful in designing amphiphilic molecules [93,94].
Since hydrogen-bonding and pi-stacking interactions are weaker than electrostatic interactions via cationic groups with DNA, it is difficult to decouple their role in directing nucleolipid-DNA complexes [95]. To understand the function of these forces, Banchelli et al. synthesized nucleolipids with a formal negative charge [96]. Two octanoyl chains were conjugated to a nucleobase at the 5′ position via an anionic phosphate linkage and an adenosine nucleotide was linked to the 1′ position (Figure 6E). The hydrogen bonding and pi-stacking interactions of these nucleolipid overcame the repulsive force between the negative charge in the headgroup and the negative charge on nucleic acids in order to complex polyuridylic acid [96]. In a similar vein, Khiati and co-workers synthesized anionic nucleotide-lipids to decouple cationic interactions from H-bonding and pi-stacking (Figure 6F) [97]. Again, the forces of H-bonding and pi-stacking dominated the anionic repulsive forces between the nucleolipid and nucleic acid to complex and transfect eGFP into HEK cells in vitro [97]. These studies show that electrostatic interactions are not necessary to complex and transfect nucleic acids in vitro, as H-bonding and pi-stacking forces can be sufficient.
The selective intermolecular binding properties of nucleolipids have been incorporated into other rationally designed lipids. In a series of papers, Ma and colleagues designed synthetic multivalent hydrogen-bonding lipids with a melamine or cyanuric acid headgroup that replace the sugar-phosphate backbone of DNA with a phospholipid [98,99]. These lipids formed large unilamellar vesicles and demonstrated complementary base pairing interactions in membrane mixing and surface plasmon resonance studies (Figure 6G,H). Polidori and colleagues extensively investigated the role of lipid shape on macromolecular structure formation of synthetic lipids with a H-bonding tris(hydroxymethyl) aminomethane (tris) moiety linked with an aminoglycerol group [100]. Increasing the length of the hydrophobic tails increased the Tm of the lipids and altered the length of the tubules they formed, their stability, and their ability to stably entrap carboxyfluorescein (CF). Substitution of an ester linkage for a carbamate linkage allowed for the formation of unstable vesicles rather than stable tubes, highlighting the lipid linkage as an important parameter in lipid design and behavior. Godeau and coworkers synthesized H-bonding glycosyl-nucleoside lipids as low-molecular-weight hydrogelators for nucleic acid delivery [101]. Lipids were first conjugated to a nucleobase using click chemistry and subsequently clicked with glycosyl groups (Figure 6I) to achieve reversible nano-fibers, hollow nanotubes, and hydrogels capable of transfecting cultured cells in serum [102]. This chemistry allows for a universal scaffold for facile lipid functionalization and engineering. Two lipid parameters dictated the formation of gels in solution: 1) H-bonding between headgroups and 2) saturation of acyl chains. H-bonding headgroups (30 fold) and saturated acyl chains (25 fold) dramatically lowered the concentration necessary for gelation [101].
While many publications have sought to simplify the synthesis, characterize the biophysics, and assess the in vitro behavior of nucleolipids and H-bonding lipids, little is published on their in vivo activity. Special attention, however, must be paid to the immunogenicity of such lipids. As previously discussed, nucleolipid headgroups may act as haptens, especially when delivering known TLR agonists such as double-stranded DNA or single-stranded RNA [103]. Such studies should be prioritized in order to understand the potential of these lipid systems for drug delivery.
4.2 Headgroups for attachment of targeting ligands
In addition to engineered lipid headgroups that rely on intrinsic molecular interactions such as H-bonding and pi-stacking, a myriad of synthetic lipid headgroups have been developed that contain specific chemistries that permit controlled covalent attachment of targeting ligands or functional groups. In particular, lipids with maleimide, avidin, ether, ester, thiol, carboxylic acid, and hydrazine moieties in the headgroup have been extensively reviewed [104,105]. The maleimide lipid is most commonly used as a functionalization point of lipids because of the reactivity of the group with a free thiol; the maleimide is typically separated from the lipid headgroup by a spacer that reduces steric hindrance in the coupling reaction. This is particularly useful, as a thiol containing ligand, such as a single chain antibody fragment, can be coupled to preformed liposomes containing a small mole fraction of maleimide lipid [106].
These covalent attachment approaches are broadly applicable but can require complex chemistry and can partially inactivate the proteins [107]. Further, the number and location of attachment sites on the ligand dictate the orientation of ligand attachment. For example, a maleimide group can form a thioether bond with any solvent exposed cysteine on a protein.
Several attempts to engineer lipid headgroups for facile non-covalent attachment have been pursued with limited success. Protein A, which interacts specifically with the Fc region of IgG, can be adsorbed to the surface of polymer nanoparticles. Addition of IgG to the nanoparticles results in uniform orientation of the antibodies and nearly 100% attachment efficiency. However, this system is limited by its stability in vivo, as the adsorbed protein is displaced by proteins in serum [108].
A number of groups have linked His-tagged proteins to liposomes using nickel chelating moieties [109–113]. His-tags are popular motifs that can be easily engineered into proteins and act as handles for protein purification or binding by nickel chelation. This interaction is reversible by stripping chelated nickel from NTA [114]. In an effort to develop a general attachment approach for liposomes, van Broekhoven and Altin [115,116] engineered trivalent nitrilotriacetic acid (tris-NTA) lipids with nanomolar affinities for polyhistidine tagged (His-tag) proteins. Our group provided an alternative synthetic route to the tris-NTA lipid [117,118]. Increasing the valency of the chelating moiety in the lipid headgroup increased the affinity for His-tagged proteins (Figure 7). Yeast cytosine deaminase (yCD) and monomeric Katushka (mKate), a far red-fluorescent protein, maintained their activity while attached to the liposomes via the tris-NTA lipid. These proteins were stably attached to the liposome in fetal calf serum and mouse plasma. However, liposome attachment via tris-NTA lipids did not enhance the circulation time of proteins in vivo, likely due to competing interactions from plasma proteins and other histidine motifs. While the in vivo applications of these chelating lipids are limited, they have shown utility in subcutaneous vaccine delivery [119] and they can effectively be used to simplify rapid screening of lipid-protein conjugates in vitro to identify binding and internalizing antibodies for use in targeted drug delivery [120].
Figure 7.
Structure of Tris-NTA lipid.
5. Lipids that direct membrane biophysics and payload release
Ideally, liposomes are designed to be stable in circulation until they reach the target site. Upon interaction with the appropriate compartment in target cells, the payload should be rapidly released, particularly if cytotoxicity is the objective. Hydrophilic liposome payload can be encapsulated in the aqueous core of the liposome or substituted for polar liposome headgroups in the bilayer as a lipid prodrug. In both scenarios, destabilization of the membrane and release of the therapeutic are critical steps in delivery.
As such, a keen understanding of membrane biophysics and stability is integral to tuning drug release. The Tm, phase, and composition of the bilayer drive its stability. At the Tm, the bilayer undergoes a gel to liquid phase transition causing lipid-packing defects that increase bilayer permeability. Lipid components can dictate the Tm and other bilayer physical properties. Generally, lipids with long saturated acyl chains have higher Tm than those with shorter or unsaturated chains. The inter- and intramolecular interactions of lipid headgroups further control Tm and membrane permeability. Regardless of the lipid composition, cholesterol can be included in bilayers to eliminate the phase transition and encourage a stable gel-like phase. In addition to intrinsic liposome parameters, microenvironmental factors such as redox state, pH, temperature, and enzyme activity have a profound impact on stability and can be used to trigger drug release [121,122].
5.1 Covalent attachment of hydrophobic moieties
Sterols are important components of natural membranes that play a critical role in regulating membrane fluidity. Cholesterol is the most common sterol in mammalian membranes and is the preferred sterol in several FDA approved liposome therapies including DaunoXome®, Myocet®, Depocyt®, Marqibo®, and Doxil® [123]. While cholesterol or cholesterol esters do not form bilayer structures on their own, their amphipathic nature allows for their inclusion in liposome bilayers. Incorporation of cholesterol into liposomes at 30 mol % eliminates the phase transition of diacylphospholipids [124,125], reduces membrane permeability [126,127], and forces the bilayer into a stable gel-like state. Because of their stability, liposomes of this composition are widely used in the formulation of chemotherapeutic drugs [128]. Below 30 mol %, cholesterol does not pack uniformly in diacylphospholipids and fails to completely eliminate their phase transition [122]. Above 50 mol %, cholesterol phase separates in the membrane and can form crystals [129]. As such, changes in cholesterol composition can have profound effects on liposome formation, membrane stability, and permeability. In vivo, free cholesterol can rapidly transfer from liposomes into biomembranes and lipoproteins [130–132]. This constant flux of cholesterol destabilizes the liposomes and promotes contents release while in circulation.
In order to prevent such transfer of cholesterol, our group synthesized a family of sterol-modified phospholipids (SMLs) by covalently attaching cholesterol to the glycerol backbone of phosphatidylcholine (Figure 8). An early example of such a cholesterol containing phospholipid was used to investigate the role of cholesterol binding on ATPase activity in a lipid micelles but had not been demonstrated to form liposomes [133]. We showed that SMLs readily formed liposomes by themselves or when mixed with diacyl PC lipids. By anchoring cholesterol in the membrane, we reduced cholesterol transfer and increased in vivo liposome stability. SMLs are easily synthesized, commercially available, and recapitulate the biophysical properties of liposomes incorporating cholesterol [134–136]. Compared to liposomes formulated with free cholesterol, SMLs eliminate the phase transition of diacylphospholipids [135], exhibit similar permeability to entrapped hydrophilic molecules [135,136] and demonstrate similar membrane fluidity [136]. SMLs maintain these properties while preventing cholesterol transfer from the bilayer [135].
Figure 8.

Structure of sterol modified lipids (SMLs).
In vivo, SML liposomes encapsulating doxorubicin demonstrate comparable efficacy to Doxil® in a C-26 colon carcinoma model [135]. Interestingly, we found that at acyl chain lengths of C16 and C18, SML liposomes are more stable in circulation than liposomes containing free cholesterol. Further, we found that these SML liposomes had improved uptake into and slower clearance from the liver and spleen compared to traditional liposomes [136]. These studies highlight the stability of SML systems in vivo and their potential utility as drug carrier systems.
In addition to cholesterol, hydrophobic moieties for therapy and diagnostics can be covalently coupled to lipids to prevent loss in circulation. Liposomal formulations of porphyrins, hydrophobic and photosensitive agents with applications in photodynamic therapy, have been used to improve their solubility. These nanoparticle systems are limited by the amount of porphyrin that can be included in the formulation (15 mol %) and by transfer of porphyrin out of the bilayer in vivo [137,138]. Anchoring of pyropheophorbide, a chlorophyll-derived porphyrin analogue, to lyso PC prevented bilayer transfer of the porphyrin and enhanced its self-quenching in liposomes [137]. Lipid systems composed of these engineered lipids along with PEG-DSPE and cholesterol were termed “porphysomes”. These nanoparticles were safe at high doses in mice, demonstrated favorable pharmacokinetics, and could be loaded with hydrophilic payload. After accumulation in tumors, these theranostic particles could be imaged for diagnostic purposes or irradiated for photothermal therapy [137]. This interesting technology has been extended to applications in triggerable systems and acoustic imaging [139,140].
5.2 Inverse zwitterlipids (IZ)
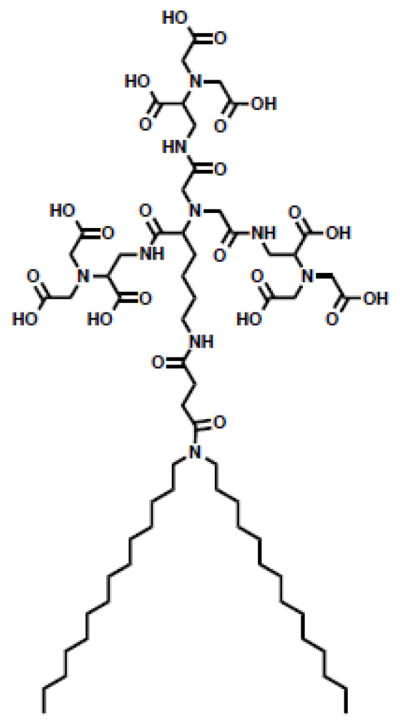
In addition to engineering the lipid hydrophobic domain, modifications to the lipid headgroup can influence membrane stability and permeability. The influence of lipid headgroup properties such as hydrogen bonding capability, size and charge on superstructure formation and molecular interactions has been rigorously examined. For example, it is well known that the relative size of the lipid headgroup to the hydrophobic tails determines the phase preference of the lipid: lipids with a small headgroup (i.e. PE) prefer an unstable hexagonal phase while those with larger headgroups (i.e. PC) prefer a stable bilayer phase [10]. However, a number of aspects of lipid headgroup architecture have not been well characterized. In particular, modifications to the zwitterionic PC headgroup have been sparsely studied despite their pivotal role in biological membranes.
The influence of the position and choice of charged moieties in zwitterionic headgroups on lipid behavior is not well understood. These headgroups are oriented with an anion proximal to, and a cation distal to, the membrane interface. In the case of the PC headgroup, the anionic phosphate group is anchored at the membrane interface while the cationic amine extends into the aqueous space (Figure 9). In a series of recent studies, our group has systematically examined modifications to the PC headgroup. We set out to understand the role of 1) the relative location of charged moieties in the headgroup and 2) the type of charged moiety on lipid behavior. To that end, we synthesized a library of lipids with an inverted architecture: the cation is proximal to the membrane while the anion extends into the aqueous space. This family includes phosphates (CP), ethyl phosphates (CPe), carboxylates (AQ), sulfonates (SB) and sulfates (CS) as the anionic moiety (Figure 9). AQ, SB, CP and CPe lipids form liposomes when formulated with cholesterol. This new class of inverse zwitterionic (IZ) lipids has distinct biophysical properties from traditional PC lipids including elevated Tm and limited interactions with divalent cations. Most importantly, these studies have highlighted the pivotal role of the charge at the bilayer interface in drug permeability and ionic interactions of liposomes. While single chain surfactants with an inverted headgroup architecture have been studied, these studies extend the understanding of headgroup charge inversion in lipid systems [141].
Figure 9. Structures of lipids with inverted headgroup architecture.
Structures of inverse zwitterlipids (IZ) lipids.
The charge at the bilayer interface dictates the ion interactions of the liposome membrane. Liposomes composed of AQ and SB lipids interact with anions according to the Hofmeister series (Figure 10A), a classification of ions according to their ability to salt in or salt out proteins. Ions with high charge densities (F−, Cl−) are strongly hydrated, while ions with low charge densities (I−, ClO4−) are weakly hydrated. Anions with low charge density interact with the bilayer of AQ and SB membranes more than those with high charge density. Further, all anions interact with AQ and SB membranes to a greater extent than PC membranes (Figure 10A). We believe that these interactions are directed by the positive charge at the membrane interface.
Figure 10. Biophysical properties of IZ lipids.
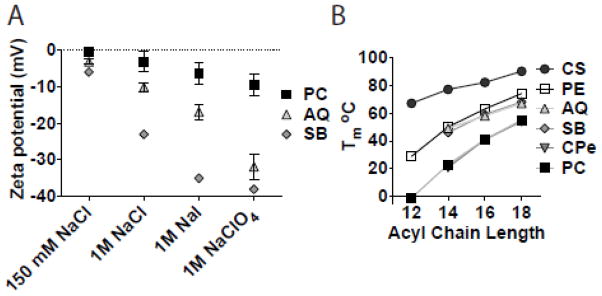
(A) IZ lipids (AQ, SB) preferentially interact with ions according to the Hofmeister series. (B) IZ lipids have elevated Tm compared to PC lipids.
While the position of the positive charge enhances interactions with anions, it reduces interactions with cations. Divalent cations, especially Ca2+, are of particular interest because they are ubiquitous in biological systems and can interact with lipid headgroups to cause membrane destabilization via aggregation, fusion, or alteration of surface charge [142]. CP, CPe, AQ and SB liposomes do not aggregate in the presence of physiological levels of Ca2+, and IZ liposomes interact with Ca2+ less than PC liposomes [143–145]. Reduced divalent cation interactions make IZ liposomes interesting candidates to investigate as in vivo delivery systems. In addition to ions found in biological environments, the charge at the membrane interface can influence the permeation of entrapped liposome cargo. CPe liposomes are more permeable to negatively charged contents and relatively less permeable to neutral contents than PC liposomes [144]. In this way, IZ lipids may be useful to tune release properties of certain entrapped liposome drugs.
The inverted headgroup architectures also alter inter- and intra- headgroup interactions. The Tm is a measure of the intramolecular forces driving hydrophilic interactions between lipid headgroups and hydrophobic and van der Waals interactions between lipid tails. Stronger interactions in these two lipid regions lead to elevated Tm. PE lipids have strong headgroup interactions because of their H-bonding capability, and as such, have higher Tm than non H-bonding PC lipids. Similarly, lipids with long saturated acyl chains have stronger hydrophobic interactions and thus higher Tm than those with shorter or unsaturated chains. IZ lipids have very high Tm characteristic of PE lipids rather than PC lipids (Figure 10B). To our knowledge, CS lipids have the highest Tm of any lipid, natural or synthetic (Figure 10B). Interestingly, these Tm are dependent on the ionic character of the surrounding solution. For SB, CS and certain chain lengths of AQ, anion interactions with the membrane according to the Hofmeister series shift the transition from PE-like to PC-like. This shift is likely driven by inter- or intra- headgroup interactions that are disrupted when anions interact with the bilayer surface.
IZ lipids have potential as triggerable in vivo delivery systems. AQ liposomes have similar pharmacokinetics and biodistribution to PC liposomes, but have dramatically different biophysical properties that may be exploited for thermo-responsive systems [145]. Further, CP and CS lipids may be useful in enzyme-triggered systems. The CP lipid headgroup can be cleaved by alkaline phosphatase, yielding a lipid with a net positive charge that may destabilize the membrane and release trapped cargo (unpublished data). Such triggerable systems may be useful in drug delivery to organs enriched in phosphatase or sulfatase activity.
5.3 Triggerable lipids
As previously described, liposomes are designed to optimize stability and circulation time in order to drive a high dose of payload to the target site. However, this stability in circulation must be balanced with the release of payload in diseased tissue. Incomplete release of drug at the site of action can limit the clinical success of therapeutics, as evidenced by the failure of liposomal cisplatin in the clinic [146]. To improve release, researchers have developed a number of triggerable liposome systems. The trigger can be an external cue (heat, light, ultrasound) or intrinsic to the disease site (pH, redox environment, enzymes). These systems have been thoroughly reviewed [147,148], but have been disappointing in practice [147]. This is especially true for systems dependent on external triggers, since only primary tumors, and not nascent metastasis, can be targeted via such cues. ThermoDox® (Celsion Corporation) is a prime example of the failure of these systems in the clinic. This heat sensitive liposome formulation incorporates lysolipid that promotes liposome degradation at mildly elevated temperatures [149] and is administered alongside radiofrequency ablation or ultrasound. However, incorporation of lysolipids can destabilize the liposome in circulation [150] and may lead to contents release before the liposome reaches the tumor site. ThermoDox® recently failed to meet its primary endpoint in a Phase III study in patients with hepatocellular carcinoma (Celsion Corporation press release January 31, 2013).
5.4 Lipid Prodrugs
Incorporation of small molecule drugs or prodrugs into self-assembling, amphiphilic molecules can improve their biodistribution, pharmacokinetics, safety profile, trafficking and stability [3,6]. In particular, lipid prodrugs can improve the characteristics of small molecule drugs that are not well suited for traditional liposomal encapsulation and delivery due to their molecular properties. In principal, a hydrophilic drug is conjugated to a hydrophobic lipid tail such that it is substituted for a polar lipid headgroup; this amphiphilic lipid prodrug can then be incorporated into a liposome. The long history of these prodrugs have been reviewed [6]. However, a number of recent clinical advances and setbacks suggest that the clinical on the development of these drugs will be challenging. In this section we review recent developments in lipid prodrugs of doxorubicin (dox), gemcitabine, cytarabine (Ara-C), mitomycin C (MMC), and paclitaxel. We focus our discussion on clinical and in vivo preclinical developments.
Dox, a topoisomerase II inhibitor, is the chemotherapeutic most often studied in nanoparticle formulations because of the thorough understanding of its pharmacology and the success of Doxil® in the clinic. Systemic doses of dox lead to myelosuppression, gastrointestinal toxicity and cardiotoxicity. These issues make doxorubicin a prime candidate for nanoparticle formulation, and a number of dox prodrugs have had preclinical success. The most promising dox derivative is not a lipid prodrug, but an albumin binding dox derivative: Aldoxorubicin® (CytRX) (Figure 11). Binding of doxorubicin to albumin improves the pharmacokinetics and efficacy of the free drug and has shown promise in early clinical trials [151]. A number of lipid prodrugs of dox are in preclinical testing. A docosahexanoic acid (DHA) dox conjugate proved more efficacious than free dox in L1210 leukemia and B16 melanoma models [152]. Further, Duhem and colleagues conjugated dox to tocopherol succinate via an amide linkage which self assembled into 250 nm macrostructures when stabilized with PEG lipid [153]. These nanoparticles have improved efficacy over free dox in CT26 tumors in vivo. While these dox derivatives are promising, future experiments should be carried out to benchmark these dox-prodrug systems to Doxil®.
Figure 11. Prodrug structures.

Selected structures of prodrugs that have advanced into the clinic.
MMC is an alkylating chemotherapeutic agent limited by toxicities such as leukopenia, thrombocytopenia, and mucous membrane toxicity [154]. A number of lipid prodrugs have been developed to improve the therapeutic index of MMC. In particular, Gabizon and colleagues have developed an MMC prodrug in which the drug is conjugated to a 1,2-distearoyl glycerol lipid via a cleavable dithiobenzyl linker (Figure 11) [155]. In this system, the MMC acts as the polar lipid headgroup and the prodrug is easily incorporated into liposome membranes. This prodrug liposome system has demonstrated impressive efficacy in a range of preclinical models [155,156], and is now advancing in a phase I dose escalation study (Promitil®, LipoMedix, NCT01705002).
Nucleoside analog chemotherapeutics, including gemcitabine and Ara-C, are a major class of chemotherapeutics. Small structural differences have a profound impact on the activity of these molecules: Ara-C is primarily used to treat hematological tumors while gemcitabine, which is structurally similar except for two fluorine atoms, is used against solid tumors. Both drugs are limited by short circulation times and can be rapidly deactivated by deamination reactions in vivo. As such, a wealth of research has been focused on lipid prodrug modifications of these compounds to prolong circulation of the active form. Gemcitabine lipid prodrugs have shown promise in preclinical studies and are comprehensively reviewed [157]. Modification of Ara-C at the 5′ position with an elaidic acid chain (Elacytarabine®, Aqualis ASA) circumvented drug resistance in vitro and resulted in an improvement in in vivo anti-tumor efficacy as compared to unmodified Ara-C (Figure 11) [158,159]. However, Elacytarabine® demonstrated no advantage in efficacy over the control arm in two phase III studies in acute myeloid leukemia and pancreatic cancer (Aqualis ASA press release November 12, 2012 and April 1, 2013). While the lipid prodrugs improved pharmacokinetic and biodistribution of Ara-C, these changes did not translate into a survival advantage in patients. Another prodrug, CP-4126, is a 5′ elaidic ester of gemcitabine that has advanced into the clinic (Figure 11) [160]. Orally administered CP-4126 was poorly absorbed and subject to metabolism before systemic exposure [160]. As such, future work with this compound will focus on intravenous administration. These studies highlight the difficulties and unknowns in moving lipidated prodrugs into the clinic.
Similar to the nucleoside analog prodrugs, paclitaxel lipid prodrugs have had disappointing clinical outcomes. Paclitaxel is highly hydrophobic and its encapsulation in liposomes is problematic. Modification of paclitaxel at the 2′ hydroxyl with DHA (Taxoprexin®, Protarga Inc.) improved anti-cancer efficacy by extending circulation [161]. However, these improvements in pharmacokinetics did not translate into improvements in efficacy, as Taxoprexin® had only modest activity in gastric and esophageal adenocarcinoma [162]. Further, a phase III trial in metastatic melanoma showed no survival advantage of Taxoprexin® versus dacarbazine [163]. Squalenoylation of paclitaxel and gemcitabine may represent a new class of prodrugs for clinical evaluation [164–166] but their promise must be viewed in the light of the challenges experience by the other lipid prodrugs reviewed above.
6. Conclusions
The large variation of lipid structures outlined in this review can be combined to form liposomes with multifunctional capabilities including increased serum stability, extended circulation, ligand targeting, optimized drug loading and triggered release. These lipids have been vital tools for probing lipid membrane biophysics, especially for understanding the role of inter- and intra-molecular interactions on liposome superstructure formation and behavior. Such studies have led to a wealth of well-characterized lipid structure function relationships that are useful to those looking to synthesize novel molecules. For this reason, systemic manipulation of lipid headgroups, linkers and hydrophobic domains to further our understanding of lipid biophysics remains a worthwhile and productive area of research.
While a number of novel lipids have been synthesized and published for applications in drug delivery, their pharmacokinetics, pharmacodynamics and immunogenicity are rarely thoroughly characterized [74]. Looking forward, such studies should be prioritized in order to realize the translational potential of these systems. Special attention should be paid to commonly overlooked areas such as lipid immunogenicity, and extreme care should be taken to ensure that liposome preparations administered in vivo are endotoxin free to avoid confounding immunological factors.
The setbacks of Lipoplatin®, Thermodox®, Elacytarabine®, and Taxoprexin® in the clinic provide insight into the critical parameters in drug delivery to solid tumors. While in circulation, liposome payload should be stably encapsulated in order to maximize accumulation in the tumor. However, once the liposome extravasates from the vasculature into the tumor, it must quickly release its payload uniformly throughout the tumor. The complex process of payload release then involves three parameters: 1) stability in circulation, 2) distribution in the tumor, and 3) payload release over the appropriate timescale. Future work should focus on the balance between these parameters in order to optimize formulations for drug delivery. As has been written elsewhere [167], the incorporation of engineered lipids into a delivery system must take into account the higher costs, complexity of manufacturing, and complicated intellectual property of the multi-component systems. In order to justify the added costs of these systems, liposomes, like other nanomedicines, must offer significant clinical advantages in both safety and efficacy.
Acknowledgments
This work was supported by NIH grants R01 GM061851, R01 EB003008, and R21AI093135 and by grant R613-CR11 from the Cystic Fibrosis Foundation (A. Verkman Principal Investigator). VJV was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number F32AI095062.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
F.C.S. declares a conflict of interest due to his involvement in a liposome company. The other authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Murphy RC, et al. A comprehensive classification system for lipids. J Lipid Res. 2005;46:839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Caffrey M. LIPIDAT A Database of Thermo Data and Association Information on Lipid Mesomorphic and Polymorphic Transitions. CRC Press; 1993. [Google Scholar]
- 3.Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63:3–23. doi: 10.1016/j.addr.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee S, Maxfield F. Role of membrane organization and membrane domains in endocytic lipid trafficking. Traffic. 2000;1:203–211. doi: 10.1034/j.1600-0854.2000.010302.x. [DOI] [PubMed] [Google Scholar]
- 5.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Reddy LH, Couvreur P. Macromolecular Anticancer Therapeutics. Springer; 2010. [Google Scholar]
- 7.Tan Y, Huang L. Overcoming the Inflammatory Toxicity of Cationic Gene Vectors. Journal of Drug Targeting. 2002;10:153–160. doi: 10.1080/10611860290016757. [DOI] [PubMed] [Google Scholar]
- 8.Duncan R, Gaspar R. Nanomedicine(s) under the Microscope. Mol Pharm. 2011;8:2101–2141. doi: 10.1021/mp200394t. [DOI] [PubMed] [Google Scholar]
- 9.Mayer LD, Bally MB, Cullis PR. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 1986;857:123–126. doi: 10.1016/0005-2736(86)90105-7. [DOI] [PubMed] [Google Scholar]
- 10.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- 11.Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 12.Stanton MG, Colletti SL. Medicinal chemistry of siRNA delivery. J Med Chem. 2010;53:7887–7901. doi: 10.1021/jm1003914. [DOI] [PubMed] [Google Scholar]
- 13.Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 1991;1068:133–141. doi: 10.1016/0005-2736(91)90201-i. [DOI] [PubMed] [Google Scholar]
- 14.Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 15.Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85:6949–6953. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabizon A, Papahadjopoulos D. The role of surface charge and hydrophilic groups on liposome clearance in vivo. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 1992;1103:94–100. doi: 10.1016/0005-2736(92)90061-p. [DOI] [PubMed] [Google Scholar]
- 17.Abuchowski A, van Es T, Palczuk NC, Davis FF. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J Biol Chem. 1977;252:3578–3581. [PubMed] [Google Scholar]
- 18.Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochimica Et Biophysica Acta (BBA) - Reviews on Biomembranes. 1992;1113:171–199. doi: 10.1016/0304-4157(92)90038-c. [DOI] [PubMed] [Google Scholar]
- 19.Gabizon A, Isacson R, Libson E, Kaufman B, Uziely B, Catane R, et al. Clinical studies of liposome-encapsulated doxorubicin. Acta Oncol. 1994;33:779–786. doi: 10.3109/02841869409083948. [DOI] [PubMed] [Google Scholar]
- 20.Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. International Journal of Nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Needham D, McIntosh TJ, Zhelev DV. Surface chemistry of the sterically stabilized PEG-liposome: General Principles. In: JAS, editor. Liposomes: Rational Design. Marcel Dekker Inc; New York: 1999. pp. 13–62. [Google Scholar]
- 23.Evans E, Klingenberg DJ, Rawicz W, Szoka F. Interactions between Polymer-Grafted Membranes in Concentrated Solutions of Free Polymer. Langmuir. 1996;12:3031–3037. [Google Scholar]
- 24.Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release. 2013;172:38–47. doi: 10.1016/j.jconrel.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 25.Dams ET, Laverman P, Oyen WJ, Storm G, Scherphof GL, van Der Meer JW, et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292:1071–1079. [PubMed] [Google Scholar]
- 26.Ishida T, Wang X, Shimizu T, Nawata K, Kiwada H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J Control Release. 2007;122:349–355. doi: 10.1016/j.jconrel.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 27.Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv Drug Deliv Rev. 2010;62:272–282. doi: 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Duncan R. Development of HPMA copolymer-anticancer conjugates: clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131–1148. doi: 10.1016/j.addr.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Etrych T, Kovář L, Strohalm J, Chytil P, Říhová B, Ulbrich K. Biodegradable star HPMA polymer-drug conjugates: Biodegradability, distribution and anti-tumor efficacy. J Control Release. 2011;154:241–248. doi: 10.1016/j.jconrel.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. International Journal of Nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 31.Zelikin AN, Such GK, Postma A, Caruso F. Poly(vinylpyrrolidone) for bioconjugation and surface ligand immobilization. Biomacromolecules. 2007;8:2950–2953. doi: 10.1021/bm700498j. [DOI] [PubMed] [Google Scholar]
- 32.Gaertner FC, Luxenhofer R, Blechert B, Jordan R, Essler M. Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s. J Control Release. 2007;119:291–300. doi: 10.1016/j.jconrel.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 33.Zalipsky S, Hansen CB, Oaks JM, Allen TM. Evaluation of blood clearance rates and biodistribution of poly(2-oxazoline)-grafted liposomes. J Pharm Sci. 1996;85:133–137. doi: 10.1021/js9504043. [DOI] [PubMed] [Google Scholar]
- 34.Torchilin VP, Trubetskoy VS, Whiteman KR, Caliceti P, Ferruti P, Veronese FM. New synthetic amphiphilic polymers for steric protection of liposomes in vivo. J Pharm Sci. 1995;84:1049–1053. doi: 10.1002/jps.2600840904. [DOI] [PubMed] [Google Scholar]
- 35.Drotleff S, Lungwitz U, Breunig M, Dennis A, Blunk T, Teßmar J, et al. Biomimetic polymers in pharmaceutical and biomedical sciences. European Journal of Pharmaceutics and Biopharmaceutics. 2004;58:385–407. doi: 10.1016/j.ejpb.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 36.Torchilin VP, Shtilman MI, Trubetskoy VS, Whiteman K, Milstein AM. Amphiphilic vinyl polymers effectively prolong liposome circulation time in vivo. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 1994;1195:181–184. doi: 10.1016/0005-2736(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 37.Maruyama K, Okuizumi S, Ishida O, Yamauchi H, Kikuchi H, Iwatsuru M. Phosphatidyl polyglycerols prolong liposome circulation in vivo. Int J Pharm. 1994;111:103–107. [Google Scholar]
- 38.Abu Lila AS, Nawata K, Shimizu T, Ishida T, Kiwada H. Use of polyglycerol (PG), instead of polyethylene glycol (PEG), prevents induction of the accelerated blood clearance phenomenon against long-circulating liposomes upon repeated administration. Int J Pharm. 2013;456:235–242. doi: 10.1016/j.ijpharm.2013.07.059. [DOI] [PubMed] [Google Scholar]
- 39.Kaneda Y, Tsutsumi Y, Yoshioka Y, Kamada H, Yamamoto Y, Kodaira H, et al. The use of PVP as a polymeric carrier to improve the plasma half-life of drugs. Biomaterials. 2004;25:3259–3266. doi: 10.1016/j.biomaterials.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi H, Kojima H, Toyoda T, Yamamoto H, Hino T, Kawashima Y. Prolonged circulation time of doxorubicin-loaded liposomes coated with a modified polyvinyl alcohol after intravenous injection in rats. European Journal of Pharmaceutics and Biopharmaceutics. 1999;48:123–129. doi: 10.1016/s0939-6411(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi H, Kojima H, Yamamoto H, Kawashima Y. Evaluation of circulation profiles of liposomes coated with hydrophilic polymers having different molecular weights in rats. J Control Release. 2001;75:83–91. doi: 10.1016/s0168-3659(01)00368-6. [DOI] [PubMed] [Google Scholar]
- 42.Roux E, Stomp R, Giasson S, Pezolet M, Moreau P, Leroux JC. Steric stabilization of liposomes by pH-responsive N-isopropylacrylamide copolymer. J Pharm Sci. 2002;91:1795–1802. doi: 10.1002/jps.10172. [DOI] [PubMed] [Google Scholar]
- 43.Riché EL, Erickson BW, Cho MJ. Novel long-circulating liposomes containing peptide library-lipid conjugates: synthesis and in vivo behavior. Journal of Drug Targeting. 2004;12:355–361. doi: 10.1080/10611860412331285279. [DOI] [PubMed] [Google Scholar]
- 44.Romberg B, Metselaar JM, Baranyi L, Snel CJ, Bunger R, Hennink WE, et al. Poly(amino acid)s: promising enzymatically degradable stealth coatings for liposomes. Int J Pharm. 2007;331:186–189. doi: 10.1016/j.ijpharm.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 45.Romberg B, Oussoren C, Snel CJ, Hennink WE, Storm G. Effect of liposome characteristics and dose on the pharmacokinetics of liposomes coated with poly(amino acid)s. Pharm Res. 2007;24:2394–2401. doi: 10.1007/s11095-007-9393-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metselaar JM, Bruin P, de Boer LWT, de Vringer T, Snel C, Oussoren C, et al. A novel family of L-amino acid-based biodegradable polymer-lipid conjugates for the development of long-circulating liposomes with effective drug-targeting capacity. Bioconjug Chem. 2003;14:1156–1164. doi: 10.1021/bc0340363. [DOI] [PubMed] [Google Scholar]
- 47.Woodle MC, Engbers CM, Zalipsky S. New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem. 1994;5:493–496. doi: 10.1021/bc00030a001. [DOI] [PubMed] [Google Scholar]
- 48.Whiteman KR, Subr V, Ulbrich K, Torchilin VP. Poly(Hpma)-coated liposomes demonstrate prolonged circulation in mice. J Liposome Res. 2001;11:153–164. doi: 10.1081/LPR-100108459. [DOI] [PubMed] [Google Scholar]
- 49.Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 50.Hong RL, Huang CJ, Tseng YL, Pang VF, Chen ST, Liu JJ, et al. Direct Comparison of Liposomal Doxorubicin with or without Polyethylene Glycol Coating in C-26 Tumor-bearing Mice Is Surface Coating with Polyethylene Glycol Beneficial? Clin Cancer Res. 1999;5:3645–3652. [PubMed] [Google Scholar]
- 51.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu CMJ, Fang RH, Copp J, Luk BT, Zhang L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotechnol. 2013;8:336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moon JJ, Huang B, Irvine DJ. Engineering Nano- and Microparticles to Tune Immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiskoot W, van Schie RMF, Carstens MG, Schellekens H. Immunological risk of injectable drug delivery systems. Pharm Res. 2009;26:1303–1314. doi: 10.1007/s11095-009-9855-9. [DOI] [PubMed] [Google Scholar]
- 55.Watson DS, Endsley AN, Huang L. Design considerations for liposomal vaccines: Influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine. 2012;30:2256–2272. doi: 10.1016/j.vaccine.2012.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kutteh WH, Hinote CD. Antiphospholipid Antibody Syndrome. Obstetrics and Gynecology Clinics of North America. 2014;41:113–132. doi: 10.1016/j.ogc.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Říhová B. Immunomodulating activities of soluble synthetic polymer-bound drugs. Adv Drug Deliv Rev. 2002;54:653–674. doi: 10.1016/s0169-409x(02)00043-1. [DOI] [PubMed] [Google Scholar]
- 58.Uemura K, Nicolotti RA, Six HR, Kinsky SC. Antibody formation in response to liposomal model membranes sensitized with N-substituted phosphatidylethanolamine derivatives. Biochemistry. 1974;13:1572–1578. doi: 10.1021/bi00705a003. [DOI] [PubMed] [Google Scholar]
- 59.Babbit B, Burtis L, Dentinger P, Constantinides P, Hillis L, McGirl B, et al. Contact-dependent, immune complex-mediated lysis of hapten-sensitized liposomes. Bioconjug Chem. 1993;4:199–205. doi: 10.1021/bc00021a003. [DOI] [PubMed] [Google Scholar]
- 60.Li WM, Bally MB, Schutze-Redelmeier MP. Enhanced immune response to T-independent antigen by using CpG oligodeoxynucleotides encapsulated in liposomes. Vaccine. 2001;20:148–157. doi: 10.1016/s0264-410x(01)00277-8. [DOI] [PubMed] [Google Scholar]
- 61.Brûlet P, McConnell HM. Lateral hapten mobility and immunochemistry of model membranes. Proc Natl Acad Sci USA. 1976;73:2977–2981. doi: 10.1073/pnas.73.9.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimura K, Arata Y, Yasuda T, Kinosita K, Nakanishi M. Location of membrane-bound hapten with different length spacers. Immunology. 1990;69:323–328. [PMC free article] [PubMed] [Google Scholar]
- 63.Hendrickson OD, Zherdev AV, Kaplun AP, Dzantiev BB. Experimental study and mathematical modeling of the interaction between antibodies and antigens on the surface of liposomes. Molecular Immunology. 2002;39:413–422. doi: 10.1016/s0161-5890(02)00175-x. [DOI] [PubMed] [Google Scholar]
- 64.Leserman LD, Weinstein JN, Moore JJ, Terry WD. Specific interaction of myeloma tumor cells with hapten-bearing liposomes containing methotrexate and carboxyfluorescein. Cancer Res. 1980;40:4768–4774. [PubMed] [Google Scholar]
- 65.van Houte AJ, Snippe H, Willers JM. Characterization of immunogenic properties of haptenated liposomal model membranes in mice. IV. induction of IgM memory. Immunology. 1981;43:627–634. [PMC free article] [PubMed] [Google Scholar]
- 66.Günther S, Hempel D, Dunkel M, Rother K, Preissner R. SuperHapten: a comprehensive database for small immunogenic compounds. Nucleic Acids Res. 2007;35:D906–D910. doi: 10.1093/nar/gkl849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schuster BG, Neidig M, Alving BM, Alving CR. Production of Antibodies Against Phosphocholine, Phosphatidylcholine, Sphingomyelin, and Lipid A by Injection of Liposomes Containing Lipid A. The Journal of \Ldots. 1979 [PubMed] [Google Scholar]
- 68.Matyas GR, Wassef NM, Rao M, Alving CR. Induction and detection of antibodies to squalene. J Immunol Methods. 2000;245:1–14. doi: 10.1016/s0022-1759(00)00268-4. [DOI] [PubMed] [Google Scholar]
- 69.Haxby JA, Kinsky CB, Kinsky SC. Immune response of a liposomal model membrane. Proc Natl Acad Sci U S A. 1968;61:300–307. doi: 10.1073/pnas.61.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matyas GR, Alving CR. Antigen-specific enhancement of natural human IgG antibodies to phosphatidylcholine, phosphatidylglycerol, phosphatidylinositol-4-phosphate, cholesterol, and lipid A by a liposomal vaccine containing lipid A. Vaccine. 2011;29:5137–5144. doi: 10.1016/j.vaccine.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 71.Phillipps KSM, Wykes MN, Liu XQ, Brown M, Blanchfield J, Toth I. A novel synthetic adjuvant enhances dendritic cell function. Immunology. 2009;128:e582–8. doi: 10.1111/j.1365-2567.2008.03038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moyle P, Toth I. Self-Adjuvanting Lipopeptide Vaccines. Current Medicinal Chemistry. 2008;15:506–516. doi: 10.2174/092986708783503249. [DOI] [PubMed] [Google Scholar]
- 73.Harmon P, Cabral-Lilly D, Reed RA, Maurio FP, Franklin JC, Janoff A. The release and detection of endotoxin from liposomes. Anal Biochem. 1997;250:139–146. doi: 10.1006/abio.1997.2216. [DOI] [PubMed] [Google Scholar]
- 74.Venditto VJ, Szoka FC. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliv Rev. 2013;65:80–88. doi: 10.1016/j.addr.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ishida T, Atobe K, Wang X, Kiwada H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: effect of doxorubicin-encapsulation and high-dose first injection. J Control Release. 2006;115:251–258. doi: 10.1016/j.jconrel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 76.Koide H, Asai T, Hatanaka K, Akai S, Ishii T, Kenjo E, et al. T cell-independent B cell response is responsible for ABC phenomenon induced by repeated injection of PEGylated liposomes. Int J Pharm. 2010;392:218–223. doi: 10.1016/j.ijpharm.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 77.Li C, Cao J, Wang Y, Zhao X, Deng C, Wei N, et al. Accelerated blood clearance of pegylated liposomal topotecan: Influence of polyethylene glycol grafting density and animal species. J Pharm Sci. 2012;101:3864–3876. doi: 10.1002/jps.23254. [DOI] [PubMed] [Google Scholar]
- 78.Laverman P, Carstens MG, Boerman OC, Dams ET, Oyen WJ, Van Rooijen N, et al. Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection. J Pharmacol Exp Ther. 2001;298:607–612. [PubMed] [Google Scholar]
- 79.Charrois GJ, Allen TM. Multiple Injections of Pegylated Liposomal Doxorubicin: Pharmacokinetics and Therapeutic Activity. J Pharmacol Exp Ther. 2003;306:1058–1067. doi: 10.1124/jpet.103.053413. [DOI] [PubMed] [Google Scholar]
- 80.Cui J, Li C, Wang C, Li Y, Zhang L, Zhang L, et al. Repeated injection of pegylated liposomal antitumour drugs induces the disappearance of the rapid distribution phase. J Pharm Pharmacol. 2008;60:1651–1657. doi: 10.1211/jpp/60.12.0011. [DOI] [PubMed] [Google Scholar]
- 81.Shek PN, Heath TD. Immune response mediated by liposome-associated protein antigens. III. Immunogenicity of bovine serum albumin covalently coupled to vesicle surface. Immunology. 1983;50:101–106. [PMC free article] [PubMed] [Google Scholar]
- 82.Shek PN, Lopez NG, Heath TD. Immune response mediated by liposome-associated protein antigens. IV. Modulation of antibody formation by vesicle-encapsulated methotrexate. Immunology. 1986;57:153–157. [PMC free article] [PubMed] [Google Scholar]
- 83.Tardi PG, Swartz EN, Harasym TO, Cullis PR, Bally MB. An immune response to ovalbumin covalently coupled to liposomes is prevented when the liposomes used contain doxorubicin. J Immunol Methods. 1997;210:137–148. doi: 10.1016/s0022-1759(97)00178-6. [DOI] [PubMed] [Google Scholar]
- 84.Oja C, Tardi P, Schutze-Redelmeier MP, Cullis PR. Doxorubicin entrapped within liposome-associated antigens results in a selective inhibition of the antibody response to the linked antigen. Biochim Biophys Acta. 2000;1468:31–40. doi: 10.1016/s0005-2736(00)00178-4. [DOI] [PubMed] [Google Scholar]
- 85.Ichikawa K, Asai T, Shimizu K, Yonezawa S, Urakami T, Miyauchi H, et al. Suppression of immune response by antigen-modified liposomes encapsulating model agents: a novel strategy for the treatment of allergy. J Control Release. 2013;167:284–289. doi: 10.1016/j.jconrel.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 86.LaManna CM, Lusic H, Camplo M, McIntosh TJ, Barthélémy P, Grinstaff MW. Charge-reversal lipids, peptide-based lipids, and nucleoside-based lipids for gene delivery. Acc Chem Res. 2012;45:1026–1038. doi: 10.1021/ar200228y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yasuhara K, Kawataki T, Okuda S, Oshima S, Kikuchi JI. Spontaneously formed semipermeable organic-inorganic hybrid vesicles permitting molecular weight selective transmembrane passage. Chem Commun (Camb) 2013;49:665–667. doi: 10.1039/c2cc36662b. [DOI] [PubMed] [Google Scholar]
- 88.Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside-lipid-based nanoparticles for cisplatin delivery. ACS Nano. 2011;5:8649–8655. doi: 10.1021/nn202291k. [DOI] [PubMed] [Google Scholar]
- 89.Gissot A, Camplo M, Grinstaff MW, Barthélémy P. Nucleoside, nucleotide and oligonucleotide based amphiphiles: a successful marriage of nucleic acids with lipids. Org Biomol Chem. 2008;6:1324–1333. doi: 10.1039/b719280k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh Y, Murat P, Defrancq E. Recent developments in oligonucleotideconjugation. Chem Soc Rev. 2010;39:2054–2070. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]
- 91.Ceballos C, Prata CAH, Giorgio S, Garzino F, Payet D, Barthélémy P, et al. Cationic nucleoside lipids based on a 3-nitropyrrole universal base for siRNA delivery. Bioconjug Chem. 2009;20:193–196. doi: 10.1021/bc800432n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ceballos C, Khiati S, Prata CAH, Zhang XX, Giorgio S, Marsal P, et al. Cationic nucleoside lipids derived from universal bases: A rational approach for siRNA transfection. Bioconjug Chem. 2010;21:1062–1069. doi: 10.1021/bc100005k. [DOI] [PubMed] [Google Scholar]
- 93.Allain V, Bourgaux C, Couvreur P. Self-assembled nucleolipids: from supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 2012;40:1891–1903. doi: 10.1093/nar/gkr681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taib N, Aimé A, Houmadi S, Castano S, Barthélémy P, Laguerre M, et al. Chemical details on nucleolipid supramolecular architecture: molecular modeling and physicochemical studies. Langmuir. 2012;28:7452–7460. doi: 10.1021/la300744x. [DOI] [PubMed] [Google Scholar]
- 95.Chabaud P, Camplo M, Payet D, Serin G, Moreau L, Barthélémy P, et al. Cationic nucleoside lipids for gene delivery. Bioconjug Chem. 2006;17:466–472. doi: 10.1021/bc050162q. [DOI] [PubMed] [Google Scholar]
- 96.Banchelli M, Berti D, Baglioni P. Molecular recognition drives oligonucleotide binding to nucleolipid self-assemblies. Angew Chem Int Ed Engl. 2007;46:3070–3073. doi: 10.1002/anie.200604826. [DOI] [PubMed] [Google Scholar]
- 97.Khiati S, Pierre N, Andriamanarivo S, Grinstaff MW, Arazam N, Nallet F, et al. Anionic nucleotide--lipids for in vitro DNA transfection. Bioconjug Chem. 2009;20:1765–1772. doi: 10.1021/bc900163s. [DOI] [PubMed] [Google Scholar]
- 98.Ma M, Gong Y, Bong D. Lipid membrane adhesion and fusion driven by designed, minimally multivalent hydrogen-bonding lipids. J Am Chem Soc. 2009;131:16919–16926. doi: 10.1021/ja9072657. [DOI] [PubMed] [Google Scholar]
- 99.Ma M, Paredes A, Bong D. Intra- and intermembrane pairwise molecular recognition between synthetic hydrogen-bonding phospholipids. J Am Chem Soc. 2008;130:14456–14458. doi: 10.1021/ja806954u. [DOI] [PubMed] [Google Scholar]
- 100.Polidori A, Michel N, Fabiano AS, Pucci B. Exotic aqueous behavior of synthetic lipids: formation of vesicular nanotubes. Chem Phys Lipids. 2005;136:23–46. doi: 10.1016/j.chemphyslip.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 101.Godeau G, Barthélémy P. Glycosyl-nucleoside lipids as low-molecular-weight gelators. Langmuir. 2009;25:8447–8450. doi: 10.1021/la900140b. [DOI] [PubMed] [Google Scholar]
- 102.Godeau G, Bernard J, Staedel C, Barthélémy P. Glycosyl nucleoside lipid based supramolecular assembly as a nanostructured material with nucleic acid delivery capabilities. Chemical Communications. 2009:5127–5129. doi: 10.1039/b906212b. [DOI] [PubMed] [Google Scholar]
- 103.Semple SC, Harasym TO, Clow KA, Ansell SM, Klimuk SK, Hope MJ. Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic Acid. J Pharmacol Exp Ther. 2005;312:1020–1026. doi: 10.1124/jpet.104.078113. [DOI] [PubMed] [Google Scholar]
- 104.Nobs L, Buchegger F, Gurny R, Allmann E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J Pharm Sci. 2004;93:1980–1992. doi: 10.1002/jps.20098. [DOI] [PubMed] [Google Scholar]
- 105.Manjappa AS, Chaudhari KR, Venkataraju MP, Dantuluri P, Nanda B, Sidda C, et al. Antibody derivatization and conjugation strategies: application in preparation of stealth immunoliposome to target chemotherapeutics to tumor. J Control Release. 2011;150:2–22. doi: 10.1016/j.jconrel.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 106.Martin FJ, Papahadjopoulos D. Irreversible coupling of immunoglobulin fragments to preformed vesicles. An improved method for liposome targeting. J Biol Chem. 1982;257:286–288. [PubMed] [Google Scholar]
- 107.Colletier JP, Chaize B, Winterhalter M, Fournier D. Protein encapsulation in liposomes: efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002;2:9. doi: 10.1186/1472-6750-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Illum L, Jones PD, Baldwin RW, Davis SS. Tissue distribution of poly(hexyl 2-cyanoacrylate) nanoparticles coated with monoclonal antibodies in mice bearing human tumor xenografts. J Pharmacol Exp Ther. 1984;230:733–736. [PubMed] [Google Scholar]
- 109.Huang Z, Park JI, Watson DS, Hwang P, Szoka FC. Facile synthesis of multivalent nitrilotriacetic acid (NTA) and NTA conjugates for analytical and drug delivery applications. Bioconjug Chem. 2006;17:1592–1600. doi: 10.1021/bc0602228. [DOI] [PubMed] [Google Scholar]
- 110.van Broekhoven CL, Altin JG. The novel chelator lipid 3(nitrilotriacetic acid)-ditetradecylamine (NTA(3)-DTDA) promotes stable binding of His-tagged proteins to liposomal membranes: potent anti-tumor responses induced by simultaneously targeting antigen, cytokine and costimulatory signals to T cells. Biochim Biophys Acta. 2005;1716:104–116. doi: 10.1016/j.bbamem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 111.Chikh G. Attaching histidine-tagged peptides and proteins to lipid-based carriers through use of metal-ion-chelating lipids. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 2002;1567:204–212. doi: 10.1016/s0005-2736(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 112.Rüger R, Müller D, Fahr A, Kontermann RE. In vitro characterization of binding and stability of single-chain Fv Ni-NTA-liposomes. Journal of Drug Targeting. 2006;14:576–582. doi: 10.1080/10611860600864018. [DOI] [PubMed] [Google Scholar]
- 113.Fischer NO, Blanchette CD, Chromy BA, Kuhn EA, Segelke BW, Corzett M, et al. Immobilization of His-tagged proteins on nickel-chelating nanolipoprotein particles. Bioconjug Chem. 2009;20:460–465. doi: 10.1021/bc8003155. [DOI] [PubMed] [Google Scholar]
- 114.Sigal GB, Bamdad C, Barberis A, Strominger J, Whitesides GM. A self-assembled monolayer for the binding and study of histidine-tagged proteins by surface plasmon resonance. Anal Chem. 1996;68:490–497. doi: 10.1021/ac9504023. [DOI] [PubMed] [Google Scholar]
- 115.Altin JG, Banwell MG, Coghlan PA, Easton CJ, Nairn MR, Offermann DA. Synthesis of NTA 3-DTDA — A Chelator-Lipid that Promotes Stable Binding of His-Tagged Proteins to Membranes. Australian Journal of Chemistry. 2006;59:302–306. [Google Scholar]
- 116.van Broekhoven CL, Altin JG. The novel chelator lipid 3 (nitrilotriacetic acid)-ditetradecylamine (NTA 3-DTDA) promotes stable binding of His-tagged proteins to liposomal membranes: Potent anti-tumor responses induced by simultaneously targeting antigen, cytokine and costimulatory signals to T cells. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 2005;1716:104–116. doi: 10.1016/j.bbamem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 117.Huang Z, Hwang P, Watson DS, Cao L, Szoka FC. Tris-nitrilotriacetic acids of subnanomolar affinity toward hexahistidine tagged molecules. Bioconjug Chem. 2009;20:1667–1672. doi: 10.1021/bc900309n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Platt V, Huang Z, Cao L, Tiffany M, Riviere K, Szoka FC. Influence of multivalent nitrilotriacetic acid lipid-ligand affinity on the circulation half-life in mice of a liposome-attached His6-protein. Bioconjug Chem. 2010;21:892–902. doi: 10.1021/bc900448f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Watson DS, Platt VM, Cao L, Venditto VJ, Szoka FC. Antibody Response to Polyhistidine-Tagged Peptide and Protein Antigens Attached to Liposomes via Lipid-Linked Nitrilotriacetic Acid in Mice. Clinical and Vaccine Immunology. 2011;18:289–297. doi: 10.1128/CVI.00425-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol. 2006;7:24–39. doi: 10.1186/1471-2172-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Torchilin VP, Weissig V. Liposomes: A Practical Approach. Oxford University Press; 2003. [Google Scholar]
- 122.New RRC. Liposomes: a practical approach. Oxford University Press; USA: 1990. [Google Scholar]
- 123.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci. 2008;97:4696–4740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 124.Mabrey S, Mateo PL, Sturtevant JM. High-sensitivity scanning calorimetric study of mixtures of cholesterol with dimyristoyl- and dipalmitoylphosphatidylcholines. Biochemistry. 1978;17:2464–2468. doi: 10.1021/bi00605a034. [DOI] [PubMed] [Google Scholar]
- 125.McMullen TP, Lewis RN, McElhaney RN. Differential scanning calorimetric study of the effect of cholesterol on the thermotropic phase behavior of a homologous series of linear saturated phosphatidylcholines. Biochemistry. 1993;32:516–522. doi: 10.1021/bi00053a016. [DOI] [PubMed] [Google Scholar]
- 126.Demel RA, Bruckdorfer KR, Van Deenen LLM. The effect of sterol structure on the permeability of lipomes to glucose, glycerol and Rb. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 1972;255:321–330. doi: 10.1016/0005-2736(72)90031-4. [DOI] [PubMed] [Google Scholar]
- 127.Papahadjopoulos D, Nir S, Oki S. Permeability properties of phospholipid membranes: effect of cholesterol and temperature. Biochim Biophys Acta. 1972;266:561–583. doi: 10.1016/0006-3002(72)90001-7. [DOI] [PubMed] [Google Scholar]
- 128.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 129.Bach D. Phospholipid/cholesterol model membranes: formation of cholesterol crystallites. Biochimica Et Biophysica Acta (BBA) - Biomembranes. 2003;1610:187–197. doi: 10.1016/s0005-2736(03)00017-8. [DOI] [PubMed] [Google Scholar]
- 130.Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochimica Et Biophysica Acta (BBA) - Reviews on Biomembranes. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 131.Hamilton JA. Fast flip-flop of cholesterol and fatty acids in membranes: implications for membrane transport proteins. Current Opinion in Lipidology. 2003;14:263–271. doi: 10.1097/00041433-200306000-00006. [DOI] [PubMed] [Google Scholar]
- 132.Kan CC, Yan J, Bittman R. Rates of spontaneous exchange of synthetic radiolabeled sterols between lipid vesicles. Biochemistry. 1992;31:1866–1874. doi: 10.1021/bi00121a040. [DOI] [PubMed] [Google Scholar]
- 133.Ding J, Starling AP, East JM, Lee AG. Binding sites for cholesterol on Ca(2+)-ATPase studied by using a cholesterol-containing phospholipid. Biochemistry. 1994;33:4974–4979. doi: 10.1021/bi00182a028. [DOI] [PubMed] [Google Scholar]
- 134.Huang Z, Jaafari MR, Szoka FC. Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed Engl. 2009;48:4146–4149. doi: 10.1002/anie.200900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang Z, Szoka FC. Sterol-Modified Phospholipids: Cholesterol and Phospholipid Chimeras with Improved Biomembrane Properties. J Am Chem Soc. 2008;130:15702–15712. doi: 10.1021/ja8065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohli AG, Kieler-Ferguson HM, Chan D, Szoka FC. A robust and quantitative method for tracking liposome contents after intravenous administration. J Control Release. 2014;176:86–93. doi: 10.1016/j.jconrel.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lovell JF, Jin CS, Huynh E, Jin H, Kim C, Rubinstein JL, et al. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat Mater. 2011;10:324–332. doi: 10.1038/nmat2986. [DOI] [PubMed] [Google Scholar]
- 138.Fretz MM, Høgset A, Koning GA, Jiskoot W, Storm G. Cytosolic delivery of liposomally targeted proteins induced by photochemical internalization. Pharm Res. 2007;24:2040–2047. doi: 10.1007/s11095-007-9338-9. [DOI] [PubMed] [Google Scholar]
- 139.Ng KK, Lovell JF, Vedadi A, Hajian T, Zheng G. Self-assembled porphyrin nanodiscs with structure-dependent activation for phototherapy and photodiagnostic applications. ACS Nano. 2013;7:3484–3490. doi: 10.1021/nn400418y. [DOI] [PubMed] [Google Scholar]
- 140.Huynh E, Lovell JF, Helfield BL, Jeon M, Kim C, Goertz DE, et al. Porphyrin Shell Microbubbles with Intrinsic Ultrasound and Photoacoustic Properties. J Am Chem Soc. 2012;134:16464–16467. doi: 10.1021/ja305988f. [DOI] [PubMed] [Google Scholar]
- 141.Tondo DW, Leopoldino EC, Souza BS, Micke GA, Costa ACO, Fiedler HD, et al. Synthesis of a new zwitterionic surfactant containing an imidazolium ring. Evaluating the chameleon-like behavior of zwitterionic micelles. Langmuir. 2010;26:15754–15760. doi: 10.1021/la102391e. [DOI] [PubMed] [Google Scholar]
- 142.Papahadjopoulos D, Nir S, Düzgünes N. Molecular mechanisms of calcium-induced membrane fusion. J Bioenerg Biomembr. 1990;22:157–179. doi: 10.1007/BF00762944. [DOI] [PubMed] [Google Scholar]
- 143.Perttu EK, Szoka FC. Zwitterionic sulfobetaine lipids that form vesicles with salt-dependent thermotropic properties. Chem Commun (Camb) 2011;47:12613–12615. doi: 10.1039/c1cc15804j. [DOI] [PubMed] [Google Scholar]
- 144.Perttu EK, Kohli AG, Szoka FC., Jr Inverse-Phosphocholine Lipids: A Remix of a Common Phospholipid. J Am Chem Soc. 2012;134:4485–4488. doi: 10.1021/ja210989h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kohli AG, Walsh CL, Szoka FC. Synthesis and characterization of betaine-like diacyl lipids: zwitterionic lipids with the cationic amine at the bilayer interface. Chem Phys Lipids. 2012;165:252–259. doi: 10.1016/j.chemphyslip.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim ES, Lu C, Khuri FR, Tonda M, Glisson BS, Liu D, et al. A phase II study of STEALTH cisplatin (SPI-77) in patients with advanced non-small cell lung cancer. Lung Cancer. 2001;34:427–432. doi: 10.1016/s0169-5002(01)00278-1. [DOI] [PubMed] [Google Scholar]
- 147.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 148.Bibi S, Lattmann E, Mohammed AR, Perrie Y. Trigger release liposome systems: local and remote controlled delivery? J Microencapsul. 2012;29:262–276. doi: 10.3109/02652048.2011.646330. [DOI] [PubMed] [Google Scholar]
- 149.Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta. 2005;1716:77–96. doi: 10.1016/j.bbamem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 150.Needham D, Park JY, Wright AM, Tong J. Materials characterization of the low temperature sensitive liposome (LTSL): effects of the lipid composition (lysolipid and DSPE-PEG2000) on the thermal transition and release of doxorubicin. Faraday Discuss. 2013;161:515–534. doi: 10.1039/c2fd20111a. [DOI] [PubMed] [Google Scholar]
- 151.Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007;16:855–866. doi: 10.1517/13543784.16.6.855. [DOI] [PubMed] [Google Scholar]
- 152.Wang Y, Li L, Jiang W, Yang Z, Zhang Z. Synthesis and preliminary antitumor activity evaluation of a DHA and doxorubicin conjugate. Bioorg Med Chem Lett. 2006;16:2974–2977. doi: 10.1016/j.bmcl.2006.02.066. [DOI] [PubMed] [Google Scholar]
- 153.Duhem N, Danhier F, Pourcelle V, Schumers J-M, Bertrand O, Leduff CS, et al. Self-Assembling Doxorubicin-Tocopherol Succinate Prodrug as a New Drug Delivery System: Synthesis, Characterization, and in Vitro and in Vivo Anticancer Activity. Bioconjug Chem. 2013 doi: 10.1021/bc400326y. [DOI] [PubMed] [Google Scholar]
- 154.Verweij J, Pinedo HM. Mitomycin C: mechanism of action, usefulness and limitations. Anticancer Drugs. 1990;1:5–13. [PubMed] [Google Scholar]
- 155.Gabizon AA, Tzemach D, Horowitz AT, Shmeeda H, Yeh J, Zalipsky S. Reduced toxicity and superior therapeutic activity of a mitomycin C lipid-based prodrug incorporated in pegylated liposomes. Clin Cancer Res. 2006;12:1913–1920. doi: 10.1158/1078-0432.CCR-05-1547. [DOI] [PubMed] [Google Scholar]
- 156.Gabizon A, Amitay Y, Tzemach D, Gorin J, Shmeeda H, Zalipsky S. Therapeutic efficacy of a lipid-based prodrug of mitomycin C in pegylated liposomes: studies with human gastro-entero-pancreatic ectopic tumor models. J Control Release. 2012;160:245–253. doi: 10.1016/j.jconrel.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 157.Moysan E, Bastiat G, Benoit JP. Gemcitabine versus Modified Gemcitabine: a review of several promising chemical modifications. Mol Pharm. 2013;10:430–444. doi: 10.1021/mp300370t. [DOI] [PubMed] [Google Scholar]
- 158.Adema AD, Losekoot N, Smid K, Kathmann I, Myhren F, Sandvold ML, et al. Induction of resistance to the lipophilic cytarabine prodrug elacytarabine (CP-4055) in CEM leukemic cells. Nucleosides Nucleotides Nucleic Acids. 2010;29:394–399. doi: 10.1080/15257771003741166. [DOI] [PubMed] [Google Scholar]
- 159.Breistøl K, Balzarini J, Sandvold ML, Myhren F, Martinsen M, De Clercq E, et al. Antitumor activity of P-4055 (elaidic acid-cytarabine) compared to cytarabine in metastatic and s.c. human tumor xenograft models. Cancer Res. 1999;59:2944–2949. [PubMed] [Google Scholar]
- 160.Stuurman FE, Voest EE, Awada A, Witteveen PO, Bergeland T, Hals PA, et al. Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Invest New Drugs. 2013;31:959–966. doi: 10.1007/s10637-013-9925-z. [DOI] [PubMed] [Google Scholar]
- 161.Bradley MO, Webb NL, Anthony FH, Devanesan P, Witman PA, Hemamalini S, et al. Tumor targeting by covalent conjugation of a natural fatty acid to paclitaxel. Clin Cancer Res. 2001;7:3229–3238. [PubMed] [Google Scholar]
- 162.Jones RJ, Hawkins RE, Eatock MM, Ferry DR, Eskens FALM, Wilke H, et al. A phase II open-label study of DHA-paclitaxel (Taxoprexin) by 2-h intravenous infusion in previously untreated patients with locally advanced or metastatic gastric or oesophageal adenocarcinoma. Cancer Chemother Pharmacol. 2007;61:435–441. doi: 10.1007/s00280-007-0486-8. [DOI] [PubMed] [Google Scholar]
- 163.Bedikian AY, DeConti RC, Conry R, Agarwala S, Papadopoulos N, Kim KB, et al. Phase 3 study of docosahexaenoic acid-paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Ann Oncol. 2011;22:787–793. doi: 10.1093/annonc/mdq438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dosio F, Reddy LH, Ferrero A, Stella B, Cattel L, Couvreur P. Novel nanoassemblies composed of squalenoyl-paclitaxel derivatives: synthesis, characterization, and biological evaluation. Bioconjug Chem. 2010;21:1349–1361. doi: 10.1021/bc100154g. [DOI] [PubMed] [Google Scholar]
- 165.Arias JL, Reddy LH, Couvreur P. Magnetoresponsive squalenoyl gemcitabine composite nanoparticles for cancer active targeting. Langmuir. 2008;24:7512–7519. doi: 10.1021/la800547s. [DOI] [PubMed] [Google Scholar]
- 166.Arias JL, Reddy LH, Othman M, Gillet B, Desmaële D, Zouhiri F, et al. Squalene based nanocomposites: a new platform for the design of multifunctional pharmaceutical theragnostics. ACS Nano. 2011;5:1513–1521. doi: 10.1021/nn1034197. [DOI] [PubMed] [Google Scholar]
- 167.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]