

Abstract
Phospho-sulindac amide (PSA) is a novel potential anti-cancer and anti-inflammatory agent. Here we report the metabolism of PSA in vitro. PSA was rapidly hydroxylated at its butane-phosphate moiety to form two di-hydroxyl-PSA and four mono-hydroxyl-PSA metabolites in mouse and human liver microsomes. PSA also can be oxidized or reduced at its sulindac moiety to form PSA sulfone and PSA sulfide, respectively. PSA was mono-hydroxylated and cleared more rapidly in mouse liver microsomes than in human liver microsomes. Of eight major human cytochrome P450s (CYPs), CYP3A4 and CYP2D6 exclusively catalyzed the hydroxylation and sulfoxidation reactions of PSA, respectively. We also examined the metabolism of PSA by three major human flavin monooxygenases (FMOs). FMO1, FMO3 and FMO5 were all capable of catalyzing the sulfoxidation (but not hydroxylation) of PSA, with FMO1 being by far the most active isoform. PSA was predominantly sulfoxidized in human kidney microsomes because FMO1 is the dominant isoform in human kidney. PSA (versus sulindac) is a preferred substrate of both CYPs and FMOs, likely because of its greater lipophilicity and masked –COOH group. Ketoconazole (a CYP3A4 inhibitor) and alkaline pH strongly inhibited the hydroxylation of PSA, but moderately suppressed its sulfoxidation in liver microsomes. Together, our results establish the metabolic pathways of PSA, identify the major enzymes mediating its biotransformations and reveal significant inter-species and inter-tissue differences in its metabolism.
Keywords: phospho-sulindac amide, cytochrome P450, liver microsomes, hydroxylation, sulfoxidation
1. Introduction
Inflammation plays critical roles at various stages of tumor development, including tumor initiation, promotion, malignant conversion, invasion, and metastasis [1]. These findings provide a foundation and rationale for the use of anti-inflammatory drugs in cancer prevention and therapy. For example, sulindac, a widely-used nonsteroidal anti-inflammatory drug, prevents colon cancer in patients with sporadic adenomas or familial adenomatous polyposis [2].
Sulindac, however, has a broad range of side effects, including gastrointestinal, central nervous system, skin rash and pruritus, and transient elevations of hepatic enzymes in plasma [3]. (Roberts and Morrow, 2001). This toxicity results from the inhibition of cyclooxygenase (COX) and the subsequent prostaglandin synthesis; the -COOH group of sulindac is critical for its binding to COX [4]. Therefore, we recently modified sulindac at its –COOH group to develop a sulindac derivative, phospho-sulindac (PS) (Fig. 1). PS exhibited more potent anti-cancer efficacy and markedly reduced toxicity than conventional sulindac in animal models [5]. However, the carboxylester bond of PS is labile, resulting in the extensive hydrolysis of PS to sulindac in vitro and in vivo [6]. We previously demonstrated that protecting PS against its hydrolysis enhanced its anti-cancer efficacy [7]. Thus, the extensive hydrolysis of PS in vivo may be detrimental to its efficacy and safety. This consideration prompted us to modify PS at its ester bond to generate phospho-sulindac amide (PSA; Fig. 1) to abrogate its undesired hydrolysis.
Fig. 1.
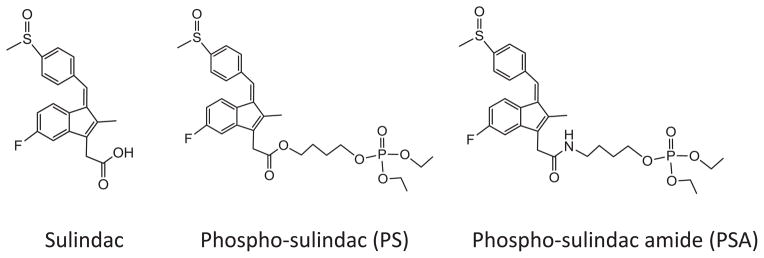
Structures of sulindac, phospho-sulindac and phospho-sulindac amide.
Drug metabolism plays a key role in defining the efficacy and toxicity of drugs. While the assessment of PSA as a novel anticancer/anti-inflammatory agent is still ongoing, herein, we report the major metabolic pathways of PSA, showing that PSA is extensively hydroxylated and sulfoxidized but not hydrolyzed by microsomes from various species and tissues. These results are essential for a better understanding of the pharmacology of PSA in animal and human studies.
2. Materials and Methods
2.1. Reagents
PSA was provided by Medicon Pharmaceuticals, Inc. (Stony Brook, NY). Sulindac, sulindac sulfone, sulindac sulfide, dithiothreitol, methimazole, and CH3CN of HPLC grade were purchased from Sigma-Aldrich (St. Louis, MO). Quinidine and ketoconazole were purchased from Toronto Research Chemicals (North York, ON, Canada). Mouse and human liver microsomes, rat liver cytosol, recombinant human CYPs (CYP1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1 and 3A4), FMOs (FMO1, FMO3 and FMO5), NADPH regenerating solution, and cryopreserved rat hepatocytes were purchased from BD Biosciences (San Jose, CA). Human intestine, kidney and lung microsomes were purchased from XenoTech LLC (Lenexa, KS).
2.2. HPLC-UV analysis
The HPLC system consisted of a Waters Alliance 2695 Separations Module equipped with a Waters 2998 photodiode array detector (328 nm) and a Thermo Hypersil BDS C18 column (150 × 4.6 mm, particle size 3 μm). The mobile phase consisted of a gradient between aqueous phase [Trifluoroacetic acid, CH3CN, H2O (0.1:4.9:95 v/v/v)] and CH3CN at a flow rate of 1 ml/min at 30°C. We applied gradient elution from 0% to 100% CH3CN in 15 min, and it was maintained at 100% CH3CN for 5 min.
2.3. LC-MS/MS analysis
The LC-MS/MS system consisted of a Thermo TSQ Quantum Access (Thermo-Fisher) electrospray ionization triple quadrupole mass spectrometer coupled to an Ultimate 3000 HPLC system (Dionex Corporation, Sunnyvale, CA). Chromatographic separations were achieved using a Luna C18 column (150 × 2 mm) and a mobile phase consisting of a gradient from 10% to 95% CH3CN.
2.4. The metabolism of PSA in mouse and human microsomes
PSA was pre-incubated at 37°C for 5 min with an NADPH-regenerating solution (1.3 mM NADP, 3.3 mM D-glucose 6-phosphate, 3.3 mM MgCl2, and 0.4 U/ml glucose-6-phosphate dehydrogenase) in 0.1 M potassium phosphate buffer (pH 7.4). The reaction was initiated by the addition of mouse or human liver microsomes (protein concentration 0.5 mg/ml) or human intestine, kidney, liver or lung microsomes (protein concentration 0.25 mg/ml) and samples were maintained at 37°C for various time periods. At each of the designated time-points, 0.1-ml aliquots were mixed with 0.2 ml of CH3CN, vortexed, and then centrifuged for 10 min at 13,000x g. The supernatants were subjected to HPLC analyses. The HPLC fractions corresponding to each metabolite of PSA were collected and analyzed by mass spectrometry.
2.5. The metabolism of PA by rat hepatocytes
Cryopreserved rat hepatocytes were thawed and incubated following the manufacturer’s protocol. Briefly, hepatocytes were incubated with PSA in 24-well tissue culture plates at a density of 2.5×105 cells/well in 5% CO2 at 37°C. At the designated time-points, the cells were mixed with 2-fold volume of acetonitrile to stop the reaction. After centrifugation, the supernatants were analyzed by HPLC.
2.6. The metabolism of PSA by rat liver cytosol
PSA (50 μM) was pre-incubated at 37°C for 5 min with 10 mM dithiothreitol in 0.1 M Tris buffer (pH 7.4). The reaction was initiated by the addition of liver cytosol (protein concentration 2 mg/mL) and samples were maintained at 37°C for various time periods. At the end of each of the incubations, 0.1mL aliquots were mixed with 0.2 mL of CH3CN, vortexed and then centrifuged for 10 min at 13, 000x g. The supernatants were analyzed by HPLC.
2.7. The metabolism of PSA by human CYP and FMO isoforms
PSA were pre-incubated at 37°C for 5 min with an NADPH-regenerating solution (1.3 mM NADP, 3.3 mM D-glucose 6-phosphate, 3.3 mM MgCl2, and 0.4 U/ml glucose-6-phosphate dehydrogenase) in 0.1 M potassium phosphate buffer (pH 7.4). The reaction was initiated by the addition of individual recombinant human CYP isoforms (25 pmol/ml) or human FMO isoforms (0.125 mg protein/ml) in a total volume of 1 ml and samples were maintained at 37°C for various time periods. At each designated time point, an aliquot was mixed with 2-fold volume of CH3CN, vortexed, and then centrifuged for 10 min at 13,000x g. The supernatants were subjected to HPLC analysis.
3. Results
3.1. The metabolism of PSA in mouse and human liver microsomes
We examined the metabolism of PSA in mouse liver microsomes (MLM) and human liver microsomes (HLM). We identified four mono-hydroxyl metabolites (henceforce referred to as mono-hydroxyl-PSA) and two di-hydroxyl metabolites using LC-MS/MS. The sodium adduct ions of the four mono-hydroxyl-PSA were observed at m/z 602.3, 602.4, 602.5 and 602.3, respectively. The sodium adduct ions of the two di-hydroxyl-PSA were observed at m/z 618.2 and 618.3, respectively. To determine the position of the hydroxyl groups of the metabolites, we treated these metabolites at pH 13 and 70°C for 3 h. These metabolites were hydrolyzed to give sulindac, suggesting that their hydroxyl groups are not located on the sulindac moiety, but on the butane-phosphate moiety. The protonated PSA sulfone was observed at m/z 580.2 (Fig. 2A), which was further fragmented to generated ion at m/z 426.06 (Fig. 2B). The identification of PSA sulfone was also supported by our observation that PSA was hydrolyzed to give sulindac sulfone at pH 13. We did not detect sulindac in the liver microsomes, indicating that the amide bond of PSA is rather stable in the liver microsomes.
Fig. 2. Identification of PSA sulfone using LC-MS/MS analysis.

A: Top, MS spectrum of protonated PSA sulfone. B: Bottom, MS2 spectrum of the above ion at m/z 580.2.
As shown in Fig. 3, PSA was rapidly hydroxylated and sulfoxidized to yield mono-hydroxyl-PSA, di-hydroxyl-PSA and PSA sulfone with the former being the major metabolite in liver microsomes. PSA generated more mono-hydroxyl-PSA in MLM than in HLM, resulting in the more rapid clearance of PSA in MLM than in HLM (Fig. 3).
Fig. 3. Kinetics of PSA metabolism in mouse and human liver microsomes.

Time course of the levels of PSA and its metabolites in PSA-treated liver microsomes. PSA (50 μM) was incubated with mouse or human liver microsomes (protein concentration 0.5 mg/ml) at 37°C for up to 3 h. PSA and its metabolites were extracted at the designated time points and assayed using HPLC.
3.2. The metabolism of PSA in human liver, intestine, kidney and lung microsomes
We compared the metabolic behavior of PSA in human liver, intestine, kidney and lung microsomes. PSA was extensively metabolized in human liver and kidney microsomes, and to a lesser extent in intestine microsomes, but not in lung microsomes (Fig. 4). While mono-hydroxyl-PSA was the major metabolite in human liver and intestine microsomes, PSA sulfone was dominant in kidney microsomes.
Fig. 4. Kinetics of PSA metabolism in human liver, intestine, kidney, and lung microsomes.

Time courses of the levels of PSA and its metabolites in PSA-treated human microsomes. PSA (50 μM) was incubated individually with human liver, intestine, kidney and lung microsomes (protein concentration 0.25 mg/ml) at 37°C for up to 4 h. PSA and its metabolites were extracted at the designated time points and assayed as described in Methods.
3.3. The metabolism of PSA and sulindac by human CYPs
Cytochrome P450s (CYPs) account for ~75% of drug metabolism [8]. Of the eight major human CYPs, CYP3A4 uniquely catalyzes the hydroxylation of PSA to form mono-hydroxyl-PSA and di-hydroxyl-PSA (Fig. 5). CYP2D6 is by far the most active isoform to catalyze the sulfoxidation of PSA at its sulindac moiety to form PSA sulfone. CYP3A4, 2C9 and 2C19 exhibited minimal activity in the sulfoxidation of PSA. In contrast, CYP1A2, 2A6, 2B6 and 2E1are inactive towards PSA (Fig. 5). In comparison, all the CYPs tested failed to appreciably sulfoxidize sulindac under the same experimental condition (data not shown), indicating that PSA (versus sulindac) is a preferred substrate of CYPs.
Fig. 5. Kinetics of the metabolism of PSA by human CYPs.

PSA (50 μM) was incubated with individual human CYP isoforms and NADPH-regenerating solution at 37°C for up to 6 h. The metabolites were extracted at the designated time points and assayed by HPLC.
3.4. The metabolism of PSA and sulindac by human FMOs
We next evaluated the metabolism of PSA by three major human flavin monooxygenases (FMOs). FMO1, FMO3 and FMO5 were all capable of catalyzing the sulfoxidation of PSA with FMO1 being by far the most active (Fig. 6). Twenty min after the initiation of the reactions, FMO1 generated 21.4 and 6.5 times higher level of PSA sulfone than FMO3 and FMO5, respectively. All the FMOs sulfoxidized PSA far more rapidly than sulindac (Fig. 6), indicating that PSA (versus sulindac) is a preferred substrate of FMOs. We did not detect hydroxyl-PSA in the reaction mixtures, indicating that FMOs do not catalyze the hydroxylation of PSA.
Fig. 6. Kinetics of the oxidation of PS versus sulindac by FMOs.

PSA or sulindac (each at 50 μM) was incubated with individual human FMOs (FMO1, FMO3 and FMO5) and NADPH-regenerating solution at 37°C for up to 20 h. The oxidized products were extracted at the designated time points and analyzed by HPLC. Results were expressed as percent of input drug.
3.5. Effect of ketoconazole and quinidine on the metabolism of PSA
We evaluated the effect of ketoconazole (CYP3A4 inhibitor) and quinidine (CYP2D6 inhibitor) on the metabolism of PSA in liver microsomes. Ketoconazole strongly inhibited the hydroxylation of PSA in MLM and HLM in a concentration-dependent manner (Fig. 7). Ketoconazole also significantly inhibited the sulfoxidation of PSA in HLM, but unexpectedly enhanced its sulfoxidation in MLM. On the other hand, quinidine (up to 100 μM) had no appreciable effect on the hydroxylation and sulfoxidation of PSA in MLM and HLM, suggesting that CYP2D6 plays a negligible role in the metabolism of PSA in liver microsomes.
Fig. 7. Effect of ketoconazole on the metabolism of PSA in liver microsomes.

PSA (50 μM) was incubated with or without ketoconazole in mouse or human liver microsomes) at 37°C for 1h. PSA metabolites were extracted and assayed using HPLC. Presented are the percentages of control at the indicated ketoconazole concentrations.
3.6. Effect of alkaline pH and methimazole on the metabolism of PSA
The activities of CYPs (but not FMOs) could be abrogated at alkaline pH (Cashman, 2005). We observed that the human CYPs were completely inactive at pH 9.5, whereas the activities of FMOs were essentially not affected by this alkaline pH. We examined the metabolism of PSA at pH 9.5 versus pH 7.4. The alkaline pH strongly inhibited the hydroxylation of PSA in MLM and HLM by 95% and 99%, respectively (Fig. 8A), confirming that the hydroxylation of PSA in the liver microsomes is catalyzed by CYPs, rather than FMOs.
Fig. 8. Effect of alkaline pH on the metabolism of PSA in microsomes.

A. PSA (50 μM) was incubated in liver microsomes at pH7.4 or pH9.5 at 37°C for 1h. PSA metabolites were extracted and assayed using HPLC. B. PSA (50 μM) was incubated in microsomes at pH7.4 or pH9.5 at 37°C for 1h. PSA metabolites were extracted and assayed using HPLC.
As is the case with ketoconazole, the alkaline pH produced opposite effects on PSA sulfoxidation in MLM and HLM (Fig. 8B). Ketoconazole and alkaline pH unexpectedly enhanced PSA sulfoxidation in MLM because they strongly inhibited PSA hydroxylation, resulting in more substrate available for PSA sulfoxidation. Indeed, ketoconazole (10 μM) did inhibit the sulfoxidation of PSA by 21% in MLM at pH 9.5. The strong sulfoxidation of PSA in human kidney microsomes was essentially not affected by the alkaline pH (Fig. 8B), indicating that this reaction in kidney is catalyzed by FMOs but not by CYPs.
We also evaluated the effect of methimazole (FMO inhibitor) on the metabolism of PSA in microsomes. Methimazole (5 mM) completely inhibited the sulfoxidation of PSA in human kidney microsomes, confirming that this reaction is catalyzed exclusively by FMOs rather than CYPs. On the other hand, methimazole partially inhibited the sulfoxidation of PSA in MLM and HLM by 48% and 73%, respectively.
3.7. The metabolism of PSA by rat hepatocytes
We examined the metabolism of PSA by cryopreserved rat hepatocytes. PSA was primarily metabolized to form mono-hydroxyl-PSA by rat hepatocytes. One hour after the incubation of PSA (35μM) with rat hepatocytes, 51.5%, 6.8% and 1.9% of PSA were converted to mono-hydroxyl-PSA, di-hydroxyl-PSA and PSA sulfone, respectively. These results are consistent with the extensive hydroxylation of PSA in liver microsomes (Fig. 3).
3.8. The metabolism of PSA in rat liver cytosol
Since conventional sulindac can be reduced to sulindac sulfide in rat liver cytosol [9], we treated PSA with rat liver cytosol at 37°C in the absence of NADPH. Two hours after the initiation of the reaction, 32% of PSA was reduced to form PSA sulfide. PSA sulfide was identified using LC-MS/MS analysis, and its sodium adduct ion was observed at m/z 570.3.
4. Discussion
This work establishes the metabolic pathways of PSA. As illustrated in Fig. 9, PSA undergoes the following metabolic reactions: 1) mono-hydroxylation at its butane-phosphate moiety to form mono-hydroxyl-PSA; 2) di-hydroxylation at its butane-phosphate moiety to form di-hydroxyl-PSA; 3) sulfoxidation at its sulindac moiety to form PSA sulfone; 4)) reduction at its sulindac moiety to form PSA sulfide. The amide bond of PSA is rather stable since we did not detect sulindac in PSA-treated liver microsomes.
Fig. 9. Overall metabolic pathways of PSA in vitro.

PSA undergoes hydroxylation reactions at its butane-phosphate moiety to form mono-hydroxyl-PSA and di-hydroxyl-PSA, which are uniquely catalyzed by CYP3A4. PSA also can be oxidized and reduced at its sulindac moiety to form PSA sulfone and PSA sulfide, respectively.
CYPs play a major role in the metabolism of PSA by catalyzing its hydroxylation reactions. Of the five major human CYPs, CYP3A4 uniquely hydroxylates PSA to generate four mono-hydroxyl metabolites and two di-hydroxyl metabolites. Similarly, CYP3A4 uniquely catalyzes the hydroxylation of phospho-ibuprofen [10] and phospho-tyrosol-indomethacin [11]. Substrate-binding to CYP3A4 induces a large increase in the volume of its active site [12], likely accounting for its preference for bulky substrates like phospho-compounds. Indeed, CYP3A4 accounts for 45–60% of the metabolism of all drugs currently used [13].
On the other hand, among the five major human CYPs, CYP2D6 is the most active in catalyzing the sulfoxidation of PSA. CYP2D6 substrate binding is uniquely governed by an ion-pair interaction between a positively charged nitrogen atom and a negatively charged Asp301 residue [14]. The partially positively charged sulfur and phosphorus atoms of PSA may interact with the Asp301 residue of CYP2D6, thereby facilitating its binding and favorable orientation in the active site. As is the case with phospho-tyrosol-indomethacin [11], PSA was hydroxylated and sulfoxidized by CYP3A4 and CYP2D6, respectively. Together, these findings demonstrate not only the important roles of CYP3A4 and 2D6 in the metabolism of phospho-compounds, but also the unique CYP-catalyzed reaction specificity within a given substrate.
We demonstrated that PSA (versus sulindac) is a preferred substrate of CYPs. Substrate lipophilicity is highly correlated with their binding affinity with CYPs [15], and PSA is more lipophilic than sulindac as evidenced by its greater LogP value (4.0 versus 2.3). Moreover, human CYPs generally prefer neutral or basic substrates. Thus, the phospho-modification of sulindac at its –COOH group enhances its lipophilicity and masks its acid group, thus promoting its binding to CYPs.
FMOs catalyze the sulfoxidation of PSA but not its hydroxylation at its C-H bonds, since FMOs act on substrates containing soft nucleophiles such as nitrogen and sulfur. FMOs oxidize PSA far more rapidly than sulindac, likely for two reasons. First, compounds containing a negative charge (e.g., sulindac) are generally poor substrates of FMOs [16]. Second, PSA is much larger than sulindac, and the rate of FMO-catalyzed reactions increases with larger size of the substrate [17].
FMO1 catalyzes the sulfoxidation of PSA far more rapidly than FMO3 and FMO5. Similar observations have been reported for other relatively large compounds, including four thioether compounds [18], lorcaserin [19] and arecoline [20]. One exception is trimethylamine, a rather small compound whose sulfoxidation reaction is predominantly catalyzed by FMO3 [21]. These observations are not surprising since FMO3 (versus FMO1) has a smaller substrate binding channel and thus prefers smaller substrates [22].
We observed significant inter-tissue differences in the metabolism of PSA by human microsomes. PSA was significantly hydroxylated in human liver and intestinal microsomes but essentially not in lung or kidney microsomes, likely because CYP3A4 (the isoform that exclusively catalyzes the hydroxylation of PSA) is abundant only in human liver and intestine [23]. On the other hand, PSA was sulfoxidized predominantly in human kidney microsomes because FMO1 (the only isoform that efficiently catalyzes the sulfoxidation of PSA) is predominantly expressed in human kidney [24]. In comparison, PSA was minimally sulfoxidized in HLM likely because human liver expresses mainly FMO3 and FMO5, but not FMO1 [25]. Human lung microsomes were essentially inactive towards PSA, and FMO2 is the predominant isoform in human lung [25]. Therefore, FMO2 should have little or no activity towards PSA.
We previously examined and documented the metabolism of PS, whose amide form is PSA (Fig. 1). As is the case with PSA, CYP2D6 is by far the most active isoform catalyzing the sulfoxidation of PS [26], suggesting that the amide bond of PSA is not critical for the CYP-mediated sulfoxidation of PSA. However, we observed significant difference in FMO-catalyzed metabolism of PS and PSA. While FMO1 catalyzes the sulfoxidation of PS and PSA at similar rates, FMO3 and FMO5 are much more active towards PS than PSA. This observation suggests that the amide bond of PSA plays an important role in FMO-mediated sulfoxidation of PSA.
The metabolic fate of PSA has important implications for its safety and efficacy. The CYP3A4-mediated hydroxylation of PSA leads to its detoxification and elevated water-solubility, thus facilitating its excretion from the body. The reduction of PSA on its sulindac moiety to form PSA sulfide may enhance its bioactivity since PS sulfide (versus PS) has been shown to be more active in inhibiting cancer growth in mice [27].
In summary, PSA undergoes significant biotransformations in vitro, generating an array of metabolites. These results will greatly help our understanding of the pharmacological effects of PSA in animal and human studies.
Acknowledgments
This work was supported by the National Institute of Health Grant R01CA139454 and the shared instrumentation grant, NIH/NCRR 1 S10 RR023680-1. We thank R. Rieger and T. Koller, Stony Brook University, for their expert LC-MS/MS analysis of our samples.
Abbreviations
- CYP
cytochrome P450
- FMO
flavin monooxygenase
- HLM
human liver microsomes
- MLM
mouse liver microsomes
- PS
phospho-sulindac
- PSA
phospho-sulindac amide
Footnotes
Statement of conflicts of interest: The authors have nothing to disclose except for BR, who has an equity position in Medicon, Pharmaceuticals, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiSArio JA, Alberts DS, Tietze CC, Khullar SK, Bohman VD, Larsen BR, et al. Sulindac induces regression and prevents progression of sporadic colorectal adenomas. Gastroenterology. 1997;112:A555–A. [Google Scholar]
- 3.Roberts J, II, Morrow J. Analgesic-antipyretic and antiinflammatory agents and drugs employed in the treatment gout. In: Hardman JG, Limbird LE, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 2001. pp. 687–731. [Google Scholar]
- 4.Piazza GA, Keeton AB, Tinsley HN, Gary BD, Whitt JD, Mathew B, et al. A Novel Sulindac Derivative That Does Not Inhibit Cyclooxygenases but Potently Inhibits Colon Tumor Cell Growth and Induces Apoptosis with Antitumor Activity. Cancer Prev Res. 2009;2:572–80. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackenzie GG, Sun Y, Huang L, Xie G, Ouyang N, Gupta RC, et al. Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe and effective in colon cancer prevention in mice. Gastroenterology. 2010;139:1320–32. doi: 10.1053/j.gastro.2010.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie G, Nie T, Mackenzie GG, Sun Y, Huang L, Ouyang N, et al. The metabolism and pharmacokinetics of phospho-sulindac (OXT-328) and the effect of difluoromethylornithine. British journal of pharmacology. 2012;165:2152–66. doi: 10.1111/j.1476-5381.2011.01705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong CC, Cheng KW, Xie G, Zhou D, Zhu CH, Constantinides PP, et al. Carboxylesterases 1 and 2 hydrolyze phospho-nonsteroidal anti-inflammatory drugs: relevance to their pharmacological activity. The Journal of pharmacology and experimental therapeutics. 2012;340:422–32. doi: 10.1124/jpet.111.188508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sweeney BP, Bromilow J. Liver enzyme induction and inhibition: implications for anaesthesia. Anaesthesia. 2006;61:159–77. doi: 10.1111/j.1365-2044.2005.04462.x. [DOI] [PubMed] [Google Scholar]
- 9.Ratnayake JH, Hanna PE, Anders MW, Duggan DE. Sulfoxide reduction. In vitro reduction of sulindac by rat hepatic cytosolic enzymes. Drug metabolism and disposition: the biological fate of chemicals. 1981;9:85–7. [PubMed] [Google Scholar]
- 10.Xie G, Sun Y, Nie T, Mackenzie GG, Huang L, Kopelovich L, et al. Phospho-ibuprofen (MDC-917) is a novel agent against colon cancer: efficacy, metabolism, and pharmacokinetics in mouse models. The Journal of pharmacology and experimental therapeutics. 2011;337:876–86. doi: 10.1124/jpet.111.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie G, Zhou D, Cheng KW, Wong CC, Rigas B. Comparative in vitro metabolism of phospho-tyrosol-indomethacin by mice, rats and humans. Biochemical pharmacology. 2013;85:1195–202. doi: 10.1016/j.bcp.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13682–7. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C, et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug metabolism and disposition: the biological fate of chemicals. 2001;29:1454–9. [PubMed] [Google Scholar]
- 14.Ellis SW, Hayhurst GP, Smith G, Lightfoot T, Wong MM, Simula AP, et al. Evidence that aspartic acid 301 is a critical substrate-contact residue in the active site of cytochrome P450 2D6. The Journal of biological chemistry. 1995;270:29055–8. doi: 10.1074/jbc.270.49.29055. [DOI] [PubMed] [Google Scholar]
- 15.Lewis DF. On the recognition of mammalian microsomal cytochrome P450 substrates and their characteristics: towards the prediction of human p450 substrate specificity and metabolism. Biochemical pharmacology. 2000;60:293–306. doi: 10.1016/s0006-2952(00)00335-x. [DOI] [PubMed] [Google Scholar]
- 16.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacology & therapeutics. 2005;106:357–87. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagata T, Williams DE, Ziegler DM. Substrate specificities of rabbit lung and porcine liver flavin-containing monooxygenases: differences due to substrate size. Chemical research in toxicology. 1990;3:372–6. doi: 10.1021/tx00016a016. [DOI] [PubMed] [Google Scholar]
- 18.Usmani KA, Karoly ED, Hodgson E, Rose RL. In vitro sulfoxidation of thioether compounds by human cytochrome P450 and flavin-containing monooxygenase isoforms with particular reference to the CYP2C subfamily. Drug metabolism and disposition: the biological fate of chemicals. 2004;32:333–9. doi: 10.1124/dmd.32.3.333. [DOI] [PubMed] [Google Scholar]
- 19.Usmani KA, Chen WG, Sadeque AJ. Identification of human cytochrome P450 and flavin-containing monooxygenase enzymes involved in the metabolism of lorcaserin, a novel selective human 5-hydroxytryptamine 2C agonist. Drug metabolism and disposition: the biological fate of chemicals. 2012;40:761–71. doi: 10.1124/dmd.111.043414. [DOI] [PubMed] [Google Scholar]
- 20.Giri S, Krausz KW, Idle JR, Gonzalez FJ. The metabolomics of (+/−)-arecoline 1-oxide in the mouse and its formation by human flavin-containing monooxygenases. Biochemical pharmacology. 2007;73:561–73. doi: 10.1016/j.bcp.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 21.Lang DH, Yeung CK, Peter RM, Ibarra C, Gasser R, Itagaki K, et al. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: selective catalysis by FMO3. Biochemical pharmacology. 1998;56:1005–12. doi: 10.1016/s0006-2952(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 22.Lomri N, Yang Z, Cashman JR. Regio- and stereoselective oxygenations by adult human liver flavin-containing monooxygenase 3. Comparison with forms 1 and 2. Chemical research in toxicology. 1993;6:800–7. doi: 10.1021/tx00036a008. [DOI] [PubMed] [Google Scholar]
- 23.Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert opinion on drug metabolism & toxicology. 2006;2:875–94. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 24.Yeung CK, Lang DH, Thummel KE, Rettie AE. Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug metabolism and disposition: the biological fate of chemicals. 2000;28:1107–11. [PubMed] [Google Scholar]
- 25.Zhang J, Cashman JR. Quantitative analysis of FMO gene mRNA levels in human tissues. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:19–26. doi: 10.1124/dmd.105.006171. [DOI] [PubMed] [Google Scholar]
- 26.Xie G, Wong CC, Cheng KW, Huang L, Constantinides PP, Rigas B. Regioselective oxidation of phospho-NSAIDs by human cytochrome P450 and flavin monooxygenase isoforms: implications for their pharmacokinetic properties and safety. British journal of pharmacology. 2012;167:222–32. doi: 10.1111/j.1476-5381.2012.01982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang L, Zhu C, Sun Y, Xie G, Mackenzie GG, Qiao G, et al. Phospho-sulindac (OXT-922) inhibits the growth of human colon cancer cell lines: a redox/polyamine-dependent effect. Carcinogenesis. 2010;31:1982–90. doi: 10.1093/carcin/bgq149. [DOI] [PMC free article] [PubMed] [Google Scholar]