

Abstract
Inflammation associated with obesity may play a role in colorectal carcinogenesis, but the underlying mechanism remains unclear. This study investigated whether the Wnt pathway, an intracellular signaling cascade that plays a critical role in colorectal carcinogenesis, is activated by obesity-induced elevation of the inflammatory cytokine tumor necrosis factor-alpha (TNF-α). Animal studies were conducted on C57BL/6 mice, and obesity was induced by utilizing a high-fat diet (60% kcal). An inflammation-specific microarray was performed, and results were confirmed with real-time polymerase chain reaction. The array revealed that diet-induced obesity increased the expression of TNF-α in the colon by 72% (P=.004) and that of interleukin-18 by 41% (P=.023). The concentration of colonic TNF-α protein, determined by ex vivo culture assay, was nearly doubled in the obese animals (P=.002). The phosphorylation of glycogen synthase kinase 3 beta (GSK3β), an important intermediary inhibitor of Wnt signaling and a potential target of TNF-α, was quantitated by immunohistochemistry. The inactivated (phosphorylated) form of GSK3β was elevated in the colonic mucosa of obese mice (P<.02). Moreover, β-catenin, the key effector of canonical Wnt signaling, was elevated in the colons of obese mice (P<.05), as was the expression of a downstream target gene, c-myc (P<.05). These data demonstrate that diet-induced obesity produces an elevation in colonic TNF-α and instigates a number of alterations of key components within the Wnt signaling pathway that are protransformational in nature. Thus, these observations offer evidence for a biologically plausible avenue, the Wnt pathway, by which obesity increases the risk of colorectal cancer.
Keywords: Obesity, TNF-α, Wnt pathway, Colon cancer
1. Introduction
The prevalence of overweight and obesity has increased alarmingly over the past several decades in North America and in other industrialized countries [1,2]. Mounting epidemiological evidence suggests that obesity is a robust risk factor of many types of cancer, and the data are particularly compelling for colorectal cancer [3]. Since the obesity epidemic shows no signs of abating and further increases in its prevalence are expected in the future, defining the underlying cellular mechanisms by which obesity enhances cancer is an important step in the development of intelligent strategies to prevent and treat obesity-associated cancer.
In the pathophysiological state of obesity, adipose tissue is a very active endocrine and metabolic organ. In addition to its lipid-storing capacity, it appears to play an important role in a number of obesity-associated diseases [4]. Adipose tissue in obese individuals is infiltrated with increased numbers of macrophages, and it is these cells that appear to be responsible for the production of many inflammatory cytokines [5]. Since the initial discovery of escalated levels of tumor necrosis factor-alpha (TNF-α) in blood and adipose tissue by Hotamisligil and colleagues in 1993 [6], many other adiposity-related inflammatory molecules, such as interferon-γ and interleukin (IL)-1, -6, -8 and -10, have been identified in the adipose tissue and, in some instances, systemically [7,8]. It is now widely accepted that obesity is associated with a state of chronic, low-grade inflammation [9], although, to date, it has not been clearly defined whether peripheral tissues (including the colon) are similarly exposed by obesity to elevated levels of these inflammatory mediators.
Epidemiologic studies have long supported a link between chronic inflammation and the development of certain solid tumors, including colorectal cancer [10]. The most overt examples are patients with chronic inflammatory bowel disease of the colon, among whom the incidence of colorectal cancer increases progressively over time, reaching 19% after 30 years of disease [11]. Animal models closely recapitulate the findings of human studies, unambiguously proving a causal link between chronic inflammation and colorectal cancer. Persistent colonic inflammation in rodents induced by exogenous agents reproducibly leads to colonic carcinomas [12]. However, whether inflammation lies on the causal pathway linking obesity to colorectal cancer remains unclear.
It is well accepted that aberrant Wnt signaling is an early event in 90% of human colorectal cancers [13,14]. Apc is the tumor suppressor gene in familial adenomatous polyposis, a hereditary syndrome associated with a substantial increase in risk of colorectal cancer [15]. However, the activation of Wnt signaling is not exclusively explained by mutations in the Apc gene. Only a small fraction of colorectal cancer occurs in an apparently inherited fashion with Apc mutation [16]. Inappropriate Wnt signaling activation may be produced by posttranslational modification of its elements. For instance, Wnt signaling activation can occur through phosphorylation of the negative regulatory elements glycogen synthase kinase 3 beta (GSK3β), which in turn causes β-catenin protein stabilization [17]. The objective of the studies in this paper is to investigate the potential role of obesity-induced inflammatory cytokines in activating Wnt signaling and thereby promoting the development of colorectal cancer.
We herein show that diet-induced obesity increases the concentration of TNF-α in the colonic mucosa. Accompanying this increase are elevated phosphorylation of GSK3β, increased steady-state levels of β-catenin and increased transcription of the downstream Wnt pathway gene, c-Myc. These observations provide compelling evidence in support of a novel mechanism by which obesity could elevate the risk of colorectal cancer.
2. Material and methods
2.1. Animals
The protocol was approved by the Institutional Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University. C57BL/6 mice were used in this study, and obesity was induced by a standard high-fat diet (60% of total kcal as fat; D12492, Research Diets Inc.) in which lard was used as the major source of fat. Two cohorts of animals were used in this study. The first cohort (10 obese and 9 lean animals) was used for the measurement of TNF-α protein concentration by ex vivo culture of the colon. The obesity in this cohort was induced by feeding the high-fat diet for 17 weeks starting at 9–12 weeks of age. All other endpoints were measured on the second cohort of mice (eight obese and eight lean). Obesity was induced in this cohort by feeding the same high-fat diet for 12 weeks (from age of 6–18 weeks). In both cohorts, the average body weights of the obese mice were 30% to 35% greater than those of the lean controls at the time of sacrifice (P<.01, data not shown).
Methods for colonic mucosa isolation have been described in a previous publication with modifications [18]. Briefly, at the time of sacrifice, the colon was opened longitudinally, flushed with iced phosphate-buffered saline (PBS) containing a protease inhibitor cocktail and placed on a glass plate lying on a bed of crushed ice. The mucosa was gently scraped off, placed in a foil packet, frozen in liquid nitrogen and subsequently used for all RNA and Western blot assays.
2.2. Inflammatory pathway-specific microarray analysis
To systematically investigate which inflammation-related genes were altered by diet-induced obesity, inflammatory pathway-specific arrays (SABiosciences, Frederick, MD, USA) were performed on pooled samples (eight mice from each of the two diet groups). This array (PAMM-011A) profiles the expression of 84 genes of inflammatory cytokines, their receptors and several other genes involved in the inflammatory response. The complete list of genes is available at http://www.sabiosciences.com. This microarray is a well-accepted assay for inflammatory pathway-specific analysis [19,20].
Briefly, RNA was extracted from the colonic scrapings using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and further purified using the RNeasy Mini Cleanup kit (Qiagen, Valencia, CA, USA). For both the obese and lean control groups, the RNA from six samples was pooled in equal amounts of RNA, and a total of 1 μg of the pooled RNA was used for cDNA synthesis using the RT2 First Strand Kit (SABiosciences, Frederick, MD, USA). Each polymerase chain reaction (PCR) array was performed on a ABI7300 real-time PCR system (Applied Biosystems, Carlsbad, CA, USA) utilizing the following thermal cycling conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. The cycle threshold (Ct) values, which were defined as the fractional cycle number at which the fluorescence passes an arbitrarily set threshold, were normalized to the average Ct of five housekeeping genes including GAPDH and β-actin.
2.3. Colon ex vivo culture and TNF-α cytokine measurement
A segment of the colon was removed, cut open longitudinally and washed in PBS containing protease inhibitors. The colon was temporarily stored in 15-ml tubes containing ice-cold RPMI 1640 medium supplemented with 1% antibiotic, antimycotic cocktail. The sample was then further cut into segments of ~1 cm and placed in 24 flat-bottom well culture plates containing RPMI 1640 medium supplemented with 1% antibiotic, antimycotic cocktail. The colon segments were incubated at 37°C in 1 ml of fresh medium for 24 h. Culture supernatants were harvested and assayed for cytokines. A similar ex vivo culture method has been previously used for inflammatory cytokine detection [21]. Protein concentrations were determined by the Bio-Rad protein assay. The colonic TNF-α level was measured by enzyme-linked immunosorbent assay (ELISA). The minimum detectable concentration of TNF-α for the kit is 4 pg/ml (Biolegend Mouse TNF-α ELISA Max).
2.4. Immunohistochemistry and immunoblotting
For immunohistochemistry, the paraffin embedding slides were deparaffinized in xylene, followed by rehydration in ethanol. Endogenous peroxidase blocking was performed with H2O2, and the antigen was retrieved by boiling. Slides were first incubated with primary antibodies. Antibodies targeting phospho-GSK3β and cleaved caspase-3 (apoptosis) were from Cell Signaling Technology (Boston, MA, USA), but anti-ki-67 (proliferation) antibody was from Abcam (Cambridge, MA, USA). Antibody was diluted at 1:200 in the antibody solution. Then following the incubation with biotinylated horse anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA, USA), the slides were treated with Vectastain Elite ABC reagent (Vector Laboratories) and hematoxylin counterstain. Scoring was performed in a blinded fashion at a 400-fold magnification. A semiquantitative immunohistochemical method was used for phospho-GSK3β and cleaved caspase-3. Positive staining of the colonic epithelium was scored as follows: 0, no staining; 1, mild staining; 2, moderate staining; and 3, intensive staining. The proliferation index (ki-67 positive staining) was calculated by determining the number of positive epithelial cells per crypt divided by the total number of crypt cells.
For immunoblotting of β-catenin, protein extracts were run out on a polyacrylamide gel with electrophoresis and transferred onto nitrocellulose membrane. Nonspecific binding was blocked with nonfat dry milk. The membrane was then probed with the primary anti-β-catenin antibody (BD Biosciences, San Jose, CA, USA) at the concentration of 1:2000 followed by a horseradish-peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA, USA). Chemifluorescence detection was achieved using ECL Plus Substrate (Amersham Biosciences, Piscataway, NJ, USA). Protein bands were quantified using a Gel-Doc image analysis system and Quantity One software (Bio-Rad). The ratio of the density for β-catenin band vs. the control GAPDH band was calculated.
2.5. Real-time PCR for gene expression
Real-time PCR was performed on total RNA isolated from colonic scrapings with Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the first-strand cDNAs were synthesized using Oligo d(T)xxx and Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). SYBR Green-based gene expression analysis were used to confirm eight inflammation-related genes, six of which were identified by the inflammatory pathway-specific microarray to have undergone at least a 50% change and also have been reported to be linked to colorectal carcinogenesis. Two additional cytokines (IL-1β and IL-6), although not displaying ≥50% change by the array, were also measured by quantitative reverse transcriptase (RT)-PCR since they are reportedly linked to colorectal cancer risk as well. The primers for these cytokines are listed in supplementary tables (table s1). The expressions of the c-myc and cyclin D1 gene were quantified using a TaqMan Gene Expression Assay and the ABI7300 real-time PCR machine (both from Applied Biosystems, Foster City, CA, USA). The statistical analyses were performed using ΔCt, and relative expression values (relative expression=2−ΔΔCt) are reported.
2.6. Statistical analysis
Analysis of the inflammatory pathway-specific microarray data was performed using the Web Portal provided by SABiosciences (http://www.SABiosciences.com). Data analyses for other endpoints were conducted using general linear regression procedure in SAS software v9.2 (SAS Institute Inc., Cary, NC, USA). Values in the text are presented as means ± S.E.M.
3. Results
3.1. Diet-induced obesity alters the transcriptional expression of inflammatory cytokines in the colon
As stated in the Introduction section, it is widely accepted that adipose tissue is an active endocrine organ responsible for the production of many inflammatory cytokines in the state of obesity [4]. Although elevated patterns of obesity-related inflammatory cytokines have been reported for serum and adipose tissue, essentially no data exist on the obesity-associated inflammatory mediators in the colon.
The analysis of the inflammatory pathway-specific array on pooled samples from obese and lean mice indicated that, among the 84 inflammation-associated genes, 11 genes were up-regulated by ≥50% and 18 genes were down-regulated by ≥50% in obese mice (Fig. 1). More details of the 29 identified genes are listed in a supplementary table (Table S2). After a search of which of the identified genes have been linked to colorectal cancer, six were identified as being both proinflammatory and highly relevant to carcinogenesis [chemokine (C-C motif) ligand 2, IL-18, TNF-α, chemokine (C-C motif) receptor 2, interferon-γ and IL-4]. These six were therefore selected for further confirmation by RT-PCR [22–26]. Moreover, we also examined two additional proinflammatory cytokines (IL-1β and IL-6) which have been linked to an increased risk of colorectal cancer [27–31]. Real-time PCR results of these eight proinflammatory genes (Fig. 2) show that diet-induced obesity increased the expression of TNF-α in the colon by 72% (P=.004), IL-1β by 44% (P=.077) and IL-18 by 41% (P=.023).
Fig. 1.
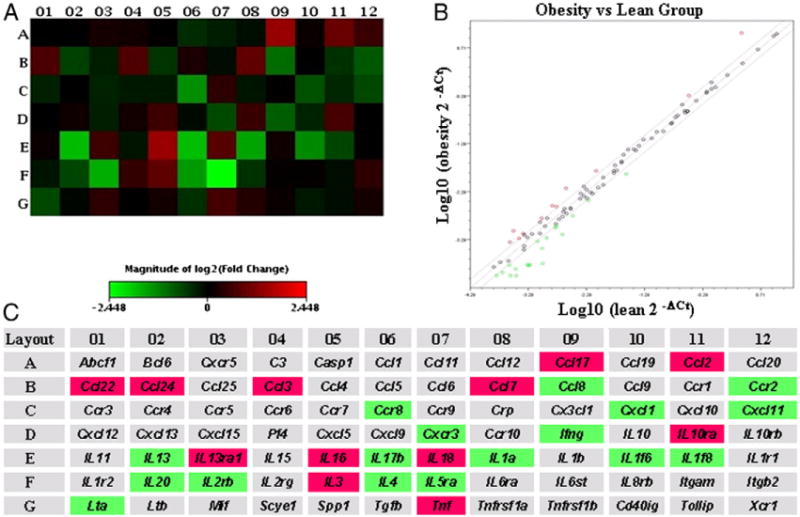
Effect of diet-induced obesity on the transcriptional expression of inflammatory cytokines. Inflammation-specific microarrays were performed on RNA pooled from eight mice/group. Heat map, scatter plot and fold change were obtained using the PCR Array Data Analysis Web Portal provided by SABiosciences (http://www.SABiosciences.com/pcrarraydataanalysis.php). (A) Heat map for the inflammatory pathway-specific microarrays. (B) A scatter plot identifies 29 genes whose expressions were up-regulated (11) or down-regulated (18) by at least 50% in the obese mice compared to the control lean mice (outside of the board line). (C) The names of the cytokines in the heat map and the identified 29 genes with ≥50% changes in expression highlighted in red (up-regulated) or green (down-regulated) when the obese group was compared to the control group. More details of the 29 genes are shown in the Supplementary Data.
Fig. 2.
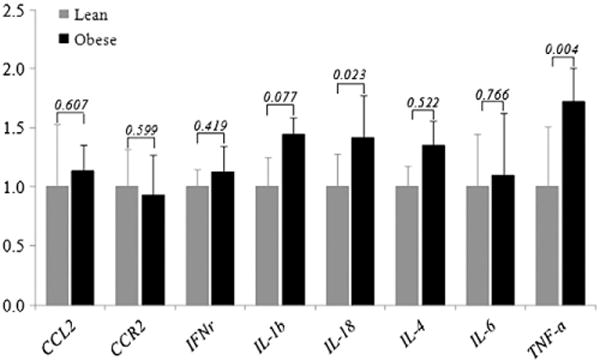
Real-time PCR was used to confirm genes identified by inflammatory pathway specific arrays that displayed at least a 50% change in expression. The values on the bars are the P value from a t test between the high-fat and the control dietary group (eight samples/group). IL-1β and IL-6, although not identified to have ≥50% change by the array, are included in the PCR confirmation since they have been linked to colorectal carcinogenesis.
3.2. TNF-α protein in the colon was significantly increased in the diet-induced obese mice
Increases of transcriptional levels of inflammatory cytokines such as the identified TNF-α indicate that obesity induces a chronic inflammation (for instance, the infiltration macrophages) in the colon, and the TNF-α, which is directly produced by infiltrated immune cells, can be determined by transcriptional expression. However, TNF-α protein produced in adipose tissue may also be circulated into colonic mucosa. Moreover, with the consideration of posttranscriptional modification, it is necessary to measure the protein level, which is even more functionally related. Therefore, colonic TNF-α protein level was also determined in this study. The TNF-α protein in the colon was assessed by an ex vivo colonic tissue culture as described above [21]. A 78% increase (P<.01) in TNF-α protein was observed in the diet-induced obese mice (Fig. 3).
Fig. 3.
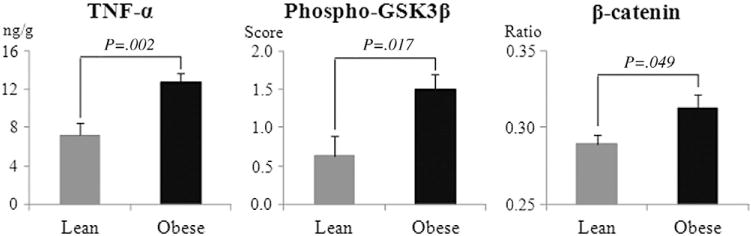
Diet-induced obesity elevated the protein level of colonic TNF-α, accompanied by increases in β-catenin and phosphorylation of GSK3β. TNF-α was measured by ex vivo culture followed by ELISA analysis of the media. The data are expressed as ng TNF-α/g of protein in the media. Phospho-GSK3β was determined by immunohistochemical staining. Positive staining of phospho-GSK3β in the colonic epithelium was scored as follows: 0, no staining; 1, mild staining; 2, moderate staining; and 3, intense staining. Total β-catenin was determined by Western blotting and normalized to GAPDH. The ratio is the band density of β-catenin divided by that of GAPDH. The P values are results from t test between the high-fat and the control dietary group. The TNF-α protein data were based on 10 mice in the obese group and 9 in the control group, but the data for phospho-GSK3β and β-catenin were based 8 samples/group.
3.3. Elevated colonic TNF-α protein level was accompanied with increased phosphorylation of GSK3β and accumulation of β-catenin
We next examined whether increases in β-catenin and the phosphorylated form of GSK3β accompany the increase of TNF-α in obese mice. The results (Fig. 3) demonstrate that dietary obesity does increase phospho-GSK3β and the accumulation of β-catenin. This is consistent with the concept that increasing activity of TNF-α induces the phosphorylation of GSK3β, thereby suppressing the proteosomal degradation of β-catenin and allowing its accumulation in the cell.
3.4. Expression of c-myc in the colon was up-regulated by diet-induced obesity
Stabilized β-catenin binds to T-cell factor/lymphoid enhancer factor-1 in the nucleus and results in increased transcriptional activation of several target genes including the c-myc proto-oncogene, which is a transcriptional factor that, in turn, activates the expression of numerous genes that have diverse procarcinogenic consequences, including increased cell proliferation [32]. Consistent with our hypothesis was an observation of significantly increased c-myc expression in the obese mice (Fig. 4). The expression of another Wnt target gene, cyclin D1, was not significantly altered. The latter observation is perhaps not surprising since the expression of cyclin D1 is well known to be influenced by a number of factors other than Wnt signaling [33–35].
Fig. 4.
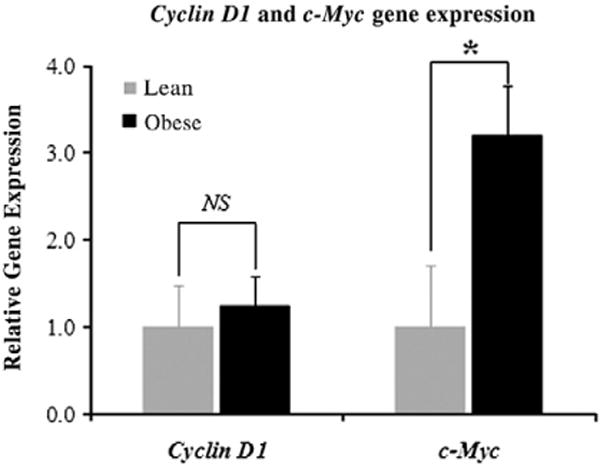
Diet-induced obesity elevated the expression of c-myc, a target gene of Wnt signaling. Student’s t test was applied to the high-fat and the control dietary group (eight samples/group).
3.5. Proliferation in the colonic epithelium was elevated by high-fat-diet-induced obesity
The canonical Wnt pathway controls cell differentiation, proliferation and apoptosis by regulating the expression of a high number of target genes [36]. c-myc, for example, is a transcription factor that regulates the expression of numerous genes involved in proliferation, cell growth, differentiation, apoptosis and neoplastic transformation [37].
Analyses of proliferation in the colonic crypts, using Ki-67 staining as the marker, indicated that diet-induced obesity significantly elevated the mean proliferation index (i.e., number of colonic epithelial cells with positively staining nuclei divided by total number of colonocytes in the crypt) by 38% (P<.05). However, the alteration of apoptosis, assessed by nuclear staining of cleaved caspase-3, was not statistically significant (Fig. 5).
Fig. 5.
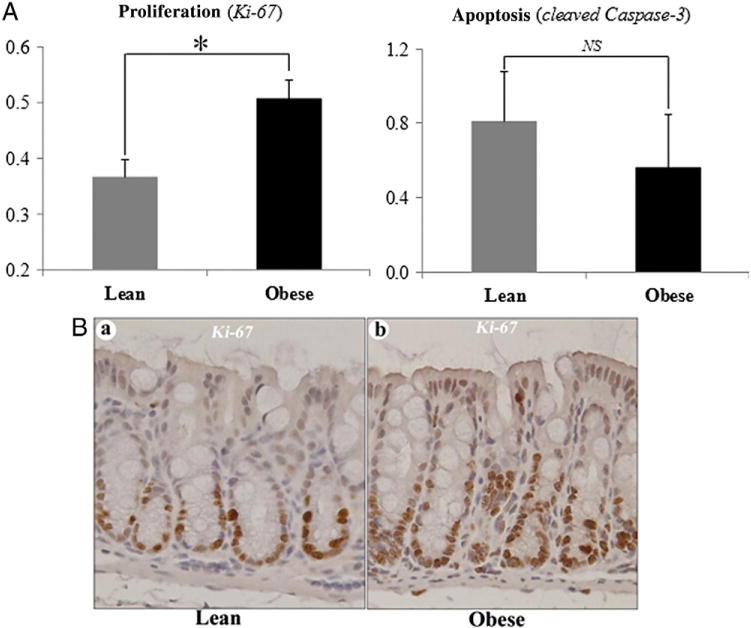
Diet-induced obesity significantly elevated the proliferation index of the colonic epithelial cells. A numerical reduction in apoptosis was observed, but this was not statistically significant (A). Representative staining for proliferation (brown) is shown in (B). *P<.05; NS, not significant; eight samples/group.
4. Discussion
Epidemiological studies continue to underscore the importance of overweight and obesity as major determinants of the risk of colorectal cancer [38], and demographic trends continue to emphasize that the problem will only become more prominent in the coming decade. Nevertheless, insights into the underlying mechanisms by which excessive weight mediates these effects — which are important in designing strategies as to how to address the problem — remain very limited.
Although it has been thought for some time that chronic inflammation, incited by obesity or other factors, might play a role in promoting colorectal carcinogenesis, the cellular pathways by which this effect is mediated have not yet been defined. TNF-α plays a prominent role in obesity-induced inflammation and has been shown to be elevated in the circulation as well as in some tissues of obese humans and rodents [7,8]. Recently, it was shown in an animal model of Helicobacter-associated gastric cancer that TNF-α activates Wnt signaling through the induction of GSK3β phosphorylation, resulting in increased nuclear localization of β-catenin, the hallmark event of canonical Wnt signaling [24]. Observations from this study support a similar concept that obesity-induced elevation of TNF-α up-regulates the procarcinogenic Wnt pathway by suppressing the activity of GSK3β in the colon.
Our observations prove that the colons of those mice who consumed the obesogenic diet underwent significant and substantial increases in the expression of multiple proinflammatory cytokines: TNF-α by 72% (P=.004), IL-1β by 44% (P=.077) and IL-18 by 41% (P=.023). Since we were particularly interested in the ability of TNF-α to mediate phosphorylation of a critical element in the Wnt pathway [24], we confirmed that the protein level of this cytokine was elevated in conjunction with elevated levels of the RNA transcript. This is consistent with a recent observation in which elevated TNF-α was observed in both rat normal colonic mucosa and tumor [39], and our data greatly extend that observation by demonstrating important downstream events that the increases in TNF-α might incite.
Moreover, because our hypothesis posits that the link between TNF-α and the Wnt pathway is via phosphorylation of GSK3β, it was important for us to demonstrate whether cellular levels of phospho-GSK3β are elevated in the obese mice, and this link was also established. Canonical Wnt signaling is unregulated when changes in upstream elements result in diminished degradation of β-catenin, thereby enabling cellular levels of β-catenin to rise and resulting in an increased translocation of this element into the nucleus. This hallmark of Wnt signaling was also demonstrated in our study. Finally, we showed that a Wnt pathway downstream gene, c-myc, and a cytokinetic readout of activated Wnt signaling, epithelial proliferation, were also significantly increased in those animals that consumed the high-fat diet. Although the design of this study falls somewhat short of proving that the apparent activation of Wnt signaling was definitively due to the increased concentrations of TNF-α, all the observations nevertheless provide evidence in support of the proposed hypothesis, establishing a solid foundation upon which more definitive proof of causality can be built. These data therefore represent initial evidence for a novel mechanism by which obesity- associated inflammation might elevate Wnt-signaling and consequently promote colorectal tumorigenesis.
Canonical Wnt signaling is a critical pathway in the regulation of tumor development [40]. In the normal intestine, a base level of Wnt signaling functions to maintain the stem-cell characteristics of the epithelial cells [41]. However, mutations in the Apc gene or other Wnt-promoting events can inappropriately activate or elevate Wnt signaling and initiate processes leading to colorectal cancer [42]. This study demonstrates that induction of obesity and of its associated chronic inflammatory state creates a microenvironment with increased levels of many cytokines, including TNF-α that apparently contributes to an elevation of Wnt signaling.
In addition to TNF-α, other humoral agents associated with obesity might also be contributing to the activation of Wnt signaling. IL-1β has also been observed to induce phosphorylation of GSK3β [43]. Also, adiponectin, which is decreased in the obese state and is not an inflammatory cytokine, can modulate GSK3β/β-catenin signaling pathway [44]. Nevertheless, there is compelling evidence that these two elements play a minor role in our study, if at all, compared to TNF-α. We did not observe a significant change in adiponectin expression in the colonic mucosa (data not shown) and only a marginal increase in IL-1β (β=0.077, Fig. 2). In contrast, significant and substantial increases in both TNF-α transcript (P=.004, Fig. 2) and protein expression (P=.004, Fig. 3) were observed. Therefore, we believe that these two elements were, at best, minor mediators of the observed alterations in Wnt signaling in the colons of the obese mice.
It is well known that obesity is accompanied by complex metabolic changes. Therefore, it is possible that multiple mechanisms may be operating in parallel and contributing to the creation of a protumorigenic milieu [45]. Nevertheless, the Wnt pathway is a pivotal tumorigenic pathway, aberrations of which appear to be important in the evolution of nearly all sporadic colorectal cancers [16]. The data presented here are supportive of a highly plausible scheme that dietary-induced obesity promotes CRC by activating the Wnt pathway through the TNF-α-mediated suppression of GSK3β.
It is noteworthy that it is not clear whether the increase in cytokines in the colon was due to the state of obesity, whether it might instead be a direct effect of the altered components of the diet or whether it is a consequence of both of them. For instance, it is known that particular fatty acids — rather than an excess of calories or the state of obesity per se — can directly modulate obesity-associated inflammation [46]. We also realize that the present studies merely demonstrate a molecular mechanism rather than provide direct evidence that links the TNF-α with the development of macroscopic tumors, but it has been shown by several studies that both TNF-α and obesity are directly associated with the development of colon cancer. Popivanova et al. demonstrated that blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis [47]. Gravaghi et al. reported that obesity enhances gastrointestinal tumorigenesis in Apc-mutant mice [48]. Nevertheless, this study demonstrated that an obesity-associated inflammatory status in the colon, particularly the elevation of TNF-α, is associated with alterations of the critical Wnt pathway.
In summary, observations from this study indicate that high-fat-diet-induced obesity is associated with a significant increase of TNF-α level in mouse colon, and in parallel with this increase are several alterations observed within Wnt signaling cascade in a pattern with activation of this pathway. These observations collectively implicate a novel molecular mechanism for obesity- associated colorectal carcinogenesis (Fig. 6): obesity induces an increase of TNF-α in the colon and in turn activates Wnt signaling for the development of colorectal cancer.
Fig. 6.
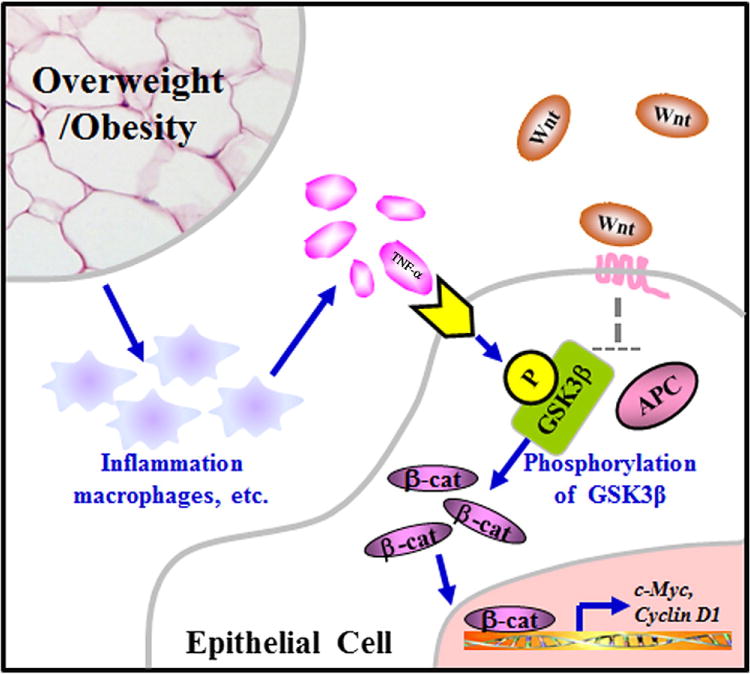
The mechanism proposed in this study. Obesity is associated with a chronic low-grade inflammation such as the infiltration of macrophages and other immune cells. Macrophages infiltrated in the colonic mucosa contribute directly to the production of colonic TNF-α, whereas TNF-α secreted from immune cells in the adipose tissue may be circulated to the colonic mucosa. Through phosphorylation of the critical GSK3β, colonic TNF-α activates Wnt signaling and thereby increases the expression of the downstream protumorigenic genes.
Supplementary Material
Acknowledgments
Supported in part by the US Department of Agriculture (USDA) Human Nutrition Research Center on Aging at Tufts University Pilot Program (Z.L.), The Prevent Cancer Foundation (Z.L.) and USDA Agricultural Research Service (Agreement No. 1950-074-01S). Any opinions, findings, conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the USDA.
Abbreviations
- TNF-α
tumor necrosis factor-alpha
- Apc
adenomatous polyposis coli
- GSK3β
glycogen synthase kinase 3 beta
Footnotes
Supplementary materials related to this article can be found online at doi:10.1016/j.jnutbio.2011.07.002.
All authors do not have any conflict of interest.
References
- 1.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–55. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Pischon T, Nothlings U, Boeing H. Obesity and cancer. Proc Nutr Soc. 2008;67:128–45. doi: 10.1017/S0029665108006976. [DOI] [PubMed] [Google Scholar]
- 3.Gunter MJ, Leitzmann MF. Obesity and colorectal cancer: epidemiology, mechanisms and candidate genes. J Nutr Biochem. 2006;17:145–56. doi: 10.1016/j.jnutbio.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Rajala MW, Scherer PE. Minireview: the adipocyte — at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–73. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 5.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 7.Fischer-Posovszky P, Wabitsch M, Hochberg Z. Endocrinology of adipose tissue — an update. Horm Metab Res. 2007;39:314–21. doi: 10.1055/s-2007-976539. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–83. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 9.Ramos EJ, Xu Y, Romanova I, Middleton F, Chen C, Quinn R, et al. Is obesity an inflammatory disease? Surgery. 2003 Aug;134:329–35. doi: 10.1067/msy.2003.267. [DOI] [PubMed] [Google Scholar]
- 10.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–9. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–22. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 14.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 15.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 16.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–20. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 17.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161–8. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perret V, Lev R, Pigman W. Simple method for the preparation of single cell suspensions from normal and tumorous rat colonic mucosa. Gut. 1977;18:382–5. doi: 10.1136/gut.18.5.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakshmanan U, Porter AG. Caspase-4 interacts with TNF receptor-associated factor 6 and mediates lipopolysaccharide-induced NF-kappaB-dependent production of IL-8 and CC chemokine ligand 4 (macrophage-inflammatory protein-1) J Immunol. 2007;179:8480–90. doi: 10.4049/jimmunol.179.12.8480. [DOI] [PubMed] [Google Scholar]
- 20.Kawaguchi A, Orba Y, Kimura T, Iha H, Ogata M, Tsuji T, et al. Inhibition of the SDF-1alpha-CXCR4 axis by the CXCR4 antagonist AMD3100 suppresses the migration of cultured cells from ATL patients and murine lymphoblastoid cells from HTLV-I Tax transgenic mice. Blood. 2009;114:2961–8. doi: 10.1182/blood-2008-11-189308. [DOI] [PubMed] [Google Scholar]
- 21.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249–54. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69:7884–92. doi: 10.1158/0008-5472.CAN-09-1451. [DOI] [PubMed] [Google Scholar]
- 23.Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–36. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008;27:1671–81. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipnik K, Naschberger E, Gonin-Laurent N, Kodajova P, Petznek H, Rungaldier S, et al. Interferon gamma-induced human guanylate binding protein 1 inhibits mammary tumor growth in mice. Mol Med. 2010;16:177–87. doi: 10.2119/molmed.2009.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toi M, Bicknell R, Harris AL. Inhibition of colon and breast carcinoma cell growth by interleukin-4. Cancer Res. 1992;52:275–9. [PubMed] [Google Scholar]
- 27.Trayhurn P. Adipose tissue in obesity — an inflammatory issue. Endocrinology. 2005;146:1003–5. doi: 10.1210/en.2004-1597. [DOI] [PubMed] [Google Scholar]
- 28.Kaler P, Galea V, Augenlicht L, Klampfer L. Tumor associated macrophages protect colon cancer cells from TRAIL-induced apoptosis through IL-1beta-dependent stabilization of Snail in tumor cells. PLoS One. 2010;5:e11700. doi: 10.1371/journal.pone.0011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–19. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut. 2008;57:1531–8. doi: 10.1136/gut.2008.159293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene. 2003;22:5707–11. doi: 10.1038/sj.onc.1206800. [DOI] [PubMed] [Google Scholar]
- 33.Germain D, Russell A, Thompson A, Hendley J. Ubiquitination of free cyclin D1 is independent of phosphorylation on threonine 286. J Biol Chem. 2000;275:12074–9. doi: 10.1074/jbc.275.16.12074. [DOI] [PubMed] [Google Scholar]
- 34.Zou Y, Ewton DZ, Deng X, Mercer SE, Friedman E. Mirk/dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J Biol Chem. 2004;279:27790–8. doi: 10.1074/jbc.M403042200. [DOI] [PubMed] [Google Scholar]
- 35.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vlad A, Rohrs S, Klein-Hitpass L, Muller O. The first five years of the Wnt targetome. Cell Signal. 2008;20:795–802. doi: 10.1016/j.cellsig.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 37.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–76. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 38.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 39.Jain SS, Bird RP. Elevated expression of tumor necrosis factor-alpha signaling molecules in colonic tumors of Zucker obese (fa/fa) rats. Int J Cancer. 2010;127:2042–50. doi: 10.1002/ijc.25232. [DOI] [PubMed] [Google Scholar]
- 40.Taketo MM. Wnt signaling and gastrointestinal tumorigenesis in mouse models. Oncogene. 2006;25:7522–30. doi: 10.1038/sj.onc.1210058. [DOI] [PubMed] [Google Scholar]
- 41.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 42.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482–6. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009;28:3892–902. doi: 10.1038/onc.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, et al. Adiponectin modulates the glycogen synthase kinase-3beta/beta-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006;66:11462–70. doi: 10.1158/0008-5472.CAN-06-1969. [DOI] [PubMed] [Google Scholar]
- 45.Renehan AG, Roberts DL, Dive C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem. 2008;114:71–83. doi: 10.1080/13813450801954303. [DOI] [PubMed] [Google Scholar]
- 46.Samuvel DJ, Sundararaj KP, Li Y, Lopes-Virella MF, Huang Y, Wada T. Adipocyte-mononuclear cell interaction, Toll-like receptor 4 activation, and high glucose synergistically up-regulate osteopontin expression via an interleukin 6-mediated mechanism. J Biol Chem. 2010;285:3916–27. doi: 10.1074/jbc.M109.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gravaghi C, Bo J, Laperle KM, Quimby F, Kucherlapati R, Edelmann W, et al. Obesity enhances gastrointestinal tumorigenesis in Apc-mutant mice. Int J Obes (Lond) 2008;32:1716–9. doi: 10.1038/ijo.2008.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.