

Abstract
Human polyclonal IgG antibodies directly against the non-human sialic acid N-glycolylneuraminic acid (Neu5Gc) are potential biomarkers and mechanistic contributors to cancer and other diseases associated with chronic inflammation. Using a sialoglycan microarray, we screened the binding pattern of such antibodies (anti-Neu5Gc IgG) in several samples of clinically-approved human IVIG (IgG). These results were used to select an appropriate sample for a multi-step affinity purification of the xeno-autoantibody fraction. The sample was then analyzed via our multi-enzyme digestion procedure followed by nanoLC coupled to LTQ-FTMS. We used characteristic and unique peptide sequences to determine the IgG subclass distribution and thus provided direct evidence that all four IgG subclasses can be generated during a xeno-autoantibody immune response to carbohydrate Neu5Gc-antigens. Furthermore, we obtained a significant amount of sequence coverage of both the constant and variable regions. The approach described here, therefore, provides a way to characterize these clinically significant antibodies, helping to understand their origins and significance.
Introduction
Sialic acids are a family of nine-carbon acidic monosaccharides mostly derived from N-acetylneuraminic acid (Neu5Ac) that are typically found as terminal sugars of glycan chains attached to cell surface glycoproteins and glycolipids and secreted glycoconjugates.1–5 Humans express predominantly Neu5Ac but not the hydroxylated form, N-glycolylneuraminic acid (Neu5Gc), due to an inactivating mutation in the enzyme synthesizing it.6 Other mammals can express both Neu5Ac and Neu5Gc.7 Despite this, human cells can metabolically incorporate Neu5Gc from exogenous sources, particularly dietary red meats. Several human tumors have also been reported to express Neu5Gc.8–12 Although most studies used immunochemical techniques, a few chemical methods have confirmed that human tumors do indeed contain Neu5Gc.13–15 Recent studies have shown that many normal human sera contain IgA, IgM, or IgG antibodies against Neu5Gc, the first example of xeno-autoantibodies.8, 16,17 Although most were of IgG type, the total levels of anti-Neu5Gc antibodies varied widely among individuals.17 Importantly, evidence indicated that weak inflammation arising from the combination of tumor-incorporated Neu5Gc and circulating anti-Neu5Gc antibodies can facilitate tumor progression and that some of these xeno-autoantibodies could be used as biomarkers of cancer.18,19
Mass spectrometry (MS) has become an essential analytical tool for the structural characterization of antibodies, due to its superior resolution over other analytical techniques. Structural features that can be determined by MS include amino acid sequences, disulfide linkages, carbohydrate structures and profiles, and many different post-translational, in-process, and in-storage modifications.20,21 In this study, we sought a better understanding of the origins of anti-Neu5Gc xeno-autoantibodies for their significance as biomarkers of cancer. To this end, MS was employed to achieve high sequence coverage in human polyclonal anti-Neu5Gc antibodies, including the variable region of both heavy and light chains. Two steps were used to achieve this goal. Firstly, we used multiple affinity purification steps to isolate the human anti-Neu5Gc antibody fragments for the mass spectrometric analysis. In approaching this, we took advantage of the fact that human IgG pooled from thousands of donors (IVIG) is commonly used for various clinical therapies, and the excess from each therapeutic dose is routinely discarded. We have analyzed several brands of IVIG using a specific sialoglycan microarray and selected the one with the broadest and strongest anti-Neu5Gc reactivity for the large-scale affinity purification. Secondly, we used a multi-enzyme approach to generate peptide sequences with good overlap, allowing us to determine and investigate all four subclasses of IgG and other significant sequences.
Experimental
IVIG Brands
IVIG leftovers were kindly provided by Dr. Richard Schwab from the UC San Diego Moores Cancer Center. Four IVIG brands were analyzed: Carimune® NF (CSL Behring, Switzerland; 10% sucrose as stabilizer), Gamunex® (Talecris Biotherapeutic, USA; 0.16–0.24 M Glycine as stabilizer), GAMMAGARD LIQUID (Baxter, USA; 0.25 M Glycine as stabilizer) and Flebogamma® (GRIFOLS, Spain; 5% D-sorbitol as stabilizer) corresponding to IVIG-A, -B, -C and -D, respectively.
Sialoglycan microarray analysis of IVIG
Each IVIG brand was tested on a sialoglycan microarray (V13) at 1 mg/mL and 0.5 mg/mL in 200 μL/sub-array with minor changes (type of printer and pins used) from the previously described.19 Arrays were fabricated by KAMTEK Inc. (Gaithersburg, MD) with VersArray ChipWriter Pro (Virtek/BioRad) on Epoxide-derivatized slides (Corning) with four SMP5B quill pins (Array It) at 8 arrays/slide. Each glycan was printed at 100 μM in printing buffer (300 mM phosphate buffer, pH 8.4) in 4 replicates. Slides were blocked with 50°C pre-warmed Ethanolamine (0.05 M) in Tris-HCl buffer (0.1 M, pH 9) for 1 hour, then washed twice with 50°C pre-warmed dH2O. Slides were centrifuge-dried at 200×g for 3 min, fitted with ProPlate™ Multi-Array slide module (Invitrogen) to divide into the sub-arrays, and then blocked with 200 μL/sub-array blocking solution 2 (PBS/OVA, 1% w/v Ovalbumin in PBS pH 7.4) for 1 hour at room temperature (RT) with gentle shaking. Next, the blocking solution was aspirated and diluted samples were added to each slide (in PBS/OVA, 200 μL/sub-array) and allowed to incubate with gentle shaking for 2 h at RT. Slides were washed three times with PBST (PBS, 1% Tween) and then one time with PBS for 10 min/wash with shaking. Bound antibodies were detected by incubating 200 μL/sub-array with Cy3-goat-anti-human IgG antibody diluted in PBS (1.5 μg/ml) at RT for 1 hour. Slides were washed three times with PBST (PBS, 1% Tween) then with PBS on time for 10 min/wash followed by removal from ProPlate™ Multi-Array slide module and immediately dipping slide in a staining dish with dH2O for 10 min with shaking, then centrifuged at 200×g for 3 min. Dry slides were vacuum-sealed and stored in dark until scanning the following day. Slides were scanned at 10 μm resolution with a Genepix 4000B microarray scanner (Molecular Devices Corporation, Union City, CA) using 450 gain. Image analysis was carried out with Genepix Pro 7.2 analysis software (Molecular Devices Corporation). Spots were defined as circular features with a variable radius as determined by the Genepix scanning software. Local background subtraction was performed.
Affinity purification of human anti-Neu5Gc IgG and generation of specific antibody-fragments
The xeno-autoantibodies anti-Neu5Gc IgG were affinity-purified from IVIG-C (GAMMAGARD LIQUID) on sequential columns with immobilized human serum and chimpanzee serum as previously described (chimpanzee serum obtained from Yerkes National Primate Research Center, Emory University, GA).19 Briefly, IVIG-C was loaded over the pre-clearance human sialo-glycoproteins column (containing only Neu5Ac-glycans), then the run-through was collected and loaded over chimpanzee sialo-glycoproteins column (containing Neu5Gc-glycans; Figure 1A). Given the high identity of human and chimpanzee serum glycoproteins, these columns differ primarily in the single oxygen atom that differentiates Neu5Gc from Neu5Ac. The bound antibodies were first eluted with glucoronic acid (similar to sialic acid contains a carboxylate group) to remove non-specific ionic-binding. Next, specific anti-Neu5Gc IgG were eluted with Neu5Gc then concentrated and filtered to remove free Neu5Gc. The columns were regenerated by wash with a weak acid. Indeed, analysis of the various fractions revealed specific-Neu5Gc reactivity only in the Neu5Gc-eluted fraction (with no Neu5Ac reactivity; Figure 1A).19 The purified full antibodies were then digested to generate 50 kDa Fc and Fab fragments (Thermo Scientific Pierce Fab-Preparation Kit according to manufacturer’s instructions) that were further separated to generate kappa-enriched or lambda-enriched Fab Fragments (Thermo Scientific Pierce NAb Protein-L Spin Columns according to manufacturer’s instructions). Briefly, 2 mg of affinity-purified xeno-autoantibodies were digested by immobilized papain (a nonspecific thiol-endopeptidase that enzymatically cleaves whole IgG just above the hinge region). The resulting Fab and Fc fragments were separated by immobilized protein-A (binds Fc fragments), Fab fragments were then further separated according to the type of the light chain by protein-L (binds kappa light chain of certain species including human; Figure 1B). All final fragments were in ~ 0.5 ml of PBS pH 7.4: Fc fragments ranged from 1.07 to 3.5 mg/mL, Fab fragment (Kappa-enriched) 0.4 mg/mL and Fab fragment (Lambda-enriched) was 0.9 mg/mL. The monoclonal antibody CNTO736 was provided by Centocor R&D (Radnor, Pennsylvania) in a buffer (10 mg/mL).
Figure 1.
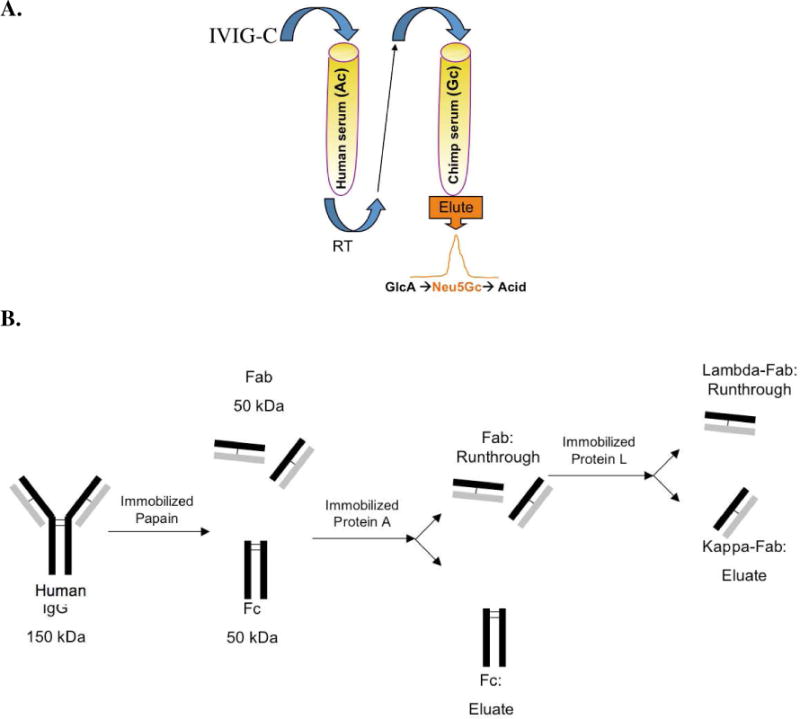
A. Affinity purification of anti-Neu5Gc antibodies from IVIG-C over sequential affinity columns with immobilized human- and chimpanzee-serum sialo-glycoproteins.19 B. Generating kappa-enriched or lambda-enriched Fab fragments from affinity-purified anti-Neu5Gc antibodies. Immobilized papain digested purified anti-Neu5Gc antibodies to create two separate Fab fragments and one Fc fragment per antibody molecule. The cleaved fragments were loaded over a Protein A column that binds only the Fc fragments, and the Fab fragments were collected in the run-through. Subsequently, the Fab fragments were loaded over a Protein L column, which binds specifically to kappa light chain, and the lambda-Fab fragments collected in the run-through.
Materials
Human IgG, dithiothreitol (DTT), iodoacetamide (IAA), guanidine hydrochloride, and ammonium bicarbonate, were obtained from Sigma-Aldrich (St. Louis, MO). Achromobacter protease I (Lys-C) was obtained from Wako (Richmond, VA). Endoproteinase Glu-C (sequencing grade) was purchased from Roche (Indianapolis, IN) and trypsin (sequencing grade) was purchased from Promega (Madison, WI). Formic acid and acetonitrile were purchased from Fisher Scientific (Fair Lawn, NJ). The HPLC-grade water used in all experiments was from J. T. Baker (Bedford, MA).
SDS-PAGE and in-gel multi-enzymatic digestion
The full-length purified human anti-Neu5Gc xeno-autoantibody samples and the Fc and Fab fragments were treated with DTT at 70°C for 10 min and loaded on a mini gel (8 × 8 cm and 4–12% Bis-Tris) for electrophoresis with subsequent Coomassie blue staining. The gel bands containing the different regions of the antibody samples were cut out. Each cut-out band was further minced into small pieces (approximately 0.5 mm2) and subjected to 2 to 3 cycles of gel dehydration with acetonitrile and rehydration with ammonium bicarbonate buffer (0.1 M, pH 8.0) in order to remove the Coomassie stain, as described in the following. Briefly, the gel slices were further washed with 300–400 μL of water for 15 min and centrifuged to remove the liquid. Acetonitrile was then added (300 μL) to the gel slice, which was placed in a microcentrifuge tube (spin for ~15 min), the liquid (acetonitrile) was removed. The gel slice was further dried in a Speed-Vac, and then rehydrated with 300 μL of 0.1 M NH4HCO3 for 10–15 min. An equal amount of acetonitrile was subsequently added, and the sample was vortexed for 15–20 min and then centrifuged to remove the liquid. This procedure was repeated up to 3 times, as necessary, or until no visible Coomassie stain remained. The remaining gel slice was then reduced with DTT by the addition of 250 μL of 10 mM DTT in 0.1 M NH4HCO3 and incubated for 30 min at 56 °C. The sample (gel slice) was subsequently alkylated at room temperature and in the dark for 60 min with 250 μL of 55 mM IAA in 0.1 M NH4HCO3. After removal of the liquid, an aliquot of 250 μL of digestion solution (containing 10 ng/μL trypsin in 50 mM NH4HCO3, pH 8.0; or 10 ng/μL Lys-C in 50 mM NH4HCO3, pH 8.0; or 10 ng/μL Glu-C in 50 mM NH4HCO3, pH 8.0) solution was added to the gel slice, and the sample was then incubated for 30–35 min at 4 °C. The incubated solution was then replaced with sufficient 50 mM NH4HCO3 to cover the gel pieces (50–100 μL) and then incubated overnight at 37 °C. In the Lys-C plus Glu-C case, after the liquid containing IAA was removed, an aliquot of 250 μL of Lys-C digestion solution (containing 10 ng/μL Lys-C in 50 mM NH4HCO3) was added to the gel slice, and the sample was incubated for 30–35 min at 4 °C. The incubated solution was then replaced with sufficient 50 mM NH4HCO3 to cover the gel pieces (50–100 μL) and then incubated for four hours at 37 °C. After that, an aliquot of 100 μL of Glu-C digestion solution (containing 10 ng/μL Glu-C in 50 mM NH4HCO3) was added to the sample, and then the sample was incubated overnight at 37 °C. The supernatant was removed and saved. The remaining gel pieces were further extracted with 5% formic acid (100 μL) at 37 °C for 5 min, and an equal amount of acetonitrile was subsequently added, and the sample was shaken for 15 min. The formic acid and acetonitrile solution, containing digested peptides, was combined with the previous supernatant and concentrated to ~10 μL. The concentrated solution (enzyme-digested peptides) was subjected to LC-MS analysis.
LC-MS analysis
The in-gel digested peptides were analyzed by on-line liquid chromatography using a linear ion trap coupled to a Fourier transfer mass spectrometer (LTQ-FT MS, Thermo Electron, San Jose, CA) with a Dionex nanoLC instrument (Ultimate 3000, Sunnyvale, CA) and a 75 μm ID × 15 cm C-18 capillary column packed with Magic C18 (3 μm, 200 Å pore size) (Michrom Bioresources, Auburn, CA). The LTQ-FT mass spectrometer was operated in the data-dependent mode to switch automatically between MS and MS2 acquisition. Survey full-scan MS spectra with 2 microscans (m/z 400–2000) were acquired in the Fourier transform ion cyclotron resonance cell with a mass resolution of 100,000 at m/z 400 (after accumulation to a target value of 2×106 ions in the linear ion trap), followed by 10 sequential LTQ-MS/MS scans throughout the 120-min separation. Dynamic exclusion was utilized with exclusion duration of 60 sec and no repeat counts. The analytical separation was carried out using a three step linear gradient, starting from 2% B to 40% B in 60 min (A: water with 0.1% formic acid; B: acetonitrile with 0.1% formic acid), increased to 60% B in 40 min, and then to 80% B in 20 min. The column flow rate was maintained at 200 nL/min.
Protein identification
Peptide sequences were first identified utilizing Bio-Works3.3.2 imbedded with Sequest (Thermo Electron) from a human database (Swiss-Prot release 52 with 15,498 protein sequences) which identified peptides with Xcorr scores above the following thresholds: ≥ 3.8 for 3+ and higher charge state ions, ≥ 2.2 for 2+ ions, and ≥ 1.9 for 1+ ions, with full trypsin/Lys-C/Glu-C specificity and up to 2 internal missed cleavages. The identified peptides were further confirmed by the high mass accuracy (≤ 5 ppm). Protein Prophet Probability software (Institute of Systems Biology, Seattle, WA) was then utilized to identify proteins based upon corresponding peptide sequences with ≥ 95% confidence. The data also further searched using the sequences of human immunoglobulins in the database for mapping the peptides at Fc, Fab, and subclasses of IgG domains.
Results and Discussion
General Approach
The isolation of IgG from patient-derived blood samples is usually achieved with Protein A or Protein G affinity columns, producing a heterogeneous mixture of immunoglobulins denoting the response to a variety of host defense or other immunological reactions that have occurred in that specific individual.22,23 Specifically, individual humans have widely varying levels of anti-Neu5Gc antibodies.18 Here, we purified these antibodies from leftovers of therapeutic human IVIG (IgG) preparations (pooled from ~10,000 donors and free of IgM and IgA) that have been discarded from the clinic. We used specific immunoaffinity purification using Neu5Gc-containing epitopes (Figure 1A) with further proteinA enrichment and separation to smaller antibodies’ fragments of the variable and constant regions (see Figure 1B). We then analyzed the affinity-purified fractions by nanoLC-MS (LTQ-FTMS) to obtain the sequences in both constant and variable regions and characterize the IgG subclass distribution based on characteristic peptides.
Sialoglycan microarray analysis of IVIG
Several IVIG brands are currently available. In order to evaluate the best source for purification of anti-Neu5Gc IgG, we tested four brands of IVIG (at a concentration-gradient of 1 mg/mL and 0.5 mg/mL in 200 μL/sub-array) using a specific sialoglycan microarray presenting multiple Neu5Gc-glycan epitopes (and Neu5Ac-glycan epitopes as controls).19 These IVIG brands showed variable binding patterns and intensities against the Neu5Gc-glycan epitopes (Figure 2A) with almost no binding to the control Neu5Ac-glycan epitopes (Figure 2B). Such differences may result from the diverse purification methods employed by different IVIG brands and/or added stabilizers. However, it may also represent different human populations with variable anti-Neu5Gc IgG reactivity. In general, none of the IVIG brands showed preference to a specific sialic acid type (Neu5Gc versus Neu5Gc9Ac) or linkage to underlying glyans (α3 versus α6) (Figure 2A). Among these brands, IVIG-C consistently showed the widest and the strongest anti-Neu5Gc-specific binding profile (Figure 2B) and was therefore used for subsequent affinity purification.
Figure 2.
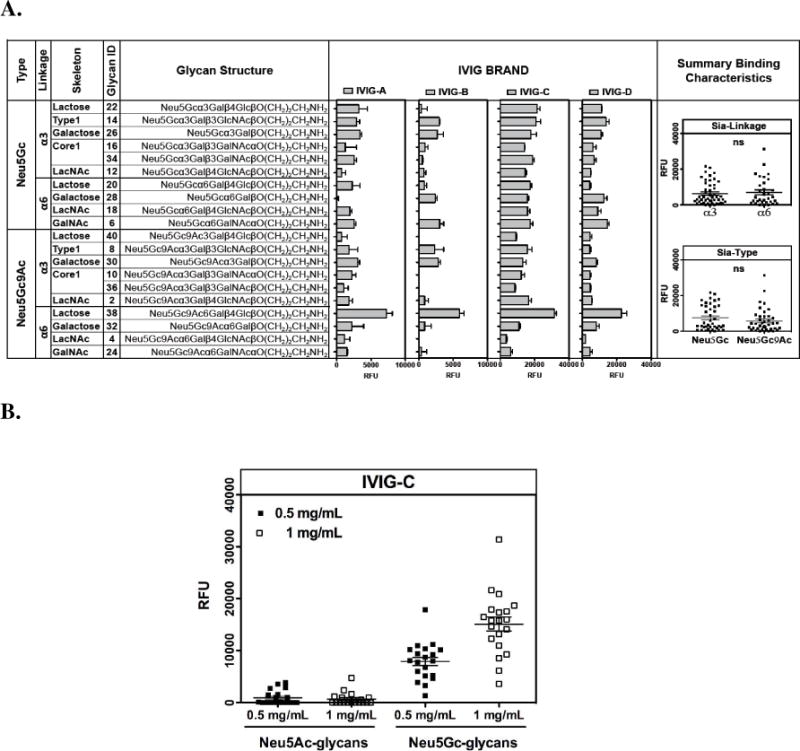
Four IVIG brands were analyzed on a sialoglycan microarray19 (as described in methods at 0.5 mg/mL and 1 mg/mL). A. Representative binding reactivity against Neu5Gc-glycans epitopes at 1 mg/mL. B. IVIG-C binds specifically to Neu5Gc-glycans epitopes and not to Neu5Ac-glycans epitopes (P<0.0001; One-Way-ANOVA with Bonferroni’s multiple comparison test) with linear binding at 0.5 mg/mL and 1 mg/mL (P<0.0001; Pearson r=0.93 for correlation test).
Purification of xeno-autoantibodies from human IVIG (IgG) preparations
Initially, human anti-Neu5Gc antibodies were isolated from IVIG-C by two approaches: 1) Neu5Gc-affinity chromatography only (Affinity-purified) and 2) Neu5Gc-affinity chromatography followed by protein A fractionation (Affinity-purified-protein-A), that enriches for purified full length IgG antibodies. The affinity purification procedure removed non-specific antibodies and specifically further concentrated the anti-Neu5Gc IgG. Indeed, the binding of the affinity-purified antibodies to the sialoglycan-miroarray showed highly specific strong binding only to the Neu5Gc-glycans and to none of the Neu5Ac-control glycans (in a much lower concentration of 20 μg/ml and 10 μg/ml; data not shown). The resulting samples were reduced and analyzed by 1D SDS-PAGE (Figure 3). In lanes 2 (Affinity-purified) and 3 (Affinity-purified-protein-A), we observed the expected banding pattern of the heavy (MW≈50 kDa) and light (MW≈23 kDa) chains. Lane 2 (Affinity-purified) shows that a significant amount of other proteins were still co-purified with the desired human anti-Neu5Gc antibodies. In contrast, the samples purified with an additional protein A step showed much less heterogeneity (Affinity-purified-protein-A; lane 3). Those gel bands with the expected MW for the light and heavy chains were cut out for in-gel trypsin digestion, and the resulting peptides were analyzed by LC-MS. In the analysis of lanes 2 and 3, only immunoglobulin species were observed, demonstrating the effectiveness of the affinity purification process. The bands containing the heavy and light chain fractions in lane 3 were further analyzed as discussed below.
Figure 3.
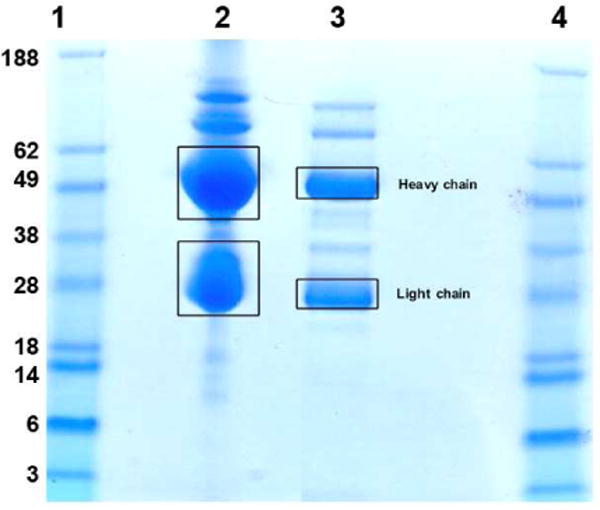
SDS-PAGE separation of purified human IVIG (IgG) preparations and the specified gel bands (with squares) are cut out for in-gel trypsin digestion. Lane 1&4: MW standards; lane 2: human anti-Neu5Gc antibodies purified by Neu5Gc-affinity chromatography only (Affinity-purified); lane 3: Affinity-purified samples followed by protein A fractionation (Affinity-purified-protein-A).
Figure 4 shows the composition (%) of different immunoglobulin species identified in both heavy and light chain fractions. In the heavy chain fraction, constant regions of IgG1, IgG2, IgG3 and IgG4 are the four most abundant species (IgG1 ≫ IgG2 > IgG4 > IgG3) with only small amount of light chain contaminants (likely incomplete dissociation of heavy and light chain species in SDS-PAGE). Similarly, in the light chain fraction we observed kappa and lambda light chains constant regions as the most abundant with only minor amount of heavy chain. Together, these findings demonstrate that in the affinity purification of anti-Neu5Gc IgG we were able to collect multiple antibodies that represent a polyclonal response to Neu5Gc in humans. Yet despite this polyclonal nature of the response, the above initial mass-spectrometry analysis revealed that we could get specific and unique peptides from individual antibodies. To gain further insight into the sequences involved in the anti-Neu5Gc response we expanded our analysis as detailed.
Figure 4.
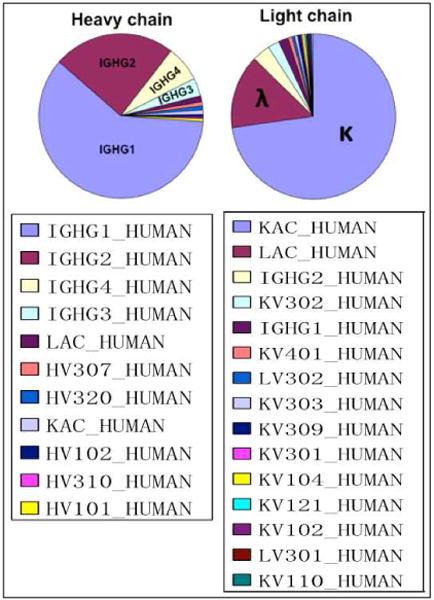
The composition (%) of gel bands shown in Figure 3, lane 3 (Affinity-purified-protein-A) as measured by spectral counting from the LC-MS analysis. The following abbreviations were used: IGHG=constant region of immunoglobulin heavy chains; LAC=constant region of immunoglobulin lambda light chain; KAC=constant region of immunoglobulin kappa light chain; HV=variable region of immunoglobulin heavy chain; LV=variable region of immunoglobulin lambda light chain; KV=variable region of immunoglobulin kappa light chain.
Determination of IgG subclass distribution
To accurately determine the IgG subclass distribution of anti-Neu5Gc xeno-autoantibodies, we selected characteristic peptides that are unique for each IgG subclass (one unique sequence per region), and that gave good mass spectrometry sensitivity and separation in the LC-MS analysis (see Table 2). As controls, the same analytical protocol was applied to a standard human IgG and CNTO736 (an IgG4 Fc fusion antibody). The extracted peak areas of these peptides were used to calculate the IgG subclass distribution in each sample. Table 1 shows the heavy chain IgG subclass distribution of human IgG, xeno-autoantibody (Affinity-purified-protein-A), and of CNTO736. While the standard human IgG subclass distribution falls in the normal range of IgG subclass distribution in humans,24 the anti-carbohydrate xeno-autoantibody (Affinity-purified-protein-A) sample seems to have increased IgG2 and IgG4 compared to the standard IgG preparation and a slightly lower amount of IgG1 (IgG1≫IgG2>IgG4>IgG3). While these differences may not seem markedly significant, these data provide strong direct evidence that human anti-carbohydrate antibodies may not be restricted to IgG2 as was previously suggested,25 and is in line with other literature.18, 26,27 As expected, in the IgG4 fusion antibody, only IgG4 subclass was observed indicating the precision of this calculation method using characteristic peptides.
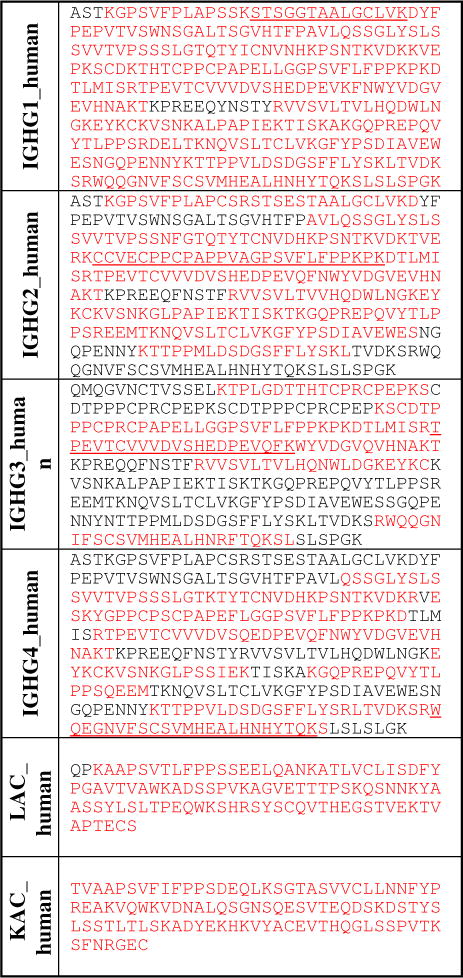
Table 2.
Sequence coverage of the constant regions (heavy and light chains) from the anti-Neu5Gc xeno-autoantibody. Red color: identified sequence; black color: deduced sequence; underlined: characteristic peptide sequences.
|
Table 1.
IgG subclass distribution of human IgG, xeno-autoantibody (Affinity-purified-protein-A) sample and CNTO736.
| IgG subclassa | IgG 1 |
IgG 2 |
IgG 3 |
IgG 4 |
|---|---|---|---|---|
|
Human IgG (total 100%) |
83% | 9% | 2% | 6% |
|
Xeno-autoantibody (total 100%) |
72% | 17% | 2% | 9% |
|
CNTO736 (total 100%) |
0% | 0% | 0% | 100% |
The percentage is calculated from the peak area of the characteristic peptides and given as an average of 3 replicas.
Determination of autoantibodies variable regions sequences
a) Analysis of intact immunoglobulin fractions
In view of the complexity of the IVIG affinity-purified human anti-Neu5Gc antibodies sample, we used a multi-enzyme digestion approach to give a greater sequence coverage of both the constant and variable regions.23 In this approach, xeno-autoantibody samples were reduced, loaded onto a mini gel, and the gel bands (heavy chain and light chain) were cut out and subjected to in gel digestion. Different proteolytic enzymes were used (trypsin, Lys-C, Glu-C) individually or together to digest the gel bands and the individual peptide mixtures were analyzed by LC-MS. Table 2 shows the resulting sequence coverage of the constant regions (heavy and light chains) from the anti-Neu5Gc xeno-autoantibody. Despite the heterogeneity of the sample, we were able to determine most of the sequences from the constant regions of the heavy chains covering all IgG subclasses -IgG1, IgG2, IgG3 and IgG4, as well as from the lambda and kappa light chains. We also obtained the sequences for the corresponding variable regions of both the heavy and light chains, albeit with less coverage, likely due to the extensive sequence diversity of these immunoglobulin regions.24
b) Proteomic analysis of Fab fragments generated by papain cleavage
As described in the Experimental section, we further digested the purified anti-Neu5Gc antibodies using immobilized papain and then obtained lambda-enriched Fab fragment, kappa-enriched Fab fragment and Fc fragment by protein A and protein L separation (Figure 1B). The resulting fragments were first reduced and then analyzed by a 1D gel (Figure 5). Only one band was observed for the Lambda-enriched Fab and Kappa-enriched Fab samples. However, three bands were observed for the Fc sample. Gel bands as shown in Figure 5 were cut out and subjected to in gel tryptic digestion and the individual peptide mixtures were analyzed by LTQ-FT MS.
Figure 5.
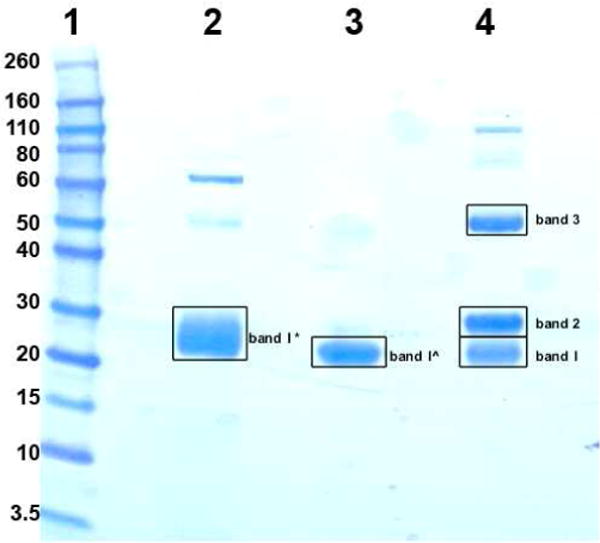
SDS-PAGE separation of Fab and Fc fragments and the specified gel bands (with squares) were cut out for in-gel tryptic digestion. Lane 1: MW standards; lane 2: Lambda-enriched Fab; lane 3: Kappa-enriched Fab; lane 4: Fc. Higher MW bands are aggregates of protein. Band 1*=runthrough from protein L in Figure 1B; band 1^=eluate from protein L in Figure 1B; bands 1–3= eluate from protein A in Figure 1B.
The LC-MS results revealed that Lambda-enriched Fab sample (band 1*) was a mixture of lambda light chain (constant plus variable regions), kappa light chain (constant plus variable regions) and heavy chain (CH1 plus variable regions); kappa-enriched Fab sample (band 1^) was relatively pure kappa light chain (constant plus variable regions) with some heavy chain (CH1 plus variable regions); band 1 from Fc sample was heavy chain (CH1 plus variable regions) and kappa light chain (constant plus variable regions), band 2 from Fc sample was glycosylated heavy chain (hinge, CH2 and CH3 regions); and band 3 from Fc sample was non-glycosylated heavy chain (undigested; variable regions, CH1, hinge, CH2 and CH3 regions).
Partial sequences from the variable regions were obtained from the Lambda-enriched and Kappa-enriched Fab samples (Table 3). For heavy chain variable region, partial sequences of nine different variants were determined; for lambda light chain variable region, partial sequence of six different variants were determined; and for kappa light chain variable region, partial sequence of sixteen different variants were determined.
Table 3.
Sequence coverage of the heavy and light chains variable region. Red color: identified sequence; black color: deduced sequence.

|
Conclusions
Anti-Neu5Gc xeno-autoantibodies from human IVIG (IgG) were characterized in this study. The antibodies were purified by a multi-affinity chromatographic strategy via bio-recognition of all Neu5Gc-glycan epitopes, and then we used FTICR mass spectrometry with our multi-enzyme digestion approach to determine the partial sequence and IgG subclass distribution of these xeno-autoantibodies. We plan to use this proof-of-concept work in follow up studies of the origin and clonality of such xeno-autoantibodies as well as methodological studies aimed at obtaining an entire sequence coverage of in vivo human antibodies, such as those with specific affinity towards Neu5Gcα2-6GalNAcα (Neu5Gc-sialyl-Tn) that were suggested to have a therapeutic potential.19
Acknowledgments
This study was supported by funding from NCI grants U01CA128427, U01CA128442, the Korean Research WCU grant R31-2008-000-10086-0, and ISEF fellowship to V.P-K. This is Contribution Number 1007 from the Barnett Institute.
References
- 1.Nguyen DH, Tangvoranuntakul P, Varki A. J Immunol. 2005;175:228–236. doi: 10.4049/jimmunol.175.1.228. [DOI] [PubMed] [Google Scholar]
- 2.Kelm S, Schauer R. Int Rev Cytol. 1997;175:137–240. doi: 10.1016/S0074-7696(08)62127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Traving C, Schauer R. Cell Mol Life Sci. 1998;(54):1330–1349. doi: 10.1007/s000180050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varki A. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angata T, Varki A. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 6.Chou HH, Takematsu H, Diaz S, Iber J, Nickerson E, Wright KL, Muchmore EA, Nelson DL, Warren ST, Varki A. Proc Natl Acad Sci USA. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muchmore EA, Diaz S, Varki A. Am J Phys Anthropol. 1998;107:187–198. doi: 10.1002/(SICI)1096-8644(199810)107:2<187::AID-AJPA5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 8.Tangvoranuntakul P, Gagneux P, Diaz S, Bardor M, Varki N, Varki A, Muchmore E. Proc Natl Acad Sci USA. 2003;100:12045–12050. doi: 10.1073/pnas.2131556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higashi H, Nishi Y, Fukui Y, Ikuta K, Ueda S, Kato S, Fujita M, Nakano Y, Taguchi T, Sakai S. Gann. 1984;75:1025–1029. [PubMed] [Google Scholar]
- 10.Hirabayashi Y, Higashi H, Kato S, Taniguchi M, Matsumoto M. Jpn J Cancer Res (GANN) 1987;78:614–620. [PubMed] [Google Scholar]
- 11.Nakarai H, Saida T, Shibata Y, Irie RF, Kano K. Int Arch Allergy Appl Immunol. 1987;83:160–166. doi: 10.1159/000234349. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi Y, Sasabe T, Nishida T, Nishi Y, Higashi H. Am J Ophthalmol. 1983;96:321–325. doi: 10.1016/s0002-9394(14)77822-5. [DOI] [PubMed] [Google Scholar]
- 13.Marquina G, Waki H, Fernandez LE, Kon K, Carr A, Valiente O, Perez R, Ando S. Cancer Res. 1996;56:5165–5171. [PubMed] [Google Scholar]
- 14.Kawai T, Kato A, Higashi H, Kato S, Naiki M. Cancer Res. 1991;51:1242–1246. [PubMed] [Google Scholar]
- 15.Devine PL, Clark BA, Birrell GW, Layton GT, Ward BG, Alewood PF, McKenzie IFC. Cancer Res. 1991;51:5826–5836. [PubMed] [Google Scholar]
- 16.Zhu A, Hurst R. Xenotransplantation. 2002;9:376–381. doi: 10.1034/j.1399-3089.2002.02138.x. [DOI] [PubMed] [Google Scholar]
- 17.Padler-Karavani V, Yu H, Cao H, Chokhawala H, Karp F, Varki N, Chen X, Varki A. Glycobiology. 2008;18:818–830. doi: 10.1093/glycob/cwn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedlund M, Padler-Karavani V, Varki N, Varki A. Proc Natl Acad Sci U S A. 2008;105:18936–18941. doi: 10.1073/pnas.0803943105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padler-Karavani V, Hurtado-Ziola N, Pu M, Yu H, Huang S, Muthana S, Chokhawala H, Cao H, Secrest P, Friedmann-Morvinski D, Singer O, Ghaderi D, Verma IM, Liu Y-T, Messer K, Chen X, Varki A, Schwab R. Cancer Res. 2011;71:3352–3363. doi: 10.1158/0008-5472.CAN-10-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Costa D, Broodman I, Vanduijn MM, Stingl C, Dekker LJ, Burgers PC, Hoogsteden HC, Sillevis Smitt PA, van Klaveren RJ, Luider TM. J Proteome Res. 2010;9:2937–2945. doi: 10.1021/pr901114w. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Wu S-L, Karger BL, Hancock WS. Anal Chem. 2010;82:6154–6162. doi: 10.1021/ac100956x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hober S, Nord K, Linhult M. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:40–47. doi: 10.1016/j.jchromb.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 23.Andrew SM, Titus JA. Curr Protoc Immunol. 2001 Chapter 2, Unit 2.7. [Google Scholar]
- 24.Jacobs & Demott laboratory test handbook. (5) :536. [Google Scholar]
- 25.Barrett DJ, Ayoub EM. Clin Exp Immunol. 1986;63:127–134. [PMC free article] [PubMed] [Google Scholar]
- 26.Hammarström L, Persson MA, Smith CI. Immunology. 1985;54:821–826. [PMC free article] [PubMed] [Google Scholar]
- 27.von Gunten S, Smith DF, Cummings RD, Riedel S, Miescher S, Schaub A, Hamilton RG, Bochner BS. J Allergy Clin Immunol. 2009;123:1268–1276. doi: 10.1016/j.jaci.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]