

Abstract
The matrix protein M1 plays a pivotal role in the budding of influenza virus from the plasma membrane (PM) of infected cells. This protein interacts with viral genetic material and envelope proteins while binding to the inner leaflet of the PM. Its oligomerization is therefore closely connected to the assembly of viral components and the formation of new virions. Of interest, the molecular details of M1 interaction with lipids and other viral proteins are far from being understood, and it remains to be determined whether the multimerization of M1 is affected by its binding to the PM and interaction with its components. To clarify the connection between M1 oligomerization and binding to lipid membranes, we applied a combination of several quantitative microscopy approaches. First, we used number and brightness (N&B) microscopy to characterize protein multimerization upon interaction with the PM of living cells. Second, we used controlled biophysical models of the PM (i.e., supported bilayers) to delve into the details of M1-lipid and M1-M1 interactions by employing a combination of raster image correlation spectroscopy (RICS), fluorescence correlation spectroscopy (FCS), and atomic force microscopy (AFM). Our results show that M1 oligomer formation is strongly enhanced by membrane binding and does not necessarily require the presence of other viral proteins. Furthermore, we propose a specific model to explain M1 binding to the lipid bilayer and the formation of multimers.
Introduction
Infection by influenza virus is a major cause of mortality around the world. All three influenza virus genera (A, B, and C) belong to the Orthomyxoviridae family of enveloped viruses (1), but only influenza A virus (IAV) is a primary concern for human health. Its capsid is formed by a lipid bilayer containing three membrane proteins: hemagglutinin (HA), neuraminidase (NA), and the proton channel M2 (2). The most abundant protein in the virus is matrix protein 1 (M1), which forms the matrix layer directly below the lipid envelope and binds the viral ribonucleoproteins (3). Therefore, in a fully formed virion, M1 has the important function of stabilizing the whole three-dimensional (3D) structure of the envelope (4–6). Acting as an endoskeleton, the M1 shell might provide anchoring points for viral membrane proteins (7–10).
In addition, M1 supposedly plays a key role during virion assembly utilizing multiple protein-lipid and protein-protein interactions (8). According to the current understanding, M1 is recruited and multimerizes at the plasma membrane (PM) of an infected cell together with M2, HA, and NA at the site of a nascent virion (8). The protein assembly process proceeds until the bilayer bends and a new viral particle is formed and released. It is yet not clear whether M1 multimerization requires or is influenced by other viral components (e.g., HA). However, it is known that M1 has the potential to form multimers in solution independently of the presence of other proteins. Zhang et al. (11) reported that M1 forms dimers and high-order multimers depending on the pH and protein concentration. Small-angle x-ray scattering measurements showed that M1 clusters in solution display an architecture similar to that of authentic virions (12). Similarities between large M1 assemblies in vitro and M1 structures in isolated viruses have also been suggested by x-ray crystallography studies (4). Furthermore, in certain cases the expression of solely M1 is sufficient to produce virus-like particles (13,14). These observations suggest that M1 multimerization has a central role in the process of new virion formation. Therefore, understanding the details of M1-M1 interaction in a simple controlled system, in the absence of other proteins, might shed light on the process of IAV assembly. On one hand, it is known that M1 binds in vitro to lipid bilayers containing phosphatidylserine (PS), probably due to electrostatic interactions (15,16). On the other hand, M1 in vivo targets internal cellular membranes (e.g., the Golgi and endoplasmic reticulum), but does not bind preferentially to the inner leaflet of the PM (17–19), despite its relatively high PS content (20). In either case, M1-M1 interaction and multimerization have not yet been explored in connection to membrane binding, leaving unresolved questions. More specifically, does the M1-lipid interaction affect the multimerization process?
To address this question, we applied the number and brightness (N&B) approach to monitor M1 multimerization at the PM of living cells. Furthermore, we used controlled model systems mimicking the PM, atomic force microscopy (AFM), and quantitative fluorescence microscopy (i.e., raster image correlation spectroscopy (RICS) and fluorescence correlation spectroscopy (FCS)) to characterize M1-M1 and M1-lipid interactions. Our results suggest a specific molecular mechanism by which M1 can bind to lipid bilayers and form high-order multimers.
Materials and Methods
Chemicals
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Alexa Fluor 647 carboxylic acid succinimidyl ester (A647) was acquired from Life Technologies (Darmstadt, Germany). PBS, medium, and supplements for cell culture were purchased from PAN Biotech (Aidenbach, Germany). The plasmid pET15b-M1 for the expression of M1 derived from A/FPV/Rostock/34 was kindly provided by Dr. N. Jungnick (Louisiana State University, Baton Rouge, LA). Bacto tryptone, Bacto yeast extract, and Bacto agar were bought from BD (Heidelberg, Germany). Ampicillin and chloramphenicol were purchased from Sigma Aldrich (Taufkirchen, Germany) and Serva (Heidelberg, Germany). NaCl, NaOH, and glucose were bought from Roth (Karlsruhe, Germany). Lysozyme, CaCl2, bovine deoxyribonuclease I (DNase), and imidazole were purchased from Sigma (Munich, Germany). Mercaptoethanol was bought from Merck (Darmstadt, Germany) and cOmplete Ultra EDTA-free protease inhibitor was bought from Roche (Basel, Switzerland). Isopropyl-1-thio-β-D-galactopyranoside (IPTG) was bought from Fermentas-Fisher Scientific (Schwerte, Germany). The unsaturated lipid analog carrying three nitrilotriacetic acid headgroups (NTA-lipid) for the binding of His-tagged proteins (21) was a generous gift from Dr. J. Piehler (University of Osnabrück, Osnabrück, Germany).
Cell culture
Madin-Darby canine kidney (MDCK) cells were seeded in 35 mm glass-bottom petri dishes 24 h before experiments were conducted, and allowed to grow to 80% confluency in full growth medium (10% fetal calf serum). The cells were transfected using TurboFect (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol and used for microscopy starting at 2–4 h posttransfection. M1 from Influenza A/FPV/Rostock/34 was cloned into plasmid pEYFP-C1 (Clontech, Mountain View, CA) yielding M1 with an N-terminal YFP tag (YFP-M1) or M1 with an N-terminal YFP tag and a C-terminal nuclear export signal (YFP-M1-NES), as described previously (17). Both plasmids were kindly provided by M. Veit and B. Thaa (Freie Universität, Berlin, Germany). The empty plasmid was used as a control.
N&B analysis
N&B analysis was performed as described previously (22,23). Briefly, confocal images were acquired on an inverted Olympus IX81 microscope equipped with a FluoView FV1000 scan and confocal detection unit (Olympus, Tokyo, Japan) coupled to a detection unit containing avalanche photodiodes (PicoQuant, Berlin, Germany). The 488 nm excitation light from a CW Argon laser (Showa Optronics, Tokyo, Japan) was focused with a 60× UPLS Aprocromat 1.2 NA water objective into the sample. The fluorescence signal passed through a 550/100 bandpass filter before detection. The laser power was chosen so as to keep the photon count below ∼1–2 MHz (i.e., typical values were ≤4 μW). Images of 128 × 128 pixels were acquired with pixel dimensions of ∼400 nm and a pixel dwell time of 100 μs. Image time stacks of 100 scans were collected at time intervals of 1.95 s per frame. The images were exported into ASCII files containing the photon counts as a function of pixel position using the PicoQuant SymPhoTime64 software. We then analyzed the time stacks using a self-written MATLAB algorithm (The MathWorks, Natick, MA), implementing the equations from Digman et al. (22) for the specific case of true photon-counting detectors (23), and thus obtained the true molecular brightness (hereafter referred to simply as brightness) as a function of pixel position. Brightness maps were filtered with a cross median filter using a 3 × 3 neighborhood.
To correct for photobleaching effects and minor cell movements, we used a boxcar filter with an eight-frame length as described previously (24). Spurious events that led to extraneous fluctuations were automatically excluded by the use of a cutoff threshold when average brightness values were calculated during the boxcar filtering (i.e., excluding the 20% of data points from top and bottom tails of the data set). We avoided saturation of detectors leading to artifactual reduction of brightness by excluding images with pixels in which photon counting exceeded ∼1–2 MHz (22). Using this selection criterion, we kept the saturation-induced brightness decrease below 10–20%. Finally, to further correct the weak residual negative correlation between brightness and pixel intensity, we measured the detector response using a reflective metal surface. The brightness-versus-intensity plot (which should be constant and equal to 0 for all intensity values) thus obtained was used to correct the actual experimental data (22).
Protein purification
The plasmid pET15b-M1 was transformed into Rosetta E. coli (DE3) pLysS-competent cells for expression of M1 with an N-terminal His6 tag. Cells were grown at 37°C in a medium containing 0.4% glucose, 50 μg/mL chloramphenicol, and ampicillin to an OD600 of ∼0.8. The culture was harvested by centrifugation after induction with 0.1 mM IPTG at 37°C for 3 h. Then the cells were resuspended in lysis buffer (PBS, pH 7.4, 250 mM NaCl with 100 μg/mL DNase, 300 μg/mL lysozyme, 5 mM mercaptoethanol, protease inhibitor cocktail, and 1 mM phenylmethylsulfonyl fluoride) at 4°C for 1 h and then lysed by sonication. Cellular membranes and debris were removed by centrifugation at 90,000 g for 20 min at 4°C. The supernatant was flowed through a chelating Talon matrix column (Clontech, Saint-Germain-en-Laye, France) and M1 was purified according to the manufacturer’s protocol. The equilibration and wash buffers were PBS, 500 mM NaCl pH 7.0, and 5 mM mercaptoethanol. The intermediate elution buffer was PBS, 500 mM NaCl pH 7.0, 5 mM mercaptoethanol, and 60 mM imidazole. The final elution buffer was PBS, 250 mM NaCl pH 7.0, 5 mM mercaptoethanol, and 250 mM imidazole. At this step, the typical protein concentration was ∼50 μM. After dilution to ∼10 μM, the protein was stored in the final elution buffer for up to 3 days at 4°C. At the beginning of each experiment, the sample was centrifuged for 5 min at 17,000 g (4°C) to remove any protein aggregate that might nevertheless have formed. When needed, M1 was labeled with A647 succinimidyl ester to obtain A647-M1. To this end, the protein was incubated with a 3-fold excess reactive dye for 24 h at 4°C (pH 7). Unbound dye was removed via gel filtration (Sephadex G-25) and the protein was stored in the final elution buffer at ∼5 μM concentration for up to 3 days at 4°C. Due to the suboptimal labeling conditions (i.e., pH 7 and protein concentration < 2 mg/mL), the typical degrees of labeling ranged between ∼0.1 and 0.3 A647 molecules per M1. Note that a low M1 concentration and neutral pH were purposely chosen to avoid the risk of protein aggregation.
Supported lipid bilayers preparation
Supported lipid bilayers (SLBs) were prepared on glass coverslips using the vesicle fusion method (25). Small unilamellar vesicles were obtained by sonication. Egg phosphatidylcholine (ePC) and PS extracted from bovine brain (bPS) were mixed in chloroform at different molar ratios. After solvent evaporation, the lipid film was rehydrated in PBS at 1 mg/mL lipid concentration and resuspended by vortexing. After sonication, 100 μl of the suspension was deposited on a clean coverslip. The sample was delimited by a plastic cylinder of 7 mm diameter. Adding CaCl2 to a final concentration of 3 mM induced vesicle fusion and the formation of a lipid bilayer. The sample volume was brought to 200 μL and after a 30 min incubation it was vigorously rinsed with PBS buffer to remove any unfused vesicle.
A647-M1 was added on top of the SLB from a ∼5 μM stock solution to a final concentration ranging from 1 to 150 nM. After 5 min, the sample was rinsed five times with 200 μL PBS and directly imaged on the microscope.
Confocal microscopy, FCS, and RICS analysis
Confocal imaging, FCS, and RICS were performed on the same setup as described in the N&B analysis section above. The 640 nm excitation light from a pulsed diode laser (PicoQuant, Berlin, Germany) was focused with a 60× UPLS Apocromat 1.2 NA water objective into the sample. The fluorescence signal passed through a 650 nm longpass filter before detection. Images of 256 × 256 pixels were acquired with pixel dimensions of 54.8 nm and a pixel dwell time of 12.5 μs. Image time stacks of 100 scans were collected at time intervals of 1.155 s per frame (4.325 ms per line). Out-of-plane fluorescence was reduced by a 100 μm pinhole in front of the detector. For each sample, we performed a total of six measurements in different positions: three time stacks using ∼0.3 μW excitation power and three time stacks using ∼8 μW. In general, for each examined sample, a total of 18 RICS measurements from three independent preparations were collected. For the measurements performed at higher laser power, to avoid bleaching artifacts, only one or two frames were analyzed. For the other measurements, all 100 frames were averaged and analyzed. The images were exported into ASCII files containing the photon counts as a function of pixel position using the PicoQuant SymPhoTime64 software. We subsequently analyzed the time stacks using a self-written MATLAB algorithm, implementing the equations from Digman et al. (26) for the 2D diffusion case. The fitting of the spatial-temporal correlation function resulted in three output parameters: the diffusion coefficient D, the lateral waist ω0, and the particle concentration C. Also, by calculating the total fluorescence intensity for the first frame of each measurement, we could calculate particle brightness as the ratio between intensity and C. The parameters, which should be independent of the laser power and protein degree of labeling (i.e., D, C, and ω0), were indeed comparable in measurements performed with different laser powers and thus were simply pooled and averaged from the 18 measurements for each sample. To take into account variations in laser power and protein labeling efficiency, the other parameters (i.e., total fluorescence intensity and brightness) were normalized to the values measured each day in a specific sample before averaging, as described in the Results section.
The same optical setup was used to perform single-point FCS. For each sample, a total of three independent protein preparations were measured at different concentrations in the same buffer solution used for RICS experiments on SLBs, using a laser power of ∼0.7 μW. Each measure consisted of a 60-s-long signal acquisition. The fluorescence signal time trace was then divided into two or more segments, which were correlated and fitted using the PicoQuantSymPhoTime64 software. To take into account variations in protein labeling efficiency, the calculated brightness values were normalized for each independent preparation on the values measured for the 50 nM M1 concentration sample. The diffusion time values were normalized to the value measured each day for the diffusion of unconjugated A647 succinimidyl ester.
AFM
For AFM, we prepared SLBs as described above, using a slightly modified protocol. A glass coverslip was attached with UV-curing glue to a glass slide to increase the mechanical stability of the sample, while maintaining the same support surface used in fluorescence-based experiments. Due to the dimensions of the AFM glass block, the total sample volume was four times larger and therefore the total lipid amount used in the preparation had to be correspondingly increased.
Before adding M1 at the required concentration, we tested for proper formation of the SLB by AFM force measurements. Bilayer piercing experiments were performed to ensure the presence of a single bilayer on the surface (27), followed by incubation with M1 as described above. For every M1 concentration, seven topography images at different positions from three independent measurements were used for further analysis.
All AFM measurements were performed in PBS at pH 7 and 24°C with a NanoWizard III (JPK, Berlin, Germany) in AC mode using a triangular shaped MLCT cantilever (Bruker, Camarillo, CA) with a nominal resonance frequency of 38 kHz. For image analysis, JPK data-processing software (JPK, Berlin, Germany) and MATLAB were employed.
Results
M1 forms multimers in proximity to the PM of living MDCK cells
To study M1 multimerization in living cells, we expressed a fluorescent YFP-M1 construct in MDCK cells. Fig. 1 A shows a confocal microscopy image of two typical cells expressing fluorescent YFP-M1. The protein appears distributed all over the cell body and, in certain cases, enriched in the perinuclear region, in line with previous observations (17). Most importantly, no strong M1 binding to the PM can be observed. Next, we used the N&B approach to measure the brightness of YFP-M1 and hence its multimerization state as a function of its position inside the cells. To this end, we collected 100 frames from each sample (total measurement time of ∼2 min) and calculated the first and second moments of the fluorescence fluctuation distribution for each pixel. The N&B analysis thus provided spatial maps of both the number and brightness of diffusing fluorescent M1 molecules (either as monomers or multimeric complexes). Multimeric protein complexes are characterized by higher brightness. Fig. 1 B shows the brightness map obtained for the same cells shown in Fig. 1 A. For comparison, Fig. 1 C shows a typical brightness map obtained for cells expressing soluble free YFP (see also Fig. S1 in the Supporting Material).
Figure 1.
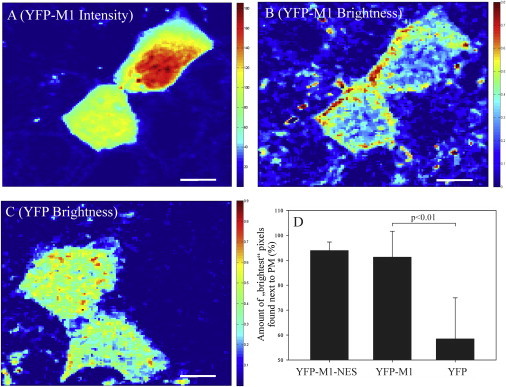
Confocal fluorescence imaging and brightness mapping of YFP-M1 expressed in living MDCK cells. (A) Confocal microscopy fluorescence image of two typical MDCK cells expressing YFP-M1. Other cells that are not expressing the fluorescent protein construct can also be observed, probably due to a weak autofluorescence signal. This image was obtained as an average of 100 frames (128 × 128 pixels) collected over ∼2–3 min. (B) Pixel brightness map obtained by applying N&B analysis to the image shown in A. Some pixels that showed out of range brightness outside the cells (e.g., due to bleaching of immobile proteins) were removed. (C) Pixel brightness map for a typical MDCK cell expressing soluble YFP. The corresponding fluorescence intensity confocal microscopy image is shown in Fig. S1. All images were collected at 24°C and scale bars are 8 μm. (D) Quantification of the intracellular spatial distribution of bright pixels. The brightest pixels are defined as the top 10% brightest pixels in each cell. For each cell, an ROI enclosing the PM and an ROI including the rest of the cell were defined. Panel D shows the relative amount of the brightest pixels found in the ROI enclosing the PM, for all of the examined samples (YFP-M1-NES (n = 8), YFP-M1 (n = 18), and soluble cytosolic YFP (n = 10)). The YFP-M1 sample and the YFP sample are significantly different (two-sample t-test, p < 0.01). Error bars represent standard deviations (SDs). To see this figure in color, go online.
The spatial distribution of pixel brightness indicates that M1 multimers (i.e., pixels with higher brightness values) are localized preferentially in proximity to the PM. This observation is specific to the expression of M1, since the expression of YFP alone (used here as a control) results in a more homogeneous brightness distribution (Fig. 1 C). To quantitatively characterize this effect, we analyzed several samples with cells expressing YFP-M1 or just YFP. First, we manually divided each cell into two regions of interest (ROIs): 1), a 5- to 10-pixel-thick stripe including the PM; and 2), a roughly circular ROI including the nucleus and most of the cytoplasm. Second, we identified the top 10% pixels with the highest brightness values (referred to as the brightest pixels) in the whole cell. Finally, we calculated how many of these brightest pixels were included in the ROI containing the PM. We applied this procedure to cells expressing YFP-M1 (18 cells, three independent samples), YFP (10 cells, three independent samples), or YFP-M1-NES (i.e., a specific construct with enhanced export from the nucleus (17); eight cells, one sample). The results in Fig. 1 D show that M1 multimers, either the wild-type or the NES construct, are reproducibly found in proximity to the PM. On the other hand, YFP brightness does not significantly correlate with PM localization.
It is worth noting that a quantitative comparison of brightness values (e.g., between Fig. 1, B and C) to precisely determine the oligomerization state of M1 cannot be provided by this approach. For example, in the simple approximation that few very large (i.e., very bright) M1 multimers are formed on the PM, and M1 monomers diffuse in the cytosol, the brightness value calculated for a single pixel including the PM would be a weighted average brightness of few multimers and many monomers (since the few-nanometers-thick PM occupies a very small volume fraction of a 400 × 400 nm2 pixel). Furthermore, to keep the total signal intensity below a reasonable threshold suitable for sensitive photon-counting detectors, cells with varying expression levels had to be imaged with varying laser power. This implies that the absolute brightness values (directly depending on laser power) might vary slightly from measurement to measurement. For the same reason, we waited only a few hours after transfection and selected cells that displayed low levels of protein expression. All of the examined cells had a number of YFP-M1 molecules between ∼102 and 4×102 per pixel, as calculated using the N&B analysis and averaging all over each cell. Cells with much higher expression levels were excluded because the total fluorescence signal would have been too high for the photon-counting detectors. The measured protein amount corresponds to a concentration with an order of magnitude of ∼10 μM, assuming a pixel size of 400 nm × 400 nm × 1 μm.
M1 multimerizes upon binding to PS-containing model membranes
Once we had assessed the potential of M1 to form large multimers in proximity to cellular PM, we delved into the details of M1-lipid and M1-M1 interactions in a well-defined and controlled system. M1 was expressed in E. coli, purified, and fluorescently labeled as described in the Materials and Methods section, yielding A647-M1. Typical protein samples consisted of a ∼1-3:10 A647-M1/M1 mixture (i.e., between one and three out of 10 M1 molecules were labeled). For the sake of simplicity, hereafter we will refer to the mixture of unlabeled M1 and labeled M1 as just A647-M1. The protein was then added at different concentrations (1–150 nM) to SLBs composed of ePC and 40 mol % bPS. The amount of bPS was similar to that used in previous studies (15). After 5 min, the unbound protein was washed away. M1 did not bind irreversibly to the membrane, as the protein could be at least partially removed with further washing (data not shown). We used confocal fluorescence microscopy and RICS to quantitatively characterize protein binding to the lipid bilayer and, at the same time, M1 multimerization. Fig. 2 A shows a typical confocal microscopy image of A647-M1 bound to the SLB. Furthermore, a RICS analysis of analogous fluorescence images provided several parameters regarding the state of the bound protein: fluorescence intensity (which is proportional to the total amount of bound protein), diffusion coefficient, concentration, and brightness (which reports the multimerization state of the protein). Note that the parameters calculated from a RICS analysis of such images (e.g., concentration) do not refer to single proteins bound to the membrane, but rather to the independent fluorescent entities that are diffusing as single objects on the bilayers. For example, if M1 multimerized on the membrane, the calculated brightness refers to the total brightness of the whole independently diffusing protein complexes.
Figure 2.
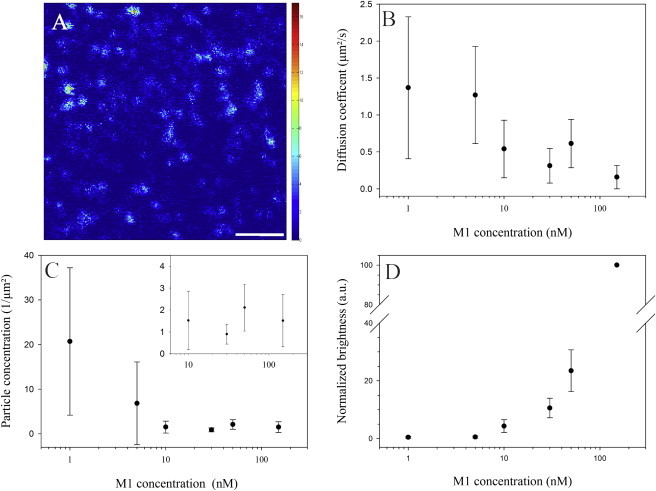
RICS analysis of M1 multimerization upon interaction with lipid bilayers as a function of protein concentration. (A) Confocal fluorescence microscopy image of a typical sample consisting of an SLB made of ePC and 40 mol % bPS after incubation with 50 nM A647-M1. The color scale indicates the average number of photons per pixel measured over 100 frames. Scale bar is 50 pixels (2.7 μm). (B–D) Protein diffusion coefficient, particle/cluster concentration, and normalized brightness, respectively, as a function of protein concentration during the binding step. Brightness normalization was performed by setting the value measured for the 150 nM M1 sample to 100. The inset in C shows the concentration measurements on a magnified scale (i.e., A647-M1 concentration between 10 and 150 nM). Error bars represent SDs of 18 measurements in three independent samples. To see this figure in color, go online.
As expected, the total amount of protein bound to the membrane increased monotonically with the concentration of M1 in solution (∼50-fold; data not shown). Fig. 2 B shows that the diffusion coefficient D of membrane-bound A647-M1 decreased 10-fold (from ∼1.5 μm2/s to 0.1 μm2/s) when the protein concentration in solution was increased from 1 nM to 150 nM. Fig. 2 C reports the concentration of M1 clusters on the bilayer, which decreased from ∼20 to ∼2 M1 clusters/μm2 (i.e., many small clusters change into fewer (possibly larger) clusters) when the M1 concentration in solution was increased from 1 to 10 nM. Above 10 nM, we did not observe further changes. Note that the values obtained for 1 and 10 nM M1 in general are associated with a larger statistical spread, simply due to the low signal/noise ratio at such low protein concentrations. Finally, Fig. 2 D shows the normalized brightness of the clusters, calculated as the total fluorescence intensity of a sample (proportional to the total amount of bound protein) divided by the total number of clusters in each image. This parameter therefore reports the degree of clustering (or relative amount of M1 molecules per cluster) as a function of M1 concentration in solution. Brightness values were normalized each day to the value measured for the 150 nM M1 sample (i.e., 100%) to take into account variations in laser power and the degree of protein labeling. Our data show that when the A647-M1 concentration in solution was increased from 1 to 150 nM, the molecular size of the protein clusters on the membrane increased of a factor ∼2 × 102 (i.e., from ∼0.5 to 100). Further washing of the sample did not significantly affect M1 multimer brightness (data not shown).
It was previously shown that M1 multimers dissociate at acidic pH (11). To verify that the brightness parameter reported by RICS is indeed sensitive to M1 multimerization similarly to other experimental approaches, we compared the brightness of M1 clusters at pH 7 and pH 5. To this end, we let M1 (50 nM) bind to an SLB as described above at pH 7. After removing the unbound protein, we measured the total bound protein (i.e., total fluorescence intensity) and its brightness. Then, we changed the pH of the sample to 5 using a small volume of a 2 M pH 5 Na-Acetate buffer (6 μl in a 200 μl sample). After 10 min and with no additional washing step, we measured the same fluorescence parameters. Our experiment showed only a marginal decrease in the amount of A647-M1 bound to the membrane, but a ∼3-fold decrease in brightness (Fig. S2).
M1 does not multimerize in solution at nanomolar concentrations
It is known that M1 tends to multimerize in solution at pH 7, especially at concentrations above ≈30 μM (11). Since the focus of this study was the role of lipid membranes in determining M1-M1 interactions, it was important to control the extent to which M1 multimers formed in solution before interacting with lipid bilayers. For this purpose, we used FCS to monitor the dynamics and brightness of A647-M1 in buffer solution, in the absence of lipid membranes. More in detail, we measured the ratio between average fluorescence intensity and number of molecules in the focal volume, thus obtaining the brightness of the diffusing molecules (or molecular complexes). Fig. 3 A shows the values obtained for different A647-M1 concentrations, normalized to the brightness value measured (each day, for each independent protein preparation) for the 50 nM M1 sample. Our results show that protein brightness (i.e., multimerization) did not increase in the concentration range in which RICS detected extensive multimerization on lipid membranes (50–150 nM), and up to 300 nM. It is worth noting that a direct comparison between the brightness measured for M1 in solution (Fig. 3 A) and that measured for M1 bound to membranes (Fig. 2 D) is not possible due to the different experimental settings (i.e., laser power, sample geometry, protein labeling efficiency, and fluorophore quantum yield) used for FCS and RICS experiments.
Figure 3.
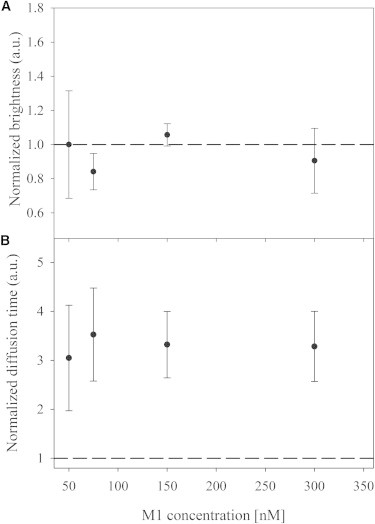
FCS measurements of A647-M1 diffusion and brightness in solution as a function of protein concentration. (A and B) Normalized brightness (A) and diffusion time (B) of A647-M1 in solution for increasing protein concentration (i.e., from 50 to 300 nM). Brightness values were normalized to what was measured for the 50 nM M1 sample. Diffusion time values were normalized to what was measured for unconjugated A647 in the same conditions. The dashed lines represent the unitary values (e.g., the value relative to unconjugated A647 for B) as a reference. Error bars are SDs (n = 3).
Similarly to the brightness measurements, Fig. 3 B shows that the dynamics of A647-M1 were not influenced by an increasing protein concentration in solution in the absence of lipids. The diffusion time of M1 (roughly corresponding to the time needed to cross the FCS focal volume and inversely proportional to the diffusion coefficient) remained constant three to four times longer than the diffusion time of unconjugated A647 succinimidyl ester (used here as a reference). Considering the molecular masses of A647 and a M1 monomer (i.e., ∼1 kDa and ∼30 kDa, respectively), our measurements are compatible with M1 diffusing as monomers or dimers (expected relative diffusion times of ∼3.1 and ∼3.9, respectively, compared with A647).
M1 total binding to membrane does not significantly affect protein multimerization
In the previous paragraphs, we have shown that increasing M1 concentrations in solution are connected to higher protein binding to the membrane and significant protein multimerization. To assess whether M1 multimerization depends directly on the extent of membrane binding, we designed two different experiments in which we modulated M1 binding to the lipid bilayer and monitored changes in protein clustering.
It is known that M1 binds to lipid bilayers containing PS (16). Therefore, we monitored M1 multimerization on bilayers containing different amounts of bPS. Fig. 4 A shows the relative fluorescence signal (i.e., total protein binding) to SLBs as a function of their bPS content. Our data show, as expected, that M1 binding increases 2- to 3-fold when bPS concentration in the membrane is increased from 0 to 40 mol %. According to our results, the M1-lipid interaction is enhanced by (but does not require) the presence of PS. Interestingly, Fig. 4 B shows that M1 cluster brightness increases only ∼20% (compared with, e.g., the 200-fold increase shown in Fig. 2 D). To take into account variations in laser power and degree of protein labeling, we normalized the brightness and fluorescence intensity values each day to the values measured for the 40 mol % bPS sample.
Figure 4.
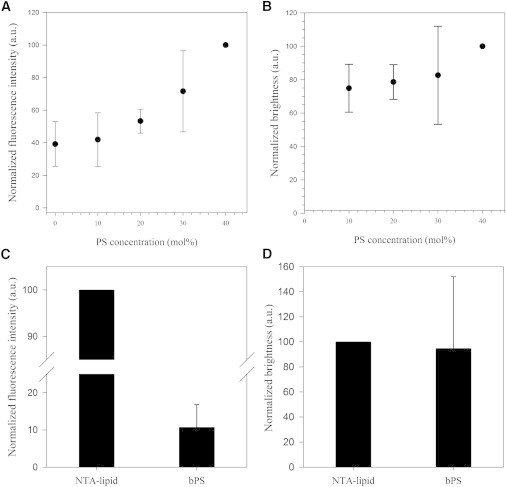
RICS analysis of M1 multimerization as a function of total protein binding. (A) Normalized total fluorescence intensity measured for A647-M1 (50 nM) bound to SLBs with varying bPS concentration. (B) Normalized brightness measured for the same samples as in A. The data in both A and B were normalized by setting the values measured for the sample containing 40 mol % bPS to 100. (C) Normalized total fluorescence intensity measured for A647-M1 (50 nM) bound to SLBs containing 95 mol % ePC and either 5 mol % NTA-lipid or 5 mol % bPS. (D) Normalized brightness measured for the same samples as in C. The data in both C and D were normalized by setting the values measured for the sample containing 5 mol % NTA-lipid to 100. The data sets shown for the two samples in D are not statistically distinguishable (two-sample t-test, p > 0.05). Error bars represent SDs of 18 measurements in three independent samples.
As a second approach, we produced SLBs containing 95 mol % ePC and 5 mol % of an unsaturated lipid analog carrying three nitrilotriacetic acid groups (NTA-lipid). NTA-lipids were shown to specifically bind His-tagged proteins (21), such as the M1 used in this study. Fig. 4 C shows that M1 binding to NTA-lipid containing SLBs was ∼10 times higher compared with SLBs containing 5 mol % bPS. As shown in Fig. 4 D, notwithstanding the dramatic increase in protein binding caused by the presence of NTA-lipids, M1 brightness (clustering) remained comparable to that observed in samples with low protein binding, such as SLBs containing 5 mol % bPS. Note that the large spread in brightness values observed for the latter sample derives from the low signal/noise ratio due to limited protein binding. Brightness and fluorescence intensity values were normalized each day to the values measured for the NTA-lipid containing samples. The diffusion coefficient of A647-M1 bound to the membrane did not vary significantly between NTA and 5 mol % bPS containing samples or among samples with varying amounts of bPS (data not shown).
AFM reveals concentration-dependent clustering of unlabeled M1
All of the experiments described thus far required labeling of M1 either with a fluorescent protein (see N&B experiments) or with a small fluorophore (see experiments with A647-M1). Furthermore, the above-mentioned optical microscopy measurements are characterized by a spatial resolution that ultimately is limited by the size of the point spread function. For example, RICS experiments have a spatial resolution of ∼300 nm. To overcome these limitations, we performed AFM measurements of M1 bound to SLBs containing 40 mol % bPS. M1 and bPS concentrations were chosen in the range used for the RICS experiments shown in Fig. 2, i.e., 10 and 50 nM.
Fig. 5, A and B, show typical topographical images of the lipid membrane surface after M1 binding (10 and 50 nM, respectively) and the subsequent removal of unbound protein. Samples incubated with 10 nM M1 already show a considerable degree of protein binding. The membrane surface is uniformly covered by objects of heterogeneous lateral shape and width, but with a well-defined height of 2.5 ± 0.6 nm (Fig. S3). Before exposure to the protein, the SLB surface appears flat and homogeneous within the sensitivity of the instrument, with only rare exceptions (Fig. S4). Therefore, it can be safely concluded that the features observed in Fig. 5 are indeed constituted of M1 molecules. Upon an increase in M1 concentration, fewer but higher objects can be observed (Fig. 5 B). Their average diameter is 93 ± 37 nm (including only the objects higher than 10 nm), with the exception of some elongated structures up to ∼1 μm long. Most importantly, M1 multimers observed in the 50 nM sample are up to ∼10–20 times higher than those observed in the 10 nM M1 sample. M1 structures with a height of ∼2.5 nm are also present in the 50 nM M1 samples in varying amounts, but cannot be easily observed with the zoom needed to visualize the larger protein clusters shown in Fig. 5 B.
Figure 5.
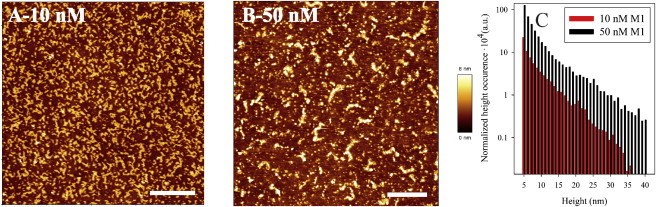
AFM imaging of M1 bound to PS-containing bilayers. (A and B) AFM topographical images of ePC bilayers containing 40 mol % bPS after treatment with 10 and 50 nM M1, respectively. As described for the previous experiments, the protein was incubated with the membrane for 5 min before extensive washing. The color code refers to the measured height and is the same for both panels. Scale bars are 2 μm. (C) Average height occurrence probability (n = 7 images from three independent preparations), referring to the frequency of observed heights between 5 and 40 nm for membranes incubated with 10 nM M1 (red) or 50 nM M1 (black). To see this figure in color, go online.
Fig. 5 C shows the observed frequency distribution of pixels with heights of 5–40 nm, calculated from an average of seven images from three independent preparations for each protein concentration. First, we calculated the pixel height histograms over the whole height range measured in the sample (see Fig. S5 for a representative 10 nM M1 sample). Such histograms are dominated by a large peak around 0 nm, representing the surface of the bilayer. A second peak can also be observed that originates from the presence of the M1 structures with a height of ∼2.5 nm. To characterize large M1 clusters that formed at a 50 nM M1 concentration and occupied a relatively small fraction of the image, we subtracted the 0 nm peak and plotted only the data between 5 and 40 nm. Fig. 5 C clearly shows that larger M1 multimers (e.g., with height > 20 nm) are observed much more frequently on SLBs incubated with 50 nm M1 than on those incubated with just 10 nM M1.
Discussion
M1 is the most abundant protein in IAV and it plays a pivotal role in the viral life cycle (3) because it is fundamentally important during the assembly of new virions from the PM of infected cells (14). According to recent hypotheses (28), M1 coordinates the assembly of viral components at the PM by binding 1), IAV genetic material; 2), lipids in the inner leaflet of the PM; 3), IAV transmembrane proteins; and 4), other M1 molecules to eventually form the 3D structure underlying the envelope. The delicate balance of protein-protein and protein-lipid interactions in a nascent virion and the precise role of M1 in regulating this process are still far from being understood. For example, although M1 was shown to have the capability to form biologically relevant structures in solution by itself (4,12), it is yet not clear whether interactions with other viral proteins are needed for M1 multimerization during viral bud formation. In vivo experiments have provided contrasting results regarding the capability of M1 expressed in cells to form virus-like-particles in the absence of other viral components (13,14,29). More generally, it was clearly shown that M1 can bind membranes containing negatively charged lipids (e.g., PS) (15), but no data are available regarding the connection between M1 binding to lipids and its multimerization, either in vivo or in vitro.
M1 forms multimers upon interaction with lipid membranes
Recent advances in quantitative fluorescence microscopy have provided powerful tools for studying protein-protein interactions in vivo as well as in vitro (30). For example, N&B analysis has been used to study the self-association of transmembrane receptors (24), membrane-associated proteins (23), and Ebola virus assembly (31). Similarly, we have investigated M1 self-association in MDCK cells expressing a fluorescent construct of the protein, YFP-M1. In line with previous observations (17), wild-type M1 associated mainly with the nucleus and perinuclear regions, whereas M1-NES accumulated in the cytoplasm and particularly at the Golgi apparatus and the endoplasmic reticulum. The lack of a specific membrane-targeting region might be responsible for the weak association with the PM (19). Our results strikingly show that in spite of the limited association with the PM, M1 could indeed form multimers in regions adjacent to the PM. The observed spatial multimerization pattern is specific to M1, since a control performed with soluble YFP displayed a uniform brightness/multimerization distribution in the cell body. This finding clearly suggests a connection between M1-lipid interaction at the PM and M1 multimerization. Interestingly, the YFP-M1 concentrations in our experiments were comparable to the physiological concentrations of M1 in infected cells (≈10 μM). An upper estimate for the M1 concentration in cells can be calculated assuming 3 × 103 M1 molecules per virus (32), up to 103–104 viruses per cell (33) and a cell volume of ∼1 pL (103 μm3).
Although N&B experiments on living cells can provide information about protein-protein interactions in a physiologically relevant context, they are limited by the complexity of the examined system itself. The presence of cellular proteins in the cytosol or PM components might in fact influence M1-M1 and M1-lipid interactions. Furthermore, N&B analysis cannot distinguish clusters of proteins diffusing in the plane of the PM from several M1 monomers bound to a single membrane vesicle in proximity of the PM. In both cases, N&B analysis would report an increase in brightness. To dissect the interaction between M1 and lipid membranes, and how it influences protein multimerization, we complemented our investigation by using model membranes in well-defined experimental conditions.
Similar to what was observed in cells, we detected the formation of M1 multimers bound to the lipid bilayer. First, we observed that the amount of bound M1 depended on the presence and amount of negatively charged lipids in the membrane (e.g., PS), as previously shown (15), and on the amount of protein in solution. Second, we observed that the amount of protein interacting with the membrane determined the size of M1 clusters. The correlation between M1 concentration and clustering efficiency was measured by image correlation spectroscopy (i.e., RICS) and AFM. When the M1 concentration was increased from 1 to 150 nM, we observed a decrease of the diffusion coefficient from ∼1.5 to ∼0.5 μm2/s, i.e., from values typical of a single lipid-anchored protein (34) to values typical of 102- to 103-nm-radius lipid assemblies (35) diffusing in the plane of a liquid membrane. Furthermore, we noticed that the number of clusters reached a constant value of ∼2 μm−1 already at 10 nM. Finally, a direct measurement of cluster brightness displayed an increase of 2 orders of magnitude. These three observations strongly suggest that already at 10 nM M1 concentration, the membrane is almost covered with M1 oligomers. This finding was directly confirmed by AFM measurements showing that the surface of the lipid bilayers was extensively covered with M1 at a 10 nM protein concentration. The height of the observed protein structures (∼2.5 nm higher than the bilayer surface) suggests at least partial penetration of M1 into the membrane, since the N-terminal monomer fragment was determined to be 3 × 4 × 3 nm3 (4,36). Increasing protein concentration does not affect the degree to which the bilayer is covered, but rather the size of the clusters, which are then characterized by high brightness and slow diffusion. This was observed with both wild-type unlabeled proteins (using AFM) and fluorescently labeled proteins. The combination of the two approaches is particularly beneficial in this case because optical methods are limited to a resolution of few hundred nanometers (AFM measurements clearly indicate a size for such clusters of ∼100 nm in diameter and up to ∼35 nm in height). We believe that these larger clusters observed via AFM in the 50 nM samples are most probably the same protein structures observed in fluorescence measurements (e.g., Fig. 2). Such protein assemblies behave as expected for M1 structures in vivo; for example, they become unstable when the pH is lowered from 7 to 5 (Fig. S2). Interestingly, pH-dependent weakening of the M1-M1 interaction was also observed in virions (37) and for M1 in solution (i.e., not bound to lipid membranes (11)). Furthermore, the M1 assemblies we observed seemed to be closely related to the structures detected in solution at higher concentration (i.e., > ∼30 μM) in previous studies (12), as suggested by the similarities in size. On the other hand, to our knowledge, the flat 2.5-nm-high M1 oligomers (which were more evident in the 10 nM samples; Figs. 6 A and S3) have not been observed before. Previous AFM investigations reported a different M1 lateral organization, but, importantly, those measurements were not performed on lipid membranes (12). Whether the flat oligomers we observed are simply the precursors for the larger aggregates or are distinct structures with physiological relevance remains to be determined. To that end, we are currently performing AFM measurements with higher magnification/resolution in our laboratory.
Figure 6.
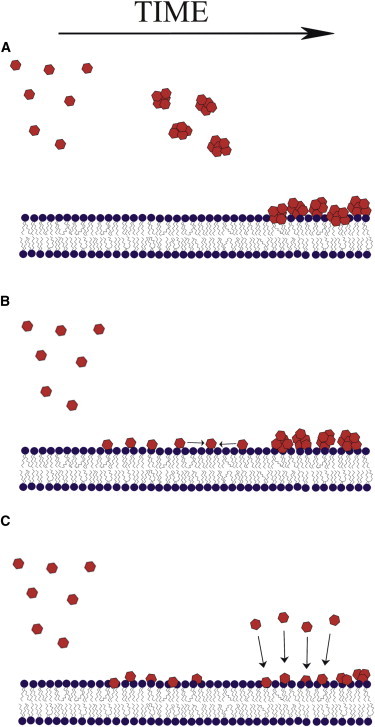
Different binding models for M1 forming multimers on lipid bilayers. Experimental evidence indicates that M1 monomers (or dimers), shown as single red particles on the left side of each panel, form multimers on the membrane surface (right side). (A) Formation of multimers might occur before interaction with the lipids. (B) Alternatively, M1 might first bind as monomer (or dimer) to the membrane and eventually cluster with the already bound protein due to increased local concentration and more likely lateral interaction. (C) Finally, M1 might first bind to the membrane as a monomer (or dimer) and subsequently interact with the remaining protein in solution to form larger clusters. M1 and lipids are not drawn to scale. To see this figure in color, go online.
M1 dimers bind to the membrane and subsequently form high-order multimers
Both the RICS (Fig. 2) and AFM data show that M1 forms multimers on the surface of the lipid membrane, ranging in size from a few to ∼103 molecules. The latter number is calculated considering the large cluster size roughly estimated from AFM images (i.e., ∼100 nm diameter and up to ∼30 nm in height) and from an estimate of M1 monomer size (i.e., ∼55 nm3 (12)). Since the technical approaches described in this work have a limited time resolution (e.g., several seconds for RICS), it is not possible to determine the temporal order of events during M1 binding and multimerization. In other words, the RICS and AFM results (Figs. 2 and 5, respectively) are compatible with at least all of the different binding modalities shown in the cartoons in Fig. 6.
One possibility is that M1 multimerizes quickly in solution and only after clustering binds to the membrane (Fig. 6 A). However, this scenario would be difficult to reconcile with the current understanding of IAV assembly in infected cells, according to which new virions are formed also via interaction between M1 and the other viral proteins that are already in the PM (i.e., HA, NA, and M2) (8). Alternatively, M1 could bind to the membrane in large amounts, at first without multimerizing (Fig. 6 B). The passage of M1 from solution to the 2D surface of the lipid bilayer would then correspond to an effective increase in local concentration, according to the principle of dimensionality reduction (38). This in turn would result in increased protein-protein lateral interaction and thus multimerization. Finally, it might be possible that M1 binds to the membrane and then acts as clustering seed for further M1 molecules recruited from solution (Fig. 6 C). Therefore, we designed specific experiments to test the different models.
To investigate the likelihood of the scenario depicted in Fig. 6 A, we performed FCS measurements of M1 diffusion and brightness in solution as a function of protein concentration. These experiments were conducted in exactly the same buffered solution used for the membrane binding and clustering measurements (e.g., the experiments shown in Fig. 2), with the only difference being the absence of lipids in the sample. FCS measurements indicated that M1 is probably in either monomeric or dimeric form in solutions, and no clustering was observed in the absence of membranes for an M1 concentration < 300 nM. This is in complete agreement with recent work showing that M1 is a dimer in solution if the protein concentration is lower than 0.1 mg/mL (3.4 μM) (11), as well as our N&B results indicating that M1 multimerizes specifically in proximity to the PM (rather than in the cytoplasm, for example). The study of Zhang et al. (11) also indicated that the smallest oligomerization state of M1 at neutral pH is a dimer. It is therefore safe to assume that M1 in our experiments (i.e., the FCS measurements in Fig. 3 and RICS measurements at the lowest concentrations in Fig. 2) is also in a dimeric state. Thus, our FCS experiments rule out the model suggesting M1 multimerization in solution followed by subsequent binding of M1 clusters to the membrane (Fig. 6 A).
To verify whether high protein surface density (as a consequence of M1 binding and dimensionality reduction) might enhance M1 multimerization, we purposely increased protein binding to the bilayer. This experiment was specifically designed to test the model presented in Fig. 6 B. Modulation of protein-membrane binding was achieved by changing the amount of PS in the bilayer (Fig. 4, A and B) or by using artificial lipids with high affinity to the His tag included in the purified M1 (Fig. 4, C and D). In either case, we observed only a minor correlation between total M1 binding to the membrane and the degree of protein clustering. This observation suggests that increased M1 surface density on the bilayer (Fig. 6 B) does not play an essential role in determining protein multimerization.
In conclusion, the results we obtained by RICS, AFM, and FCS indicate that M1 binds to the membrane at first probably as a dimer and eventually multimerizes when its concentration in solution is above ∼10 nM. Multimer growth seems to occur mostly via interaction between already bound M1 and M1 that are still in solution (see Fig. 6 C). The situations depicted in Fig. 6, B and C, are not necessarily mutually exclusive, but our results do not support the hypothesis that lateral M1-M1 interactions play a major role. Of interest, the model described in Fig. 6 B implies that a higher amount of M1 expression in an infected cell might be required (compared with the model described in Fig. 6 C) to reach sufficiently high surface concentrations at the PM and trigger M1 multimerization. Such a requirement would probably lower the efficiency of the viral reproduction process. On the other hand, we speculate that the preferential interaction between unbound M1 and lipid-bound M1 (rather than that between lipid-bound M1 molecules, for example) might derive from the protein-lipid interaction itself. Once bound to the bilayer, M1 might undergo a conformational change or, for example, stably expose a side that favors interaction with unbound M1 molecules. In fact, ongoing circular dichroism spectroscopy experiments in our laboratory suggest that the M1-lipid interaction is accompanied by secondary structure rearrangements (N. Jungnick and S.C., unpublished data). This issue will be further explored in future studies.
Conclusions
The experiments described in this work demonstrate the tendency of M1 to form high-order multimers when interacting with PS-containing lipid membranes, in the absence of other proteins. Similarly, two recent studies described the formation of M1 self-assemblies that also formed spontaneously (although in the absence of lipid membranes and at concentrations ∼100 times higher than those used in this study) (11,12). Our data provide for the first time, to our knowledge, information about the role of lipid membranes in the process of M1 multimerization. This information is particularly interesting in a biological context because M1 supposedly multimerizes at the PM of infected cells (8). According to our results, the M1-M1 interaction is triggered and enhanced by the presence of the lipid membrane already at nanomolar concentrations. M1 dimer binding to the surface of the bilayer is followed by further binding of the protein still in solution and the formation of large complexes (see Fig. 6 C).
How precisely the M1 structures reported by us and other groups are directly connected to those that form during IAV assembly in vivo will be subject of further investigation. For example, the protein concentrations that we explored in our work were 2–3 orders of magnitude lower than that calculated for the in vivo case. Although this observation might suggest that the M1-lipid interaction is strong and that IAV assembly might require very small amounts of M1 to be initiated, it is important to note that IAV assembly in the cellular environment is far more complex. In a living cell, both the cytoplasm and the PM are crowded with proteins and other biological macromolecules (39) that could interfere with the above-mentioned M1-lipid interaction. It is also possible that M1 assembly at the PM is modulated in particular by the presence of spike proteins, e.g., through interactions between M1 and the cytosolic moieties of HA or NA. The model systems and the technical approaches described in this work will most probably provide an opportunity to investigate these intriguing possibilities.
Acknowledgments
We thank B. Thaa for a critical review of the manuscript. The MATLAB routines used to calculate matrix autocorrelation were based on those written by D. Kolin. NTA-lipids and the M1 plasmids were kindly provided by J. Piehler and M. Veit, respectively.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through SFB 740 and grant CH 1238/3-1.
Supporting Material
References
- 1.Veit M., Thaa B. Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011;2011:370606. doi: 10.1155/2011/370606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nayak D.P., Balogun R.A., Barman S. Influenza virus morphogenesis and budding. Virus Res. 2009;143:147–161. doi: 10.1016/j.virusres.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruigrok R.W., Calder L.J., Wharton S.A. Electron microscopy of the influenza virus submembranal structure. Virology. 1989;173:311–316. doi: 10.1016/0042-6822(89)90248-1. [DOI] [PubMed] [Google Scholar]
- 4.Harris A., Forouhar F., Luo M. The crystal structure of the influenza matrix protein M1 at neutral pH: M1-M1 protein interfaces can rotate in the oligomeric structures of M1. Virology. 2001;289:34–44. doi: 10.1006/viro.2001.1119. [DOI] [PubMed] [Google Scholar]
- 5.Calder L.J., Wasilewski S., Rosenthal P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA. 2010;107:10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaap I.A., Eghiaian F., Veigel C. Effect of envelope proteins on the mechanical properties of influenza virus. J. Biol. Chem. 2012;287:41078–41088. doi: 10.1074/jbc.M112.412726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee K.K. Architecture of a nascent viral fusion pore. EMBO J. 2010;29:1299–1311. doi: 10.1038/emboj.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossman J.S., Lamb R.A. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris A., Cardone G., Steven A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen B.J., Leser G.P., Lamb R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008;82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang K., Wang Z., Liu W. Dissection of influenza A virus M1 protein: pH-dependent oligomerization of N-terminal domain and dimerization of C-terminal domain. PLoS ONE. 2012;7:e37786. doi: 10.1371/journal.pone.0037786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shtykova E.V., Baratova L.A., Svergun D.I. Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH. PLoS ONE. 2013;8:e82431. doi: 10.1371/journal.pone.0082431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bialas K.M., Desmet E.A., Takimoto T. Specific residues in the 2009 H1N1 swine-origin influenza matrix protein influence virion morphology and efficiency of viral spread in vitro. PLoS ONE. 2012;7:e50595. doi: 10.1371/journal.pone.0050595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gómez-Puertas P., Albo C., Portela A. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 2000;74:11538–11547. doi: 10.1128/jvi.74.24.11538-11547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruigrok R.W., Barge A., Whittaker G.R. Membrane interaction of influenza virus M1 protein. Virology. 2000;267:289–298. doi: 10.1006/viro.1999.0134. [DOI] [PubMed] [Google Scholar]
- 16.Baudin F., Petit I., Ruigrok R.W. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 2001;281:102–108. doi: 10.1006/viro.2000.0804. [DOI] [PubMed] [Google Scholar]
- 17.Thaa B., Herrmann A., Veit M. The polybasic region is not essential for membrane binding of the matrix protein M1 of influenza virus. Virology. 2009;383:150–155. doi: 10.1016/j.virol.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Ali A., Avalos R.T., Nayak D.P. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000;74:8709–8719. doi: 10.1128/jvi.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., Harmon A., Li F. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J. Virol. 2010;84:4673–4681. doi: 10.1128/JVI.02306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiessling V., Wan C., Tamm L.K. Domain coupling in asymmetric lipid bilayers. Biochim. Biophys. Acta. 2009;1788:64–71. doi: 10.1016/j.bbamem.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beutel O., Nikolaus J., Piehler J. High-fidelity protein targeting into membrane lipid microdomains in living cells. Angew. Chem. Int. Ed. Engl. 2014;53:1311–1315. doi: 10.1002/anie.201306328. [DOI] [PubMed] [Google Scholar]
- 22.Digman M.A., Dalal R., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crosby K.C., Postma M., Gadella T.W. Quantitative analysis of self-association and mobility of annexin A4 at the plasma membrane. Biophys. J. 2013;104:1875–1885. doi: 10.1016/j.bpj.2013.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellriegel C., Caiolfa V.R., Zamai M. Number and brightness image analysis reveals ATF-induced dimerization kinetics of uPAR in the cell membrane. FASEB J. 2011;25:2883–2897. doi: 10.1096/fj.11-181537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brian A.A., McConnell H.M. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc. Natl. Acad. Sci. USA. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Digman M.A., Brown C.M., Gratton E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 2005;89:1317–1327. doi: 10.1529/biophysj.105.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dufrene Y.F., Boland T., Lee G.U. Characterization of the physical properties of model biomembranes at the nanometer scale with the atomic force microscope. Faraday Discuss. 1998;79:137–157. doi: 10.1039/a807637e. [DOI] [PubMed] [Google Scholar]
- 28.Imai M., Watanabe S., Odagiri T. Influenza B virus BM2 protein is a crucial component for incorporation of viral ribonucleoprotein complex into virions during virus assembly. J. Virol. 2004;78:11007–11015. doi: 10.1128/JVI.78.20.11007-11015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen B.J., Leser G.P., Lamb R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007;81:7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Digman M.A., Stakic M., Gratton E. Raster image correlation spectroscopy and number and brightness analysis. Methods Enzymol. 2013;518:121–144. doi: 10.1016/B978-0-12-388422-0.00006-6. [DOI] [PubMed] [Google Scholar]
- 31.Adu-Gyamfi E., Digman M.A., Stahelin R.V. Investigation of Ebola VP40 assembly and oligomerization in live cells using number and brightness analysis. Biophys. J. 2012;102:2517–2525. doi: 10.1016/j.bpj.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamb R.A., Krug R.M. Orthomyxoviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields’ Virology. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 487–531. [Google Scholar]
- 33.Stray S.J., Air G.M. Apoptosis by influenza viruses correlates with efficiency of viral mRNA synthesis. Virus Res. 2001;77:3–17. doi: 10.1016/s0168-1702(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 34.Chiantia S., Ries J., Schwille P. Combined AFM and two-focus SFCS study of raft-exhibiting model membranes. ChemPhysChem. 2006;7:2409–2418. doi: 10.1002/cphc.200600464. [DOI] [PubMed] [Google Scholar]
- 35.Petrov E.P., Schwille P. Translational diffusion in lipid membranes beyond the Saffman-Delbruck approximation. Biophys. J. 2008;94:L41–L43. doi: 10.1529/biophysj.107.126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arzt S., Baudin F., Ruigrok R.W. Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology. 2001;279:439–446. doi: 10.1006/viro.2000.0727. [DOI] [PubMed] [Google Scholar]
- 37.Fontana J., Steven A.C. At low pH, influenza virus matrix protein M1 undergoes a conformational change prior to dissociating from the membrane. J. Virol. 2013;87:5621–5628. doi: 10.1128/JVI.00276-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rich A., Davidson N. W. H. Freeman and Company; San Francisco: 1968. Reduction of Dimensionality in Biological Diffusion Processes; pp. 198–215. [Google Scholar]
- 39.Dix J.A., Verkman A.S. Crowding effects on diffusion in solutions and cells. Annu. Rev. Biophys. 2008;37:247–263. doi: 10.1146/annurev.biophys.37.032807.125824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.