

Abstract
Phylloseptin-1, -2, and -3 are three members of the family of linear cationic antimicrobial peptides found in tree frogs. The highly homologous peptides encompass 19 amino acids, and only differ in the amino acid composition and charge at the six most carboxy-terminal residues. Here, we investigated how such subtle changes are reflected in their membrane interactions and how these can be correlated to their biological activities. To this end, the three peptides were labeled with stable isotopes, reconstituted into oriented phospholipid bilayers, and their detailed topology determined by a combined approach using 2H and 15N solid-state NMR spectroscopy. Although phylloseptin-2 and -3 adopt perfect in-plane alignments, the tilt angle of phylloseptin-1 deviates by 8° probably to assure a more water exposed localization of the lysine-17 side chain. Furthermore, different azimuthal angles are observed, positioning the amphipathic helices of all three peptides with the charged residues well exposed to the water phase. Interestingly, our studies also reveal that two orientation-dependent 2H quadrupolar splittings from methyl-deuterated alanines and one 15N amide chemical shift are sufficient to unambiguously determine the topology of phylloseptin-1, where quadrupolar splittings close to the maximum impose the most stringent angular restraints. As a result of these studies, a strategy is proposed where the topology of a peptide structure can be determined accurately from the labeling with 15N and 2H isotopes of only a few amino acid residues.
Introduction
In an era of increasing resistance of pathogens against the commonly used antibiotics the development of new bactericidal and fungicidal compounds has become an urgent need. One strategy consists in searching the plant and animal kingdoms for new compounds, understanding their function, and using them as template structures to develop new treatments (1–3). In this context it is notable that host defense peptides are found in abundant variety in nature and some of them are part of the innate immune systems of animals (4–6) and humans (7), where they are stored in exposed tissues or synthesized upon induction, thereby assuring a fast response when infections occur (6,8,9). There is good evidence that many of these peptides act on the membrane of sensitive organisms, by mechanisms that are quite different from the commonly used compounds and thereby less prone to resistance. Therefore, understanding how they selectively kill pathogens provides new avenues to design alternative therapeutic strategies (2,10,11). By searching the natural environments or by design, >2000 polypeptide sequences have been discovered and are listed in the corresponding databases (12).
It is thought that by destabilizing the integrity of bacterial and fungal membranes many of these peptides disrupt the energy metabolism of sensitive organisms (reviewed in (8,13,14)). Alternatively, by interacting with membranes they enter the cell interior where actions ultimately leading or enhancing antimicrobial effects can take place (1,15). Most of the peptides investigated so far exhibit a very dynamic character where the detailed structure adapts to the environment. It is therefore essential to understand how they interact with and/or cross liquid phase phospholipid bilayers and how the interplay between amino acid sequence and lipids can explain their biological activities. Such knowledge will provide the basis to design more efficient, serum-resistant, and cheaper molecules (2,16,17).
Phylloseptins (PS) are antimicrobial peptides that have been discovered in the skin secretions of Phyllomedusa genus (anura), tree frogs inhabiting the tropical forests of South and Central America (4). They consist of 19 to 20 highly conserved amino acid residues belonging to the family of linear amphipathic peptides. All of them carry histidines at residues 7 and 18 as well as an amidated carboxy-terminus. From the viewpoint of structure-function relationship they are of special interest as they exhibit high homology within their N-terminal domains but a high degree of variation involving the last six residues (Table 1). PS, like other linear cationic peptides, exhibit a broad range of antimicrobial activities including against gram-positive and gram-negative bacteria (Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Acinetobacter calcoacetius, and Streptococcus agalactiae) and yeast (Candida albicans), where the detailed spectrum of antibiotic and antifungal activities depends on the specific amino acid sequence (18). This may allow the species producing these peptides to adapt to their microenvironment in the best possible manner, and by storing cocktails of several different sequences being prepared to fight infections by a wider variety of pathogens (19–21). Minimal inhibitory concentrations in the 2–10 μM range have been observed for bacteria and yeast, whereas the PS show little hemolytic activity at similar concentrations (4,18).
Table 1.
The primary sequences of the PS investigated in this work
| PS-1 | FLSLI PHAIN AVSAI AKHN-NH2 |
| PS-1(B) | FLSLI PHAIN AVSAI AKHN-NH2 |
| PS-2 | FLSLI PHAIN AVSTL VHHF-NH2 |
| PS-3 | FLSLI PHAIN AVSAL ANHG-NH2 |
The positively charged residues are shown in bold, the 15N-labeled sites are underlined, and the 3,3,3-2H3-labeled Ala residues are doubly underlined.
When the primary and secondary structures of the three PS investigated here are compared to each other the cationic character of residues 17 and 18 is quite different where lysine-histidine and histidine-histidine correlates well with an extension of the helix segment near the C-terminal region of phylloseptin-1 (PS-1) and PS-2, respectively (18). In contrast, in the presence of asparagine-histidine at positions 17 and 18 the helix of PS-3 extends only from residue 5 to 15, not involving the very C-terminus. Overall, the structures of these PS are characterized by degrees of helicity in membrane mimetic environments of 74% for PS-1, 79% for PS-2, and 58% for PS-3, an observation that has been explained with the stabilizing interactions between the cationic residues and the helical dipole moment as well as possible helix capping and aromatic interactions (18). Additional differences arise as the hydrophobicity of the carboxy-terminal nonpolar amino acids of PS-2 are somewhat different from those of PS-1, -and -3 (Table 1).
Multidimensional solution-state NMR structures have been obtained in trifluoroethanol/water and the helix content that is found in this hydrophobic environment agrees well with the circular dichroism spectroscopic secondary structure analysis in the presence of phospholipid vesicles (18). The data provide a useful prerequisite to investigate in detail the structure, dynamics, and topology of these closely related sequences in phospholipid bilayers (22). Thereby, the phylloseptin sequences provide a rather unique possibility to investigate if and how the subtle structural differences between PS-1, PS-2, and PS-3 have an effect on the interactions and membrane alignment of the helical domains. In particular, the PS provide a test case on how the effect of changes in the cationic properties is translated in differences in peptide topology. Therefore, the focus of this work is on exploring the solid-state NMR approaches for an accurate analysis of topological differences and the composition of the membranes was chosen to provide a stable and reproducible environment. Indeed 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and POPC/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) membranes have proven to provide valuable information on the mechanisms of action of several cationic linear antimicrobial peptides of similar size (23) even though interesting structure-function correlations have been revealed in the pioneering work by the Straus team when other membrane compositions were evaluated in a systematic manner by comparing biophysical experiments and antimicrobial assays (24–26).
Solid-state NMR spectroscopy has the proven potential to investigate polypeptides associated with extended lipid bilayers with high accuracy (27–29). In particular, static oriented solid-state NMR provides valuable information about the membrane interactions, structure, topology, and dynamics of polypeptides including antimicrobial peptides (22,30–34). In static solids or in semisolid samples the spectral features such as the chemical shift frequency are strongly dependent on the molecular alignments relative to the magnetic field of the spectrometer (Bo) and this can be used to obtain valuable information about the orientation of bonds and molecules relative to the membrane normal. Whereas a few angular constraints are sufficient to characterize the membrane topology of known structures in considerable detail (22), or to follow changes in topology at high precision, the high-resolution structure of membrane-associated polypeptides can be derived from the analysis of many labeled sites (35).
Whereas the 15N chemical shift or the 15N-1H dipolar interaction alone suffices to determine the approximate alignment of α-helical polypeptides relative to the membrane surface (36,37) additional angular information can be obtained from deuterium solid-state NMR spectroscopy of 2H3-labeled alanines (29). Together the two measurements provide highly accurate tilt and rotational pitch information, which reveals minor changes in alignment (±1°) or can be used to characterize the mosaicity of sample orientation (38). Furthermore, at sample alignments with the normal perpendicular to Bo, information about the rotational diffusion of the labeled sites and thereby oligomerization of the peptides is obtained (39). Here, we use oriented 15N and 2H solid-state NMR spectroscopy to investigate in detail the topology of the helical domains of three closely related PS in phospholipid bilayers. The investigations thereby also test the precision of some of the relatively novel, to our knowledge, approaches in the field.
Materials and Methods
Materials
POPC and POPS were purchased from Avanti Polar Lipids (Birmingham, AL). Labeled and unlabeled amino acid derivatives for peptide synthesis were from Cambridge Isotope Laboratories (Andover, MA) and NovaBiochem-Merck (Darmstadt, Germany), respectively. TFA, triisopropylsilane, trifluoroethanol, chloroform and dichloromethane were obtained from Sigma-Aldrich (Saint Louis, MO), N,N′-diisopropylcarbodiimide from Fluka (Steinheim, Germany), 1-hydroxybenzotriazole from NovaBiochem-Merck (Darmstadt, Germany), N,N-dimethylformamide and diisopropyl ether from Vetec (Duque de Caxias, Brazil), acetonitrile from JT Baker (Center Valley, PA). If not mentioned otherwise analytical grade solvents were used.
Peptide synthesis
The PS, with amidated C-terminus, were obtained by solid-phase peptide synthesis on a Rink amide resin (NovaBiochem-Merck) (0.61 mmol.g−1) by using the Fmoc/t-butyl strategy (40). Couplings were performed with N,N′-diisopropylcarbodiimide (Fluka)/1-hydroxybenzotriazole (NovaBiochem-Merck) in N,N-dimethylformamide (Vetec, PA) for 60–120 min. Cleavage and final deprotection were conducted with trifluoroacetic acid (TFA, Sigma-Aldrich):triisopropylsilane (Sigma-Aldrich):water (95:2.5:2.5, v:v:v) for 120 min at room temperature. The crude peptides were precipitated with diisopropyl ether (Vetec, PA), extracted with water, and lyophilized. PS-1 was prepared twice to obtain a different labeling scheme (Table 1). 3,3,3-2H3 Ala residues were incorporated into the peptide sequences at positions 8 or 11 as indicated in Table 1. 15N-labeled amino acid residues were incorporated at Ala-14 for PS-1, Leu-15 for PS-2, and Val-12 for PS-3 (see Table 1).
Peptide purification
Synthetic peptides were purified by reversed phase-high performance liquid chromatography (Varian Pro Star 210, Walnut Creek, CA) using a semipreparative C18 column (Vydac 250 × 10 mm, 5 μm, Albany, OR), high performance liquid chromatography grade solvents and a gradient from 5% to 95% of acetonitrile (JT Baker) containing TFA (Sigma-Aldrich) 0.1% (solvent B). Solvent A was ultrapure H2O (Mili-Q Plus, Millipore, Billerica, MA) containing TFA 0.1%. The identities of each of the purified peptides have been confirmed by electrospray (ESI) and/or matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (ESI-Q-ToF Micro Micromass, Manchester, UK and MALDI-ToF/ToFMS, Bruker Daltonics, Billerica, MA). The experimentally observed and theoretical m/z values of [M+H]+ are, respectively: 2019.53 and 2019.137 for PS-1; 2119.72 and 2119.168 for PS-2; 1948.44 and 1948.063 for PS-3.
Solid-state NMR sample preparations
200 mg of POPC and the respective amounts of peptides were dissolved in trifluoroethanol (TFE; Sigma-Aldrich) at a peptide/lipid ratio of 1/99 mol/mol. The resulting solutions were mixed with naphthalene (Sigma-Aldrich) dissolved in chloroform (Sigma-Aldrich) respecting a 1:1 molar ratio of naphthalene: (lipid and peptide) as described in (41). The solvent volume was reduced by exposure to a nitrogen stream and the viscous mixtures spread onto 27 ultrathin coverglasses (9 × 22 mm2, Marienfeld, Lauda-Königshofen, Germany). The samples on glass plates were first dried in air and thereafter in high vacuum for 4 days. Thereafter, the membranes were equilibrated at 93% relative humidity for 6 days, the glass plates stacked on top of each other, and the samples sealed with Teflon tape and plastic wrappings (42). An additional sample composed of 1 mol % PS-2 in POPC/POPS (3:1) was prepared in a similar fashion, but POPS was dissolved in dichloromethane (Sigma Aldrich) before mixing with the peptide. Using this approach the pH of the samples was around 5.5. Notably, control experiments on PS-2 at higher pH did not show topological differences.
Solid-state NMR measurements
Proton-decoupled 15N solid-state NMR spectra were acquired on a Bruker AMX400 wide-bore NMR spectrometer using a commercial double-resonance solid-state NMR probe modified with flattened coils of inner dimensions 15 × 4 × 9 mm3 (42,43). Measurements were carried out at two different sample orientations, either with the membrane normal parallel or perpendicular to the magnetic field direction (tilted coil). A cross-polarization sequence with adiabatic shapes on both channels (44) was applied with the following typical acquisition parameters: 8 μs 90° pulse width, 700 μs contact time, 3.5 s recycle delay, 512 data points, 18,000 number of acquisitions (30,000 for tilted coil), and 33 kHz spectral width. NH4Cl was used as external reference (41.5 ppm).
Proton-decoupled 31P solid-state NMR spectra of the oriented phospholipid samples were recorded at 162.0 MHz on a Bruker AMX400 wide-bore NMR spectrometer using a commercial double-resonance solid-state NMR static probe. A Hahn echo pulse sequence (45) was employed for spectral acquisitions. The following spectral parameters were used: 75 kHz spectral width, 13.6 ms acquisition time, 2048 time domain data points, 2.5 μs 90° pulse width, 40 μs echo delay, 5 s recycle delay, 64 scans. H3PO4 at 85% was used as external reference (0 ppm).
Deuterium solid-state NMR spectra were recorded on a Bruker Avance DSX-500 wide-bore NMR spectrometer using a commercial triple resonance static probe. A quadrupolar echo pulse sequence was used (42,46) with the following parameters: 2H 50 kHz B1 field, 40 μs echo delay, 100 kHz spectral width, 4096 data points, 80,000 acquisitions, and a repetition time of 1 s. The spectra were referenced relative to 2H2O (0 ppm).
15N-1H polarization spin exchange at the magic angle (PISEMA) spectra (37,47) were acquired on a Bruker AMX400 wide-bore NMR spectrometer using a commercial double-resonance solid-state NMR probe modified with flattened coils (43) of inner dimensions 15 × 4 × 9 mm3. Measurements were carried out for sample orientations with the membrane normal parallel to the magnetic field direction. The following typical acquisition parameters have been used: F1 and F2 spectral widths of 22,502 and 8993 Hz, respectively, 8 μs 90° pulse width, 800 μs spin lock time, radiofrequency field strengths of 45 kHz, 3.5 s recycle delay, 512 data points, and 33 kHz spectral width. 26 t1 increments were collected with 12,288 transients of 512 points for each free induction decay. NH4Cl was used as external reference (41.5 ppm).
Simulations
At room temperature the methyl group of alanine residues exhibits fast rotational motions around the Cα-Cβ bond in such a way that the three 2H nuclei of the labeled alanine methyl groups are magnetically equivalent with the main axis of the resulting deuterium quadrupolar interaction tensor being aligned parallel to the Cα-Cβ bond.
When the peptides undergo fast rotational diffusion around the membrane normal the quadrupolar splitting ΔνQ is calculated as
| (1) |
where e2qQ/h is the quadrupolar coupling constant (48,49), Θ the angle between the Cα-Cβ bond and the membrane normal, and Ω the angle between the sample normal and the external magnetic field direction (49). From inspection of Eq. 1 it becomes obvious that sample orientations with Bo perpendicular to the membrane normal (Ω = 90°) provide quadrupolar splittings of half the size (−1/2) observed for sample orientations with Bo parallel to the membrane normal (Ω = 0°).
The 1H-15N dipolar splitting (DHN) obtained from PISEMA spectra as a scaled value, is given by the following equation:
| (2) |
where μoγ1γ2ħ/4π represents the 1H-15N dipolar coupling constant, rHN the respective H-N bond length, and Ψ the angle between the H-N bond vector and the external magnetic field direction (50). Similar to Eq. 1, Ω represents the angle between the sample normal and the external magnetic field direction.
The observed chemical shift value is given by Eq. 3, where σ11, σ22, and σ33 are the chemical shift main tensor elements (51) and ϕ and θ are the Euler angles representing respectively rotations around z and y′ axes (36).
| (3) |
To determine the topology of PS in lipid bilayers the solution NMR structures of PS-1, PS-2, and PS-3 (18) were used for the simulations of σzz, ΔνQ, and DHN. Each molecular geometry was previously aligned (52), in such a way that the 15N-labeled atom (N-Ala-14 for PS-1, N-Leu-15 for PS-2, and N-Val-12 for PS-3) was set at the origin and then a second atom was chosen to define the z axis, which would lie parallel to the helix main axis (the backbone N of His-7 for PS-1, N of Ala-8 for PS-2, and the carbonyl C of Leu-4 for PS-3). Finally, the internal coordinate system was defined by setting the carbonyl carbon of the peptide bond involving the 15N-labeled site (C-Ser-13 for PS-1, C-Thr-14 for PS-2, and C-Ala-11 for PS-3) at the xz-plane to make the y axis orthogonal to the plane of peptide bond carrying the 15N-labeled site.
After having defined the coordinate system the peptide structures were successively rotated (180 × 180 steps) around the helix main axis and the y axis. The origins of the topological analysis (tilt/rotation pitch angles 0/0) were chosen such that helices 5–15 of all three PS are aligned in a similar manner. The values of σzz, ΔνQ, and DHN were evaluated for each alignment as a function of the tilt and rotational pitch angles. σzz was evaluated from Eq. 3 corrected by two terms representing small angle (obtained directly from the geometrical data) rotations around the y and x axes to perfectly align the local coordinate system with the main axes system, because it is known that σ33 lies on the peptide bond plane covering an angle of ∼18° with the N-H bond vector ((51) and references cited therein). The coordinates of Cα and Cβ of the 2H-labeled sites were used to evaluate cos2Θ, and then, from Eq. 1, the values of ΔνQ were obtained. The term cos2Ψ has been evaluated from the coordinates of the H-N bond atoms, and then, from Eq. 2 the value of DHN was determined for the respective orientations. Both the geometry alignments and the simulations of σzz, ΔνQ, and DHN were performed using a FORTRAN program developed in one of our laboratories.
Results
PS were prepared by solid-phase peptide synthesis, labeled with stable 2H and 15N isotopes at selected positions, and reconstituted into mechanically oriented POPC phospholipid bilayers. Thereafter, the samples were inserted into the NMR spectrometer with the glass plate normal parallel to the magnetic field direction. The proton-decoupled 15N solid-state NMR spectra of (15N-Leu-15)-PS-2 and (15N-Val-12)-PS-3 exhibit resonances at 74 and 73 ppm, respectively (see Fig. 1, B and C). The narrow line shapes and the chemical shifts smaller than 100 ppm indicate that the labeled sites of the peptides exhibit a quite homogenous alignment of these helices approximately parallel to the bilayer surface (cf. below). The 15N resonance of (15N-Ala-14)-PS-1 exhibits a chemical shift of 104 ppm (Fig. 1 A), a value that is significantly higher than the ones observed for the other two peptides. This value suggests a somewhat more titled alignment when compared with PS-2 and PS-3. Finally, the resonance signal observed on the 15N spectrum of (15N-Leu-15)-PS-2 reconstituted into POPC/POPS (3:1) bilayers (see Fig. 1 G) presented within experimental error the same chemical shift as the one observed on the spectrum obtained for the peptide reconstituted into POPC bilayers (Fig. 1 B).
Figure 1.
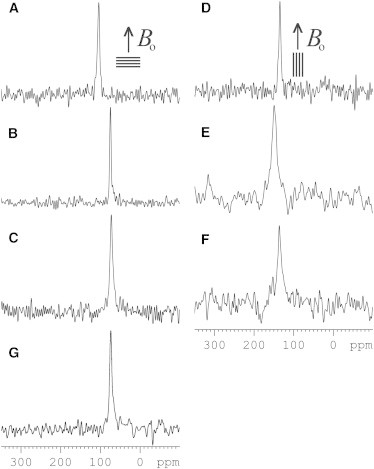
Proton-decoupled 15N solid-state NMR spectra of PS at peptide/lipid ratios of 1.0 mol % in uniaxially oriented lipid bilayers. The spectra are from (3,3,3-2H3-Ala-11, 15N-Ala-14)-PS-1 (A and D), (3,3,3-2H3-Ala-8, 15N-Leu-15)-PS-2 (B, E, and G), and (3,3,3-2H3-Ala-11, 15N-Val-12)-PS-3 (C and F). A–C and G were recorded at sample orientations of the membrane normal parallel to Bo and D–F at alignments of the membrane normal perpendicular to Bo. A–F represent spectra of peptides reconstituted into oriented POPC lipid bilayers, whereas G was obtained in the presence of POPC/POPS (3:1 mol/mole).
When 15N spectra are recorded for sample orientations with the membrane normal perpendicular to Bo, the 15N resonances are observed at 134 ppm, 149 ppm, and 136 ppm for (15N-Ala-14)-PS-1, (15N-Leu-15)-PS-2, and (15N-Val-12)-PS-3, respectively (Fig. 1, D–F). The observation of sharp resonances and the values of the experimental 15N chemical shifts indicate fast rotational motion of the helices around the membrane normal (39).
The 2H solid-state NMR spectra of PS peptides in POPC lipid bilayers are shown on Fig. 2 and Fig. 3, A and C. The (3,3,3-2H3-Ala-11)-PS-1, (3,3,3-2H3-Ala-8)-PS-2, and (3,3,3-2H3-Ala-11)-PS-3 sequences reconstituted into oriented and hydrated phospholipid bilayers each presents a signal splitting of ≤ 2.5 kHz from membrane-associated water molecules (2H at natural abundance) and a well-defined pair of resonances from the labeled peptide sites. The latter define quadrupolar splittings of 6 kHz (Fig. 2 A), 45 kHz (Fig. 3 A), and of ∼8.5 kHz (Fig. 2 C), respectively. To compare the effect of introducing negatively charged phospholipids PS-2 was also studied in POPC/POPS (3:1) bilayers (Fig. 3, B and D). When compared to (3,3,3-2H3-Ala-8)-PS-2 in POPC bilayers (Fig. 3 A) the lines exhibit a similar quadrupolar splitting (46 kHz), although each of the quadrupolar transitions exhibits a narrower line shape (Fig. 3 B). This observation suggests that the alignment of the peptide helix is more homogenous in POPC/POPS (1° mosaic spread) when compared to POPC bilayers (2° mosaic spread), and indicative of well-aligned helices in both environments (38).
Figure 2.
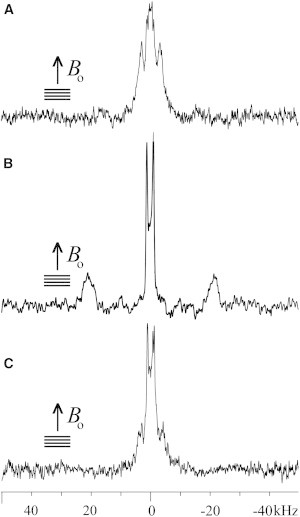
2H solid-state NMR spectra of PS-1 and -3 at 1.0 mol % in uniaxially oriented POPC lipid bilayers. The spectra are from (3,3,3-2H3-Ala-11, 15N-Ala-14)-PS-1 (A), (3,3,3-2H3-Ala-8, 15N-Ala-14)-PS-1 (B), and (3,3,3-2H3-Ala-11, 15N-Val-12)-PS-3 (C). The samples were oriented with the normal parallel to Bo.
Figure 3.
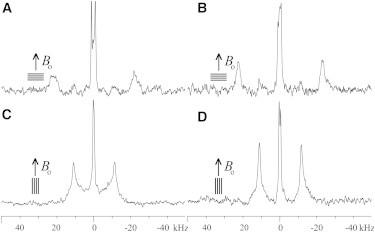
2H solid-state NMR spectra of (3,3,3-2H3-Ala-8, 15N-Leu-15)-PS-2 at 1.0 mol % reconstituted in uniaxially oriented bilayers of POPC (A and C) or POPC/POPS (3:1) (B and D). (A) and (B) were recorded for sample orientations with the glass plate normal parallel to Bo, (C) and (D) with sample orientations perpendicular to Bo.
Also visible in the PS-2 2H spectra are less intense deuterium intensities that are characterized by quadrupolar splittings of 22 kHz and 23 kHz in POPC and POPC/POPS (3:1) bilayers, respectively (Fig. 3, A and B). These values are within experimental error the same ones obtained for sample orientations with the membrane normal perpendicular to Bo (cf. below) and correspond to the maximal peak intensities that would be observed for nonoriented membranes, or for peptides associated with supramolecular assemblies where parts of the membrane are aligned perpendicular to the sample normal (53).
When the PS-2 samples shown in Fig. 3, A and B, are tilted by 90° the spectra shown in Fig. 3, C and D, are obtained. The observed splittings (22.4 kHz and 22.7 kHz) represent within experimental error half of the values obtained for sample orientations with the membrane normal parallel to Bo (Fig. 3, C and D), a result that is in good agreement with Eq. 1. These quadrupolar splittings as well as the well-defined 2H line shapes confirm once more the fast rotational motion around the membrane normal for PS peptides on the 10−5 s timescale of the methyl group deuterium quadrupolar interaction (39).
In a next experiment, we investigated the effects of the peptides on the macroscopic alignment and phase of the phospholipid membranes and the lipid head group conformation (26,49,54). To this end, the proton-decoupled 31P solid-state NMR spectra of the same oriented phospholipid-peptide samples were recorded. The 31P NMR spectra of all samples show a predominant peak close to 30 ppm (Fig. 4) characteristic for liquid crystalline phase POPC bilayers where the lipid long axes are aligned parallel to the magnetic field direction. Small 31P resonance intensities are observed reaching from 30 to −15 ppm when 200 mg phospholipid in the presence of 1 mol % PS-2 was spread between 27 glass plates (Fig. 4 A). These contributions are indicative of misalignment of bilayers locally (55) or globally or conformational changes at the level of the lipid headgroup (49,54). When the amount of sample per glass plate is reduced fivefold these intensities are reduced and disappear in the noise (Fig. 4 B) suggesting that membrane disturbances induced by the peptide propagate from one bilayer to the next and are thereby enhanced in the thicker stacks (56).
Figure 4.
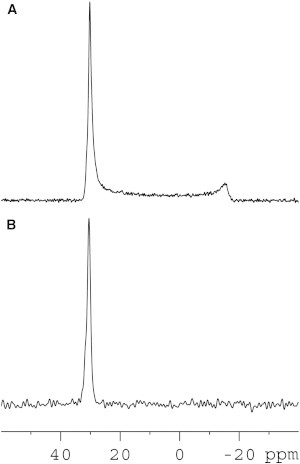
Proton-decoupled 31P solid-state NMR spectra of POPC lipid bilayers containing PS-2 at 1.0 mol %. (A) 200 mg of lipid spread on 27 glass plates and (B) 10 mg of lipid spread on 7 glass plates.
Although it is not possible to determine precisely the peptide orientation from a single 15N chemical shift or the 2H quadrupolar splitting alone, the two parameters furnish highly complementary topological restraints that can be used to obtain a more accurate definition of the orientation of the peptides in bilayers (49). This is shown in Fig. 5 where the solution NMR structures (18) were used to evaluate for each sequence the angular restraints that are obtained from the 15N chemical shift and the 2H quadrupolar splitting. The traces in black represent orientations in which the simulated 15N chemical shift agrees with the experimental data (uncertainty of ±2 ppm) and the red lines trace the angular pairs where the 2H quadrupolar spectra shown in Fig. 2, A and C, and Fig, 3, A and B, agree with experimental values (within ±1 kHz). Only a small number of topologies are in agreement with both measurements and are represented by the intersections highlighted by blue circles. Nine possible angular regions are obtained for PS-1, whereas four possibilities remain for PS-2, and six for PS-3. Next to each contour plot are shown the respective orientations of the NMR structures (18) relative to the bilayers for PS-1, PS-2, and PS-3, respectively (Fig. 5, D–F). The hydrophilic residues are shown in green and the hydrophobic ones in blue.
Figure 5.
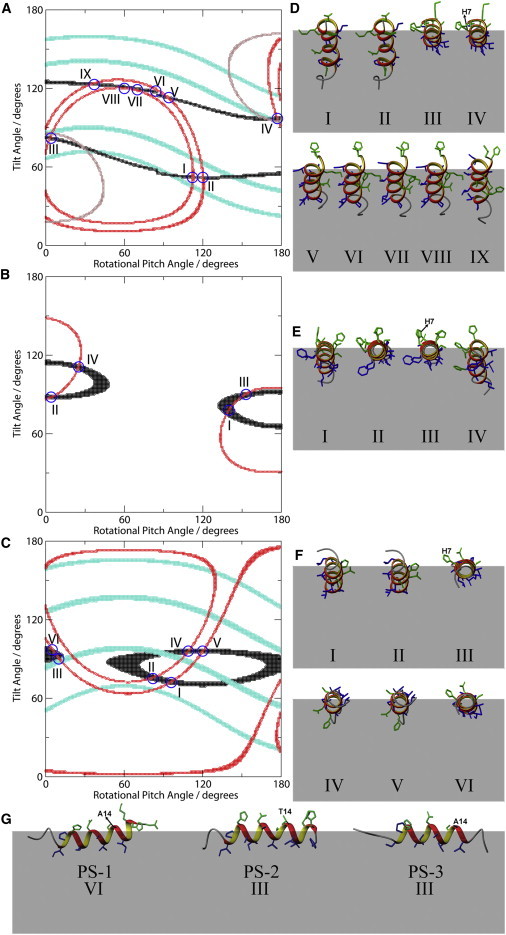
Alignments of PS-1 (A), PS-2 (B), and PS-3 (C) in lipid bilayers shown as functions of the tilt and rotational pitch angles. Black points represent orientations that are in agreement within ±2.0 ppm with the experimental 15N chemical shift and red points represent alignments that agree within ±1.0 kHz the experimental deuterium quadrupolar splitting. The intersection shown by blue circles indicate orientations that simultaneously agree with both 15N chemical shift and deuterium quadrupolar splitting. In panel A are also included topologies obtained from measuring in addition the 2H quadrupolar splitting of (3,3,3-2H3-Ala-8)-PS-1 (43 kHz; brown traces). In panels A and C, points in turquoise represent orientations that agree with 1H-15N dipolar splittings obtained from PISEMA spectra of (3,3,3-2H3-Ala-11, 15N-Ala-14)-PS-1 and (3,3,3-2H3-Ala-11, 15N-Val-12)-PS-3, respectively. Next to each contour plot are shown the peptide structures (PDB 2JQ0, 2JPY, and 2JQ1 (18);) at orientations that represent the intersection points in the contour plot of PS-1 (D), PS-2 (E), and PS-3 (F), with the N-termini shown in the back. Rotated views of the most stable alignments of all three peptides are shown in panel G. The N-terminus of the peptides is on the left, the C-terminus on the right. Hydrophilic residues are shown in green, whereas hydrophobic residues in blue. The gray areas represent approximately the hydrophobic thickness of a monolayer. The His-7 residues are highlighted on topologies IV, III, and III, presented on panels D, E, and F, respectively. It should be noted that the solid-state NMR data do not reveal the peptide penetration depth, which has been estimated on energetic grounds for the representations shown here.
Although energetic considerations could help in resolving this ambiguity (57,58) we decided to perform additional experiments that further restrict the alignment of PS-1. First, we recorded a two-dimensional separated-field experiment (37) from the same oriented sample used to obtain the 15N spectrum shown in Fig. 1, A and D. In addition to the 15N chemical shift this two-dimensional solid-state NMR experiment also reveals a 1H-15N dipolar splitting of 7.4 ± 1 kHz (Fig. 6 A), which results in the orientational constraints shown in turquoise in Fig. 5 A. The orientational constraints of the 15N chemical shift and the 1H-15N dipolar coupling resemble each other in that they both restrict the tilt angle but not the angular pitch (29). Nevertheless, the intersection of the turquoise (1H-15N dipolar splitting), black (15N chemical shift), and red traces (2H quadrupolar splitting) indicates that the topology IV fits all three NMR measurements, with I, II, and III being close to solutions that could come into play when conformational variations of the PS-1 structure (18), other sources of systematic error (59) such as the uncertainty in defining the solid-state NMR interactions tensors (51) or the dynamic properties of the peptide are to be considered (22).
Figure 6.
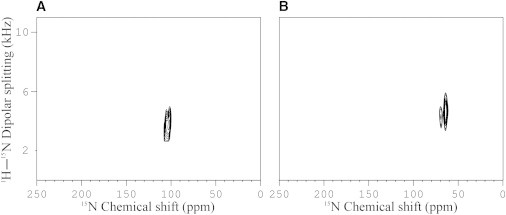
15N-1H separated local field spectra of (A) (3,3,3-2H3-Ala-8, 15N-Ala-14)-PS-1 and (B) (3,3,3-2H3-Ala-11, 15N-Val-12)-PS-3 at peptide/lipid ratios of 1 mol % in uniaxially oriented POPC lipid bilayers. The spectra were recorded at sample orientations with the normal parallel to Bo. Only the top half of the symmetric spectra are shown.
We therefore simulated the conformational restraints from other isotopic labeling positions that could be used with the goal to provide optimal complementarity to the existing data set (29). These calculations predict that, provided that the peptide adopts an approximately in-planar alignment, most of the ambiguity would be removed with a 2H3-label of the alanine-8 position of PS-1 (29). Therefore, the peptide (3,3,3-2H3-Ala-8, 15N-Ala-14)-PS-1 was prepared, reconstituted into POPC-oriented bilayers, and the 2H solid-state NMR spectrum obtained (Fig. 2 B). The 2H quadrupolar splitting of 43 kHz results in the topological constraints shown in brown in Fig. 5 A. The data confirm alignment IV with tilt and rotational pitch angles 98°/177° and clearly exclude peptide orientations such as I or II. Although alignment IV is the only topology that fits all four measurements, it is almost identical with alignment III in particular when experimental and systematic errors during the topological analysis are taken into consideration (59).
In a next step, we considered the topological restraints that derive from the 1H-15N dipolar coupling of 10.6 kHz observed for (15N-Val-12)-PS-3 (Fig. 6 B). These are shown in turquoise in Fig. 5 C and the intersection from three NMR parameters leaves only topology III for PS-3 with tilt and rotational pitch angles 90°/10°. Finally, the alignment III of PS-2 was restricted on energetic grounds as this is the topology that best matches the amphipathic character of the helical conformation with the bilayer interface. Notably, this topology is between the alignments of the closely related sequences PS-1 and PS-3 and thereby analogous to their membrane alignment. Therefore, the tilt and rotational pitch angles are 98°/177° (PS-1, IV), 90°/153° (PS-2, III), and 90°/10° (PS-3, III). When Fig. 5, E and F, are compared to each other it becomes obvious that although PS-2 and PS-3 align parallel to the surface their azimuthal alignment is different by 37°. To better follow the different azimuthal angles the His-7 residues are highlighted in the illustrations of topologies IV, III, and III for PS-1, -2, and -3, respectively (Fig. 5, D–F).
Interestingly, the 2H solid-state NMR spectra of (3,3,3-2H3-Ala-8)-PS-2 in POPC and POPC/POPS bilayers exhibit the same quadrupolar splitting within experimental error, although the method is quite sensitive to small changes in topology (29,60). However, the small difference in line width seem to suggest that the peptide exhibits less conformational and/or topological variation in the presence of the anionic lipid suggesting that electrostatic interactions stabilize the protonation state of the three histidines and/or anchor the helix in a more stable manner.
When the contour plots resulting from the 2H quadrupolar splitting are investigated in more detail (Fig. 5, A–C), it becomes obvious that the quadrupolar splittings of (2H3-Ala-8) in PS-1 or PS-2 (43 kHz and 46 kHz; brown trace in Fig. 5 A and red trace in Fig. 5 B, respectively) restrict the possible alignments more than the splittings obtained for (2H3-Ala-11) in PS-1 or PS-3 (6 kHz and 8.5 kHz, red traces in Fig. 5, A and C, respectively). As a consequence, due to the high selectivity of the 2H3-Ala-8 label the two 2H and one 15N chemical shift measurements for PS-1 (brown, red, and black traces in Fig. 5 A) are sufficient to unambiguously determine the topology of this peptide without the need for additional dipolar coupling information (turquoise trace).
Indeed, the simulations summarized in Fig. 7 show that the number of tilt/rotational pitch angular pairs that agree with a given quadrupolar splitting is maximal when ΔνQ approaches (3/2)(e2qQ)/h)/2, whereas for higher splittings the number of topologies is significantly reduced. Furthermore, a relatively large number of angular pairs is also obtained for splittings < (3/2)(e2qQ)/h)/2. This feature can be useful in situations where the peptide orientation is already known to the first approximation but needs to be refined and/or confirmed experimentally as was the approach chosen here for PS-1 (Fig. 5 A). When it is feasible to label with 2H3 an alanine exhibiting a predictably large quadrupolar splitting highly selective orientational restraints are obtained.
Figure 7.
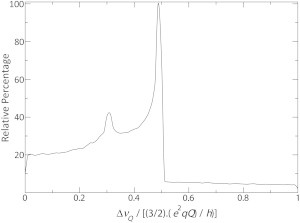
Relative percentage of the maximum number of tilt/rotational pitch angular pairs for PS-1 that agree with a given quadrupolar splitting (±1 kHz). The graph was obtained from a simulation considering an increment of 1° for both tilt and rotational pitch angles.
For instance, PS-1 carries four alanine residues in its primary sequence and Table 2 shows the number of solutions (representing selectivity) for each 3,3,3-2H3-Ala residue (where a range of ΔνQ ±1 kHz has been taken into account) as well as the expected quadrupolar splittings for the alignment IV shown in Fig. 5, A, D, and G. Clearly, a higher selectivity is obtained from labeling Ala-8 rather than Ala-11. Notably, without prior knowledge of the peptide orientation, labeling the Ala-14 or the Ala-16 sites with 3,3,3-2H3 would have produced even smaller selectivity. As a consequence, combining the restraints obtained from (15N-Ala14)-PS-1 (σzz) and ΔνQ observed for (3,3,3-2H3-Ala-11)-PS-1 results in nine intersection points (topologies I-IX), whereas only two possible alignments (IV and the topology represented by the tilt/rotational pitch angles 75°/27°) are obtained from the intersection of σzz and ΔνQ observed for 3,3,3-2H3-Ala-8-PS-1. Combining the two quadrupolar splittings, an approach that is used in the GALA method (61), results in four intersections of the brown and the red traces (Fig. 5 A).
Table 2.
Expected 2H quadrupolar splittings for different alanine-labeled sites of PS-1 and the number of respective orientations that result from simulations of topology IV (Fig. 5D)
| Amino-acid residue | Expected |ΔνQ|/kHz | Number of orientations |
|---|---|---|
| Ala-8 | 43 | 246 |
| Ala-11 | 6 | 915 |
| Ala-14 | 38 | 2503 |
| Ala-16 | 36 | 1853 |
The number of orientations are obtained from a simulation considering an increment of 1° for both tilt and rotational pitch angles. A range of ±1 kHz was taken into account for the deuterium quadrupolar splitting ΔνQ.
Discussion
Here, we investigated the membrane topology of three phylloseptin antimicrobial peptides with closely related sequences and structures (18). In membrane mimetic environments the three peptides have been found to adopt helical conformations encompassing residues 5–18 (PS-1), 5–19 (PS-2), and 5–15 (PS-3). The differences in three-dimensional structures correlate with the sequence variations within residues 14–19 (Table 1), and in particular their cationic nature of residues 17 and 18, which decreases in the sequence PS-1 (HK) > PS-2 (HH) > PS-3 (NH), where the chemical shifts are suggestive that the histidine side chains were partially charged (18).
The solid-state NMR investigations presented in this work indicate that residues 8, 11, and 14 of PS-1, 8 and 15 of PS-2, and 11 and 12 of PS-3 are located within folded regions as otherwise the chemical shift, dipolar, and quadrupolar couplings would occur close to their isotropic values (∼120 ppm for the 15N chemical shifts (51), and 0 kHz for the couplings). The well-oriented spectra obtained in lipid bilayers (Figs. 1–3) are thereby in agreement with the outlines of the helical domains observed in TFE/water environments (18). The tilt angles observed for PS-2 and PS-3 correspond to perfect alignments along the membrane surface, in agreement with the amphipathic nature of the helical domains. For example, among the four possibilities obtained for PS-2, only orientation III avoids the insertion of polar residues inside the hydrophobic membrane interior (Fig. 5 E). In particular, the orientations I and IV (representing tilt angles of 78° and 111°, respectively) are characterized by the deep insertion of polar residues, including His-7 into the hydrophobic part of the membrane, a thermodynamically unfavorable situation.
When compared to the other two PS, PS-1 adopts a slightly tilted alignment with the N-terminus being located below the C-terminus. This deviation from a perfect in-plane alignment is reflected in the increased 15N chemical shift value (104 ppm; Fig. 1 A) and confirmed by the detailed topological analysis (Fig. 5, A and D, IV). Fig. 5 G indicates that by adopting a more tilted topology the C-terminus of PS-1 including the charged lysine-17 and the hydrophilic asparagine-19 residues are moved further away from the membrane interior. Although snorkeling of the latter probably avoids direct exposure to the hydrophobic environment, moving the cationic charges away from the membrane reduces the passage of electrical field lines through the hydrophobic membrane interior (thereby reducing image charges; (62)).
Similar considerations apply to the differences in azimuthal angles, which assure that the cationic charge of lysine-17 of PS-1 is moved the furthest away from the membrane interface as possible (Fig. 5 D, IV). For the histidine residues on the other hand the possibility exists that their pK values shift in the membrane proximal regions and that these side chains are in average only partially charged or not charged at all (63,64). Interestingly, the azimuthal angle of PS-2 is such that the histidines 7, 17, and 18 are about equidistant from the membrane surface (Fig. 5 E, III). In contrast, the topology of residues 16–19 of PS-3 could not be determined from the experiments described here as these amino acids may not be part of the helical domain (18). In addition, for this peptide the His-7 residue is localized in the aqueous environment (Fig. 5 F, III). Furthermore, the peptide topologies shown in Fig. 5 assure that threonine-14 of PS-2 is well exposed to the aqueous environment, whereas the corresponding alanines of PS-1 and PS-3 are in contact with the membrane interface (Fig. 5 G). Taken together, the data reveal that the detailed secondary structures and topologies of these peptides are tightly interlinked and modulated not only by interactions between the cationic side chains and the helix dipole (18), but also the hydrophobicity and hydrophobic moment of the amphipathic helices and the positioning of individual side chains relative to the membrane interface (cf. above; Fig. 5).
Interestingly, in the case of aureins, a family of antimicrobial peptides isolated from Australian frogs, the propensity to form tilted/inserted alignments has been correlated with differences in their antibacterial activities (25,26). When the three PS peptides are compared to each other the differences in minimal inhibitory concentrations are relatively small and which of the three sequences is the most potent analog depends on the bacterial strains investigated. To test if the observed differences in membrane interactions of the three PS peptides correlate with their antimicrobial activities it would be interesting to compare their detailed topology in membranes that correspond closely to the different bacterial membrane compositions, similar to the biophysical investigations that have been presented previously for aureins (24–26,55,65).
The helical structure and the in-planar alignment of the three PS agree with previous observations on other linear cationic antimicrobial peptides, although they carry less net positive charge than, e.g., magainins (23), suggesting that similar models that have been developed previously also apply for PS. In short, these models suggest that amphipathic peptides of this class partition into the membrane interface thereby disordering the packing of the fatty acyl chains. Depending on the lipid composition, the peptide concentrations and environmental factors the interactions between the lipids and the peptides can result in many different local and global membrane morphologies including the carpet-, torroidal pore-, and other models, that may all be needed to describe the peptide-membrane interactions but are present in different regions of a phase diagram (66,67). The resulting membrane destabilization result in the killing of sensitive cells and organisms (8) or the entry of peptides into the cells where they can reach internal targets (15). In this work, this global view is refined in revealing how relatively small changes in the amino acid compositions result in subtle topological and conformational alterations of the peptides when interacting with membranes.
Acknowledgments
J.M.R., R.M.V., and D.P.V. acknowledge grants from CNPq. We thank CNPq, FAPEMIG, Vaincre la Mucoviscidose, the Agence Nationale de la Recherche (projects Membrane DNP, TRANSPEP, and LabEx Chemistry of Complex Systems), the University of Strasbourg, the CNRS, the Région Alsace and the RTRA International Center of Frontier Research in Chemistry, CAPES (project 63-2010), RQ-MG and PRPq-UFMG for financial support. We acknowledge the support by the Brazilian–French program CAPES-COFECUB (487/05) who promoted this collaboration by initially funding several transatlantic visits.
References
- 1.Hancock R.E., Sahl H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 2.Brotman Y., Makovitzki A., Viterbo A. Synthetic ultrashort cationic lipopeptides induce systemic plant defense responses against bacterial and fungal pathogens. Appl. Environ. Microbiol. 2009;75:5373–5379. doi: 10.1128/AEM.00724-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giuliani A., Rinaldi A.C. Beyond natural antimicrobial peptides: multimeric peptides and other peptidomimetic approaches. Cell. Mol. Life Sci. 2011;68:2255–2266. doi: 10.1007/s00018-011-0717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leite J.R., Silva L.P., Bloch C., Jr. Phylloseptins: a novel class of anti-bacterial and anti-protozoan peptides from the Phyllomedusa genus. Peptides. 2005;26:565–573. doi: 10.1016/j.peptides.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Brand G.D., Leite J.R., Bloch C., Jr. Novel dermaseptins from Phyllomedusa hypochondrialis (Amphibia) Biochem. Biophys. Res. Commun. 2006;347:739–746. doi: 10.1016/j.bbrc.2006.06.168. [DOI] [PubMed] [Google Scholar]
- 6.Pukala T.L., Bowie J.H., Tyler M.J. Host-defence peptides from the glandular secretions of amphibians: structure and activity. Nat. Prod. Rep. 2006;23:368–393. doi: 10.1039/b512118n. [DOI] [PubMed] [Google Scholar]
- 7.Yang D., Chertov O., Oppenheim J.J. The role of mammalian antimicrobial peptides and proteins in awakening of innate host defenses and adaptive immunity. Cell. Mol. Life Sci. 2001;58:978–989. doi: 10.1007/PL00000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 9.Boman H.G. Antibacterial peptides: basic facts and emerging concepts. J. Intern. Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]
- 10.Violette A., Fournel S., Guichard G. Mimicking helical antibacterial peptides with nonpeptidic folding oligomers. Chem. Biol. 2006;13:531–538. doi: 10.1016/j.chembiol.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Mason A.J., Moussaoui W., Bechinger B. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine-rich amphipathic cationic peptides. J. Biol. Chem. 2009;284:119–133. doi: 10.1074/jbc.M806201200. [DOI] [PubMed] [Google Scholar]
- 12.Wang G., Li X., Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009;37:D933–D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bechinger B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1999;1462:157–183. doi: 10.1016/s0005-2736(99)00205-9. [DOI] [PubMed] [Google Scholar]
- 14.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 15.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 16.Hwang P.M., Vogel H.J. Structure-function relationships of antimicrobial peptides. Biochem. Cell Biol. 1998;76:235–246. doi: 10.1139/bcb-76-2-3-235. [DOI] [PubMed] [Google Scholar]
- 17.Mason A.J., Gasnier C., Bechinger B. Enhanced membrane disruption and antibiotic action against pathogenic bacteria by designed histidine-rich peptides at acidic pH. Antimicrob. Agents Chemother. 2006;50:3305–3311. doi: 10.1128/AAC.00490-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Resende J.M., Moraes C.M., Bechinger B. Solution NMR structures of the antimicrobial peptides phylloseptin-1, -2, and -3 and biological activity: the role of charges and hydrogen bonding interactions in stabilizing helix conformations. Peptides. 2008;29:1633–1644. doi: 10.1016/j.peptides.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 19.Giovannini M.G., Poulter L., Williams D.H. Biosynthesis and degradation of peptides derived from Xenopus laevis prohormones. Biochem. J. 1987;243:113–120. doi: 10.1042/bj2430113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaz Gomes A., de Waal A., Westerhoff H.V. Electric potentiation, cooperativity, and synergism of magainin peptides in protein-free liposomes. Biochemistry. 1993;32:5365–5372. doi: 10.1021/bi00071a011. [DOI] [PubMed] [Google Scholar]
- 21.Salnikov E.S., Bechinger B. Lipid-controlled peptide topology and interactions in bilayers: structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys. J. 2011;100:1473–1480. doi: 10.1016/j.bpj.2011.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michalek M., Salnikov E., Bechinger B. Structure and topology of the huntingtin 1-17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 2013;105:699–710. doi: 10.1016/j.bpj.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bechinger B. Insights into the mechanisms of action of host defence peptides from biophysical and structural investigations. J. Pept. Sci. 2011;17:306–314. doi: 10.1002/psc.1343. [DOI] [PubMed] [Google Scholar]
- 24.Pan Y.L., Cheng J.T.J., Straus S.K. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2007;92:2854–2864. doi: 10.1529/biophysj.106.097238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng J.T.J., Hale J.D., Straus S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2009;96:552–565. doi: 10.1016/j.bpj.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng J.T.J., Hale J.D., Straus S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta. 2011;1808:622–633. doi: 10.1016/j.bbamem.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 27.Hong M., Su Y. Structure and dynamics of cationic membrane peptides and proteins: insights from solid-state NMR. Protein Sci. 2011;20:641–655. doi: 10.1002/pro.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weingarth M., Baldus M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc. Chem. Res. 2013;46:2037–2046. doi: 10.1021/ar300316e. [DOI] [PubMed] [Google Scholar]
- 29.Bechinger B., Resende J.M., Aisenbrey C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: established concepts and novel developments. Biophys. Chem. 2011;153:115–125. doi: 10.1016/j.bpc.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Verardi R., Traaseth N.J., Scaloni A. Probing membrane topology of the antimicrobial peptide distinctin by solid-state NMR spectroscopy in zwitterionic and charged lipid bilayers. Biochim. Biophys. Acta. 2011;1808:34–40. doi: 10.1016/j.bbamem.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thennarasu S., Tan A., Ramamoorthy A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010;98:248–257. doi: 10.1016/j.bpj.2009.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouellet M., Voyer N., Auger M. Membrane interactions and dynamics of a 21-mer cytotoxic peptide: a solid-state NMR study. Biochim. Biophys. Acta. 2010;1798:235–243. doi: 10.1016/j.bbamem.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez D.I., Sani M.A., Separovic F. Interactions of a synthetic Leu-Lys-rich antimicrobial peptide with phospholipid bilayers. Eur. Biophys. J. 2011;40:471–480. doi: 10.1007/s00249-010-0660-5. [DOI] [PubMed] [Google Scholar]
- 34.Perrin S., Jr., Tian B.S., Cotton M.L. High-Resolution Structures and Orientations of Antimicrobial Peptides Piscidin 1 and Piscidin 3 in Fluid Bilayers Reveal Tilting, Kinking, and Bilayer Immersion. J. Am. Chem. Soc. 2011;136:3491–3504. doi: 10.1021/ja411119m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cross T.A. Solid-state nuclear magnetic resonance characterization of gramicidin channel structure. Methods Enzymol. 1997;289:672–696. doi: 10.1016/s0076-6879(97)89070-2. [DOI] [PubMed] [Google Scholar]
- 36.Bechinger B., Sizun C. Alignment and structural analysis of membrane polypeptides by 15N and 31P solid-state NMR spectroscopy. Concepts Magn. Reson. 2003;18A:130–145. [Google Scholar]
- 37.Ramamoorthy A., Wei Y., Lee D. PISEMA solid-state NMR spectroscopy. Annu. Rep. NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 38.Aisenbrey C., Bechinger B. Tilt and rotational pitch angles of membrane-inserted polypeptides from combined 15N and 2H solid-state NMR spectroscopy. Biochemistry. 2004;43:10502–10512. doi: 10.1021/bi049409h. [DOI] [PubMed] [Google Scholar]
- 39.Aisenbrey C., Bechinger B. Investigations of polypeptide rotational diffusion in aligned membranes by 2H and 15N solid-state NMR spectroscopy. J. Am. Chem. Soc. 2004;126:16676–16683. doi: 10.1021/ja0468675. [DOI] [PubMed] [Google Scholar]
- 40.Chan W.C., White S.H. Oxford University Press; Oxford, UK: 2000. Fmoc Solid-Phase Peptide Synthesis: A Practical Approach. [Google Scholar]
- 41.Hallock K.J., Henzler Wildman K., Ramamoorthy A. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys. J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aisenbrey C., Bertani P., Bechinger B. Solid-state NMR investigations of membrane-associated antimicrobial peptides. In: Guiliani A., Rinaldi A.C., editors. Antimicrobial Peptides. Humana Press, Springer; NY: 2010. pp. 209–233. [DOI] [PubMed] [Google Scholar]
- 43.Bechinger B., Opella S.J. Flat-coil probe for NMR spectroscopy of oriented membrane samples. J. Magn. Reson. 1991;95:585–588. [Google Scholar]
- 44.Hediger S., Meier B.H., Ernst R.R. Adiabatic passage Hartmann-Hahn cross polarization in NMR under magic angle sample spinning. Chem. Phys. Lett. 1995;240:449–456. [Google Scholar]
- 45.Rance M., Byrd R.A. Obtaining high-fidelity spin-1/2 powder spectra in anisotropic media: phase-cycled Hahn echo spectroscopy. J. Magn. Reson. 1983;52:221–240. [Google Scholar]
- 46.Davis J.H., Jeffrey K.R., Higgs T.P. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 1976;42:390–394. [Google Scholar]
- 47.Aisenbrey C., Michalek M., Bechinger B. Solid-state NMR approaches to study protein structure and protein-lipid interactions. In: Kleinschmidt J.H., editor. Lipid-Protein Interactions: Methods and Protocols. Springer; New York: 2013. pp. 357–387. [DOI] [PubMed] [Google Scholar]
- 48.Seelig J. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 1977;10:353–418. doi: 10.1017/s0033583500002948. [DOI] [PubMed] [Google Scholar]
- 49.Bechinger B., Salnikov E.S. The membrane interactions of antimicrobial peptides revealed by solid-state NMR spectroscopy. Chem. Phys. Lipids. 2012;165:282–301. doi: 10.1016/j.chemphyslip.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 50.Straus S.K., Scott W.R., Watts A. Assessing the effects of time and spatial averaging in 15N chemical shift/15N-1H dipolar correlation solid state NMR experiments. J. Biomol. NMR. 2003;26:283–295. doi: 10.1023/a:1024098123386. [DOI] [PubMed] [Google Scholar]
- 51.Salnikov E., Bertani P., Bechinger B. Analysis of the amide (15)N chemical shift tensor of the C(alpha) tetrasubstituted constituent of membrane-active peptaibols, the alpha-aminoisobutyric acid residue, compared to those of di- and tri-substituted proteinogenic amino acid residues. J. Biomol. NMR. 2009;45:373–387. doi: 10.1007/s10858-009-9380-5. [DOI] [PubMed] [Google Scholar]
- 52.Goldstein H. Addison-Wesley; MA: 1959. Classical Mechanics, Reading. [Google Scholar]
- 53.Wi S., Kim C. Pore structure, thinning effect, and lateral diffusive dynamics of oriented lipid membranes interacting with antimicrobial peptide protegrin-1: 31P and 2H solid-state NMR study. J. Phys. Chem. B. 2008;112:11402–11414. doi: 10.1021/jp801825k. [DOI] [PubMed] [Google Scholar]
- 54.Scherer P.G., Seelig J. Electric charge effects on phospholipid headgroups. Phosphatidylcholine in mixtures with cationic and anionic amphiphiles. Biochemistry. 1989;28:7720–7728. doi: 10.1021/bi00445a030. [DOI] [PubMed] [Google Scholar]
- 55.Kim C., Spano J., Wi S. Evidence of pores and thinned lipid bilayers induced in oriented lipid membranes interacting with the antimicrobial peptides, magainin-2 and aurein-3.3. Biochim. Biophys. Acta. 2009;1788:1482–1496. doi: 10.1016/j.bbamem.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 56.Verly R.M., de Moraes C.M., Bechinger B. Structure and membrane interactions of the antibiotic peptide dermadistinctin K by multidimensional solution and oriented 15N and 31P solid-state NMR spectroscopy. Biophys. J. 2009;96:2194–2203. doi: 10.1016/j.bpj.2008.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aisenbrey C., Sizun C., Tampé R. Structure and dynamics of membrane-associated ICP47, a viral inhibitor of the MHC I antigen-processing machinery. J. Biol. Chem. 2006;281:30365–30372. doi: 10.1074/jbc.M603000200. [DOI] [PubMed] [Google Scholar]
- 58.Resende J.M., Moraes C.M., Bechinger B. Membrane structure and conformational changes of the antibiotic heterodimeric peptide distinctin by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2009;106:16639–16644. doi: 10.1073/pnas.0905069106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salnikov E., Aisenbrey C., Bechinger B. Investigations of the structure, topology and dynamics of membrane-associated polypeptides by solid-state NMR spectroscopy. In: Separovic F., Naito A., editors. New Developments in NMR: Advances in Biological Solid-State NMR, Proteins and Membrane-Active Peptides. Royal Society of Chemistry; 2014. pp. 214–234. [Google Scholar]
- 60.Bechinger B., Aisenbrey C., Bertani P. The alignment, structure and dynamics of membrane-associated polypeptides by solid-state NMR spectroscopy. Biochim. Biophys. Acta. 2004;1666:190–204. doi: 10.1016/j.bbamem.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 61.van der Wel P.C., Strandberg E., Koeppe R.E., 2nd Geometry and intrinsic tilt of a tryptophan-anchored transmembrane alpha-helix determined by (2)H NMR. Biophys. J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McLaughlin S. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 1989;18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- 63.White S.H., Wimley W.C. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 64.Bechinger B. Towards membrane protein design: pH-sensitive topology of histidine-containing polypeptides. J. Mol. Biol. 1996;263:768–775. doi: 10.1006/jmbi.1996.0614. [DOI] [PubMed] [Google Scholar]
- 65.Ambroggio E.E., Separovic F., Bagatolli L.A. Direct visualization of membrane leakage induced by the antibiotic peptides: maculatin, citropin, and aurein. Biophys. J. 2005;89:1874–1881. doi: 10.1529/biophysj.105.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bechinger B., Lohner K. Detergent-like action of linear cationic membrane-active antibiotic peptides. Biochim. Biophys. Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 67.Bechinger B. Rationalizing the membrane interactions of cationic amphipathic antimicrobial peptides by their molecular shape. Curr. Opin. Colloid Interface Sci. 2009;14:349–355. [Google Scholar]