

Abstract
Using Escherichia coli as a model organism, we studied how water is recruited by a bacterial swarm. A previous analysis of trajectories of small air bubbles revealed a stream of fluid flowing in a clockwise direction ahead of the swarm. A companion study suggested that water moves out of the agar into the swarm in a narrow region centered ∼30 μm from the leading edge of the swarm and then back into the agar (at a smaller rate) in a region centered ∼120 μm back from the leading edge. Presumably, these flows are driven by changes in osmolarity. Here, we utilized green/red fluorescent liposomes as reporters of osmolarity to verify this hypothesis. The stream of fluid that flows in front of the swarm contains osmolytes. Two distinct regions are observed inside the swarm near its leading edge: an outer high-osmolarity band (∼30 mOsm higher than the agar baseline) and an inner low-osmolarity band (isotonic or slightly hypotonic to the agar baseline). This profile supports the fluid-flow model derived from the drift of air bubbles and provides new (to our knowledge) insights into water maintenance in bacterial swarms. High osmotic pressure at the leading edge of the swarm extracts water from the underlying agar and promotes motility. The osmolyte is of high molecular weight and probably is lipopolysaccharide.
Introduction
When some bacteria grow in a rich medium on the surface of moist agar, cells elongate, multinucleate, grow flagella, and swarm across the surface in coordinated packs (1–3). Swarming promotes the invasiveness of bacterial pathogens as shown in a wide range of clinical isolates, including Pseudomonas aeruginosa, Proteus mirabilis, Bacillus cereus, and Salmonella enterica (4–7). In 1994, Harshey and Matsuyama (8) discovered that strains of Escherichia coli K-12 swarm on Eiken agar (from Japan) rather than on Difco agar, presumably because Eiken agar is more wettable. Many bacteria produce surfactants as they swarm, which influences the patterns of expansion (2,3). However, these surfactants are not essential; in particular, there is no indication that E. coli produces surfactants (9). The absence of surfactants simplifies efforts to establish the role played by osmotic flow in swarm expansion.
An E. coli swarm consists of an actively expanding rim of cells followed by a relatively inactive interior (10,11). The cells swim in a thin film of fluid that extends some 10–20 μm ahead of the leading edge of the swarm. Tracking of microbubbles prepared from the surfactant Span 83 indicated that this region of fluid streams clockwise in front of the swarm at a rate ∼3-fold greater than that of the swarm advance (12). This chiral flow is driven by the counterclockwise rotation of the flagella of cells stalled at the edge of the swarm (12,13). Patterns of flow of fluid within the swarm also were inferred from the motion of microbubbles. The data could be fit by a model in which a large amount of fluid moved from the agar into the swarm near its leading edge, while a smaller amount moved from the swarm back into the agar ∼100 μm farther behind (10). The bulk fluid in a swarm provides the environment for flagellar rotation and for transportation of nutrients and signaling molecules. The only source for this fluid is the underlying agar. An intriguing question is, how is water extracted from the agar? Presumably, this occurs because the osmolarity of the fluid within the swarm is higher than that within the agar.
In this work, we probed the change in osmolarity within E. coli swarms with osmolarity-sensitive liposomes, inspired by the work of Jayaraman et al. (14) (see below). First, we deposited liposomes in spots ∼450 μm in diameter and collected fluorescence signals from entire spots as swarms ran over them. To improve the spatial resolution, we made liposome pads that were five times larger in diameter than the spots, allowed the swarms to run over them, and then scanned the pads with an excitation beam only 20 μm in diameter. We found that the osmolarity rose abruptly near the leading edge of the swarm and then dropped to a level close to that of the virgin agar ∼100 μm behind. The intervening region is one of high cell density. Evidently, growth in this region generates a substantial concentration of soluble osmolytes, which draw fluid out of the underlying agar. As the swarm expands, some of these osmolytes diffuse into the agar, raising its osmolarity above the initial value. Cells following behind the band of high cell density move over the region that was formerly covered by these cells and experience an osmolarity inversion that draws some fluid back into the agar. Thus, one would expect to see the biphasic flow profile predicted by the fluid flow measurements. The excess fluid that remains in the swarm fuels its spreading.
Materials and Methods
Swarm plates
The swarm agar contained 1% Bacto peptone, 0.3% beef extract, and 0.5% NaCl (the swarm medium) and 0.48% Eiken agar. To test the influence of plate osmolarity on swarming velocity, 0.5–0.55% NaCl and 0.48–0.55% Eiken agar were used. The agar was autoclaved or melted in a microwave oven and cooled to ∼60°C, and then 0.5% filter-sterilized arabinose was added. Then 25 ml aliquots were pipetted into Petri plates (150 mm diameter × 15 mm deep). The plates were swirled gently to spread the agar over the entire plate, cooled for 15 min inside a Plexiglas box, and then inoculated.
Cell growth
Bacterial strains AW405 (15) and HCB1668 (16) were grown under conditions described previously (10). Both strains were used for preliminary work, and AW405 was used for the final experiments. In brief, single bacterial colonies were cultivated in LB broth overnight at 30°C and diluted 10−5 with swarm medium. Then 1 μl of this suspension was dispensed ∼3 cm from the edge of a swarm plate and the plate was dried in the Plexiglas box for another 30 min, until the inoculum was completely absorbed. Plates were incubated at 30°C and ∼100% relative humidity.
Liposome preparation
Lipids (in chloroform) were purchased from Avanti Polar Lipids. The liposomes were prepared from 20 mg L-α-phosphatidylethanolamine (from E. coli), 6 mg 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000], 6 mg 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000], and 3 mg cholesterol (molar ratio of 1:0.04:0.04:0.3). The mixture was dried with gently flowing nitrogen gas in a rotating 50 ml round-bottom flask for 30 min, followed by vacuum evaporation for 3 h. The lipid film was hydrated with 1 ml of dye solution under argon with agitation at 160 rpm at room temperature for 2 h. To prepare the dye solution, 18.67 mg calcein (Life Technologies) and 1.21 mg sulforhodamine-101 (Life Technologies) were dissolved in Buffer I, which contained 0.27 mM KCl, 0.147 mM KH2PO4, 1.5 mM Na2HPO4, 0.2 mM Tris, 102 mM NaOH, and 5 mM NaCl. The final pH was adjusted to 7.4 with 1 M HCl, and the osmolarity was adjusted to 240 mOsm with 1 M NaCl. Osmolarity was measured with an Osmette II osmometer (Precision Systems, Natick, MA). Then 0.5 ml aliquots of the multilamellar lipid vesicles generated by this procedure were stored in argon at –20°C, where they were stable for a few months (typically, they were used within 1 month).
The dye-encapsulated multilamellar vesicles were converted to unilamellar vesicles by five cycles of freezing in dry ice for 5 min and thawing at 50°C for 15 min. The unilamellar vesicles were then passed through a 0.8 μm Nuclepore Track-Etch membrane (Whatman) 21 times at 50°C with a Mini-Extruder (Avanti Polar Lipids) that was preconditioned with Buffer I at 50°C for 20 min. This generated liposomes ∼0.5 μm in diameter. The liposomes were cooled to room temperature for 5 min before they were loaded onto a Sephadex G-50 coarse gel-filtration column to remove free dyes. To prepare the column, the Sephadex beads were swelled with Buffer II at 4°C for 2 days. Buffer II differs from Buffer I in that it contains an additional 0.5 mM CaCl2 and 0.5 mM MgCl2. The Sephadex slurry loaded on a column (1 cm × 25 cm) was washed with 4 volumes of swarm medium. The liposomes were eluted with the same medium (osmolarity = 245 mOsm) and collected in glass vials. The liposomes were stable for up to a week when stored at 4°C under argon, and typically were used within 3 days. Approximately 0.5 ml of liposome suspension could be generated in one preparation. Aliquots were stored in eight vials. Each vial was used no more than three times; otherwise, a new G/R ratio osmolarity calibration was performed. The liposomes were quite hardy. They did not fuse when they collided with one another. Nor did they break when they were pipetted onto the surface of agar or struck by swimming cells. Had they done so, the dyes would have diffused into the underlying agar.
Fluorescence detection and imaging
The swarm plates were mounted on the temperature-controlled stage of an upright microscope (Nikon Optiphot2) as described previously (10) except that a 20× bright-phase objective was used. The entire setup, except for the photomultiplier power supplies and computer, was enclosed by a black foamboard box. Fluorescence was excited by a cold white LED (Thorlabs) via an FITC/Texas red fluorescence cube (Chroma). The fluorescence from calcein (G) and sulforhodamine-101 (R) was detected by photon-counting photomultipliers (H7421; Hamamatsu) connected to a data-acquisition board (USB6211; National Instruments) and quantified with custom software written in LabView (National Instruments), which provided the G/R ratio.
The liposome suspension (10 nL) was dispensed with a 0.5 μl Neuros syringe (Hamilton, Reno, NV) and formed a spot ∼450 μm in diameter, similar to the size of the microscope’s field of excitation. Liposome spots were placed at locations ahead of the swarm according to the estimated swarm expansion rate, so that the bacteria would arrive 1.5 h later. Data acquisition was started 30 min after the suspension was dispensed. The liposomes ended up on the top of the agar and were overrun by the advancing swarm.
We recorded phase-contrast videos using a Hi-res Exvision CCD camera connected to a digital videocassette recorder before and after recording the fluorescence signal as described previously (10). We then determined the swarming speed after importing the images to a computer as described previously (10). We converted the times on the traces to the distance from the swarm edge using the swarm expansion rate. To monitor the bacterial behavior on the liposome spots, we recorded continuous video clips at the proximal edge, the distal edge, and in the middle of the spots. Alternatively, we took snapshots of a few seconds during a fluorescence recording session by transiently blocking the light path to the photomultipliers and activating the video system.
Scanning swarms moving on liposome pads
All scanning experiments were performed with standard swarm plates and a 40× bright-phase objective. Liposome suspensions (50 nl) were dispensed with the Neuros syringe to form liposome pads that were ∼2.3 mm in diameter. The liposome pads were placed at locations where a swarm would arrive in ∼2 h, based on the swarm expansion rate. The liposome pads had a thick edge, ∼100 μm wide. Only fluorescence signals from uniformly distributed liposomes found within this edge were collected. The microscopic field of excitation was reduced to a diameter of 20 μm by closing the excitation beam field iris.
The observation field for phase contrast (∼500 μm in diameter) was centered ∼100 μm away from the proximal edge of the liposome pad, between the pad and the advancing swarm. The swarm movement was monitored with the Hi-res Exvision CCD camera, using the microscope tungsten source. When the bacteria reached the liposome pad, the light path was switched to the photomultipliers and fluorescence signals were recorded. After 10–40 s, the swarm plate was pushed by an Intelligent Picomotor (New Focus, San Jose, CA) at 10, 20, or 30 μm/s, so that the excitation beam scanned the swarm in the swarm’s direction of motion. Scanning was continued until the excitation beam reached the far edge of the pad, and then the plate was quickly pulled back manually and a second scan was initiated. We determined the location of the interface between the monolayer and multilayer region of the swarm (see Fig. 1 of Wu and Berg (10)) by briefly looking through the microscope during the scanning process. The swarming speed on agar was determined from the phase-contrast video taken before the fluorescence scanning. The swarming speed on liposome spots or pads was based on videos taken on swarms that were not subjected to fluorescence recording. We superimposed the traces of the G/R ratios of the two scans after determining the distances from the monolayer/multilayer interface, which was taken as position 0.
Calibration curves
To convert the G/R ratio to osmolarity, we plotted two kinds of standard curves (Fig. 1). The G/R ratio in solutions of different osmolarities was determined by mixing 1 μl liposome suspension with 44 μl standard swarm medium or standard solution in a silicon-grease well on a microscopic slide. Two 0.46 mm spacers made from double-sided Scotch tape and the paper disks supplied with Millipore filters were placed outside the grease ring to fix its depth, and a narrow channel was included through its wall to relieve the pressure when a coverslip was added to seal the top. This channel was sealed with grease before the measurements were made. Standard swarm media were prepared by adding different amounts of NaCl to swarm media, and their osmolarities were measured with the Osmette II osmometer. Standard solutions were either purchased from Precision Systems or made by adding 1 M NaCl to Buffer I. The slide was prewarmed for 5 min at 30°C in the dark and the fluorescence signal was recorded for 500 s at 30°C. The averaged values of G/R ratios obtained between 300 s and 350 s were used to plot the calibration curves. The part after 250 s was fitted by linear regression to determine the photobleaching rate.
Figure 1.
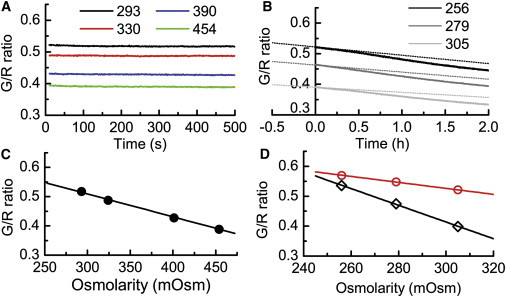
Calibration of osmolarity-sensitive liposomes. (A) G/R fluorescence ratio recorded over an interval of 500 s when liposomes prepared at 245 mOsm were suspended in swarm media at the osmolarities indicated on the plot. Note the minimal effect of fluorescence bleaching on the G/R ratio. (B) G/R ratios from liposomes that were loaded on agar surfaces 0.5 h before recording. Osmolarities of the swarm media used to prepare the plates were measured after the media and agar were separated by centrifugation. These values are shown on the plot. The dotted lines show the data after correction for photobleaching. The changes that remain are due to evaporation. (C) G/R ratios at medium osmolarities recorded between 300 and 350 s. (D) Comparison of the G/R ratios measured in suspension (red circles) and those measured on an agar surface (black diamonds) as a function of the osmolarity of the swarm media.
Swarm plates with different osmolarity were prepared by adding different amounts of NaCl to the swarm agar. A 10 nL liposome suspension was dispensed on the agar plate. After 30 min incubation in the dark, fluorescence signals were recorded for 2 h. Then 0.5 mL agar was scratched from the plate and centrifuged at 16,000 g for 20 min. Clean supernatant (50 μl) was taken out to determine osmolarity with the Osmette II osmometer. The G/R traces were then fitted by linear regression and corrected for photobleaching. The curves corrected for photobleaching were extrapolated back 30 min to obtain the values for the G/R ratios free of the influence of evaporation. These values were used to plot the standard curves for liposomes on an agar surface. The calibration curves of the scanning experiments were obtained via a procedure similar to the real experiment, but on control plates with different osmolarities. The liposome pads were equilibrated 30 min before scanning so that dense packing of liposomes would not occur.
Results and Discussion
Osmolarity-sensitive liposomes
We prepared liposomes using a recipe adapted from Jayaraman et al. (14), as described in Materials and Methods. We encapsulated two kinds of fluorescent dyes: one that self-quenches when liposomes shrink (G, the green dye calcein, emitting at 519 nm) and one that does not (R, the red dye sulforhodamine-101, emitting at 619 nm). Therefore, the G/R ratio reflects the osmolarity of the surrounding fluid. The lipid composition of the liposomes described by Jayaraman et al. (14) was not well defined and the liposome response curves were nonlinear. We tested different combinations of diacyl glycerophospholipids and their polyethylene glycol-modified derivatives. Liposomes made from mixtures of E. coli L-α-phosphatidylethanolamine, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000], 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000], and cholesterol worked well at 30°C. They responded to the change in osmolarity linearly over a range of ∼250 mOsm, as shown in Fig. 1 C. We prepared the liposomes at 245 mOsm so they could be used to measure up to 500 mOsm. These liposomes were stable up to a week when stored in argon at 4°C and up to 5 h when applied to a swarm plate in air at 30°C.
Since we used photon counting, the intensity of the excitation light was low and the effect of photobleaching was small (Fig. 1 A). Photobleaching was estimated by fitting the G/R ratio recorded in liquid suspension by linear regression. The data for the first 250 s were excluded to allow time for equilibration. The averaged slope of the fitting curves was (3.75 ± 0.7) × 10−6 per second (mean ± SD, n = 9). This amount of photobleaching led to a small correction on all measured curves. The G/R ratios recorded between 300 s and 350 s were averaged to plot the standard curves in suspension (Fig. 1 C); within this short time, the amount of photobleaching was small enough to be ignored.
To obtain the calibration curves on plates, the G/R values were corrected for photobleaching and then extrapolated to the time of dispensing, so that evaporation through our temperature-control apparatus could be ignored (Fig. 1 B). The calibration curve thus generated had a steeper slope than that found for liposomes in suspension (Fig. 1 D). The two curves crossed at the point where the osmolarity of the agar matched that of the medium in which the liposomes were prepared. The liposomes formed a multilayer disk on the agar surface. It is likely that if the agar surface and the liposomes were isotonic, the liposomes would remain spherical and the interliposome space would be similar to that in suspension, whereas on a hypertonic agar surface the shrunken liposomes would be more densely packed, like flattened balls. If so, the loss of light by absorption or diffraction might be more severe in the green than in the red, leading to a decrease in the G/R ratio. Whatever the reason, liposome packing caused a reduction of signal equal to 6.42 mOsm per 0.01 G/R ratio. The calibration curves for the scanning experiments were obtained with liposome pads made from larger aliquots of liposomes equilibrated for shorter periods of time, before dense packing became significant, so these curves were similar to those obtained with liposomes in suspension.
Monitoring osmolarity in swarms
We first tried to monitor the change in osmolarity in real time by collecting fluorescence signals from a fully illuminated fixed liposome spot ∼450 μm in diameter as an advancing swarm ran over it (Fig. 2). Although we used the same swarm medium to prepare the plates and the liposomes, the medium in the liposome suspension needed more than 1 h to equilibrate with the medium in the agar. After equilibration for >1.5 h, the traces of the G/R ratios showed a quick decrease upon the arrival of bacteria, followed by slow recovery to a level slightly lower than the baseline before the invasion (Fig. 2 B, with the long-time end of the trace not shown). In cases in which the swarms expanded rapidly, traces recorded after a period of >1.0 h were occasionally used to calculate the changes in osmolarity.
Figure 2.
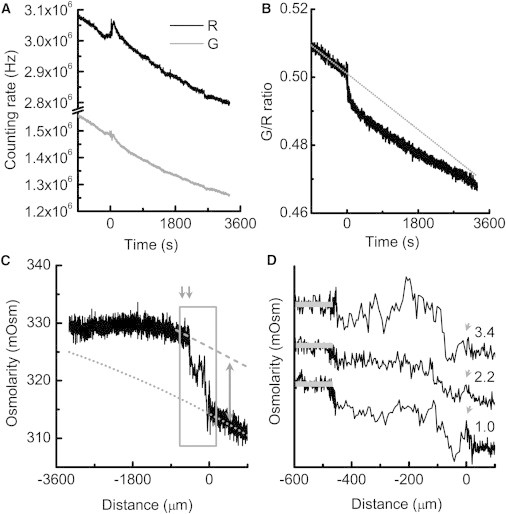
Change of osmolarity on swarm plates measured with fixed liposome spots. (A) Fluorescence signals of sulforhodamine 101 (R) and calcein (G) recorded on a swarm. Time zero corresponds to the time when bacteria first invaded the liposome spot. (B) G/R ratio after correction for photobleaching. The gray dotted line is the reference baseline for osmolarity on the agar surface, which changed because of evaporation. The time required for the measurement, set by the dimensions of the swarm divided by its spreading rate, was relatively long. (C) The trace after the G/R ratio was converted to osmolarity and time was converted to distance (negative toward the swarm center). The gray dotted line is the reference baseline. It curves down because the evaporation rate was constant but the swarming speed slowed down. The initial swarming speed was 1.0 μm/s. The gray broken line indicates the osmolarity maximum and the double-headed arrow indicates the difference in osmolarity between the maximum and the baseline. The difference between the osmolarity trace and the baseline was maximal between the two gray down-pointing arrows. (D) Enlarged view of the boxed region in C with two other traces aligned on top of it (with swarm speeds in μm/s noted at the right ends of the traces). The small peak at time zero is indicated by gray arrows. The osmolarity maxima are highlighted by gray bars superimposed on the traces.
At the beginning of each trace, there was a section where the fluorescence signals were recorded before the arrival of the swarm (Fig. 2). These signals provided a baseline for subsequent changes in osmolarity. The calibration curve measured on plates (Fig. 1 D, black open diamonds) was applied. Extrapolating the baseline to the time when the liposomes were dispensed allowed us to estimate the osmolarity that supports active swarming under our experimental conditions (∼296.8–316.2 mOsm). By changing the concentrations of NaCl and Eiken agar, we could obtain swarms that expanded at velocities ranging from 1.0 μm/s to 4.3 μm/s (Fig. S1 in the Supporting Material). There was an inverse correlation between swarming speed and agar osmolarity.
We observed a transient increase of both the green and red signals when the swarm invaded the liposome spot (Fig. 2 A). The swarm drew water out of the agar and lifted the liposomes from the packed state to the suspended state, and the motionless liposomes began jiggling around when the bacteria arrived. This process suddenly increased the interliposome space, decreasing the loss of light by absorption or diffraction. Thereafter, a small number of liposomes were carried away by the swarm fluid, causing a slow decrease of both fluorescence signals. However, this process would not change the G/R ratio. It is the decrease of green fluorescence caused by self-quenching that is responsible for the persistent change of the G/R ratio (Fig. 2 B).
After bacterial invasion, the calibration curve measured on agar would only be applicable to some liposomes within the excitation field, and the curve in suspension would be applicable to the other liposomes. The measurements were corrected for the fraction of liposomes in the packed state versus those in suspension, and the bona fide osmolarity is plotted in Fig. 2 C. The distance inside the swarm (negative toward the swarm center) was determined by multiplying the time after invasion by the instantaneous velocity of the swarm front. The location of the leading edge of the swarms was at position 0.
Before the large increase of osmolarity shown in Fig. 2 C occurred, a small peak of width 51.0 ± 18.3 μm (n = 6) always appeared (Fig. 2 D, gray arrows). It was followed by a brief, sharp biphasic dip with recovery. We believe this peak was due to the arrival of the stream of fluid that flows in a clockwise sense in front of the swarm (12) (Fig. S2 A). This small amount of fluid caused an increase of osmolarity 2.8 ± 0.9 mOsm. Within ∼10 s the bacteria arrived, and a large amount of fluid flooded the spot.
The actively expanding rim of a swarm can be divided into four regions (10,11). The outermost region is a monolayer of cells, many of which are stuck on the agar surface. These cells are released when the cells of the second multilayer region, which are vigorously motile and at high density, catch up. The cell density decreases in the third region, dubbed the falloff, leading to the plateau, where motility remains at a relatively low level. When the monolayer cells (and some from groups in the high-density region) rushed onto the liposome spot (Fig. S2 A), the cells dispersed into a band 130–150 μm wide and the cell density decreased from ∼0.18 cells/μm2, as in the normal swarm monolayer, to ∼0.03 cells/μm2. This dispersed monolayer contained the same number of cells, but covered 4.5 times as large an area as the monolayer on agar. The front edge of this dispersed monolayer was irregular in shape, with dynamic protrusions and invaginations.
The osmolarity increased steadily upon the arrival of the cells, and then the rate of increase slowed down until an osmolarity maximum was reached (Fig. 2 D). The rapid increase took ∼35 s, whereas the slow increase that followed was swarm-speed dependent. The whole process of this osmolarity jump took place within a distance slightly longer than the diameter of the liposome spot, 480 ± 22 μm (n = 6). The dispersed monolayer and multilayer regions moved rapidly forward at 6.0 ± 1.0 μm/s (n = 5) until the dispersed monolayer reached the far edge of the liposome spot. The osmolarity jump (i.e., the difference between the osmolarity maxima and the baseline (Fig. 2 C, double-headed arrow)) was 11.4 ± 5.0 mOsm (n = 15). The steady level reached at the end of the experiment was 3.7 ± 0.6 mOsm (n = 15) higher than the baseline. This value reflects the osmolarity of the swarm interior. When the monolayer cells reached the far edge of the liposome spot, the cells stalled and the multilayer caught up with decreasing velocity (Fig. S2 A). Eventually, the swarm ran over the far edge of the liposome spot and continued advancing on the agar. Because the swarm expanded faster on the liposome spot than on the adjacent agar, a hump formed at the swarm front that eventually smoothed out.
Scanning across swarms moving on liposome pads
The fixed spots did not provide the spatial resolution required to evaluate the fluid-flow model (10). Therefore, we performed another set of experiments in which the liposome pads were five times larger in diameter (Fig. 3). The excitation beam (20 μm in diameter; not shown) was held fixed while the plate was pushed by a picomotor, so that the swarm was scanned in its direction of motion, as shown by the small arrows in Fig. 3. The fast expansion rate of the swarm on the liposome pad made the swarm spread out, so that the monolayer and multilayer regions of the swarms were cleanly separated from their counterparts on the agar surface (Fig. 3). Because the front of the dispersed monolayer was not uniform, we used the position of the monolayer/multilayer interface as our reference point (position 0).
Figure 3.
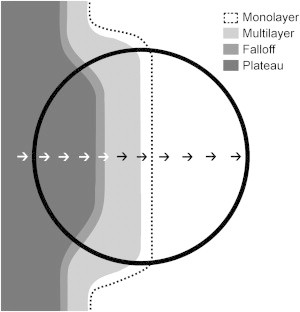
Diagram showing a swarm that has arrived at the center of a liposome pad 2.3 mm in diameter. Different grayscale values represent different swarm regions as labeled in the figure. The black dotted line indicates the swarm front. Arrows show the direction of the scan.
To convert measurement time to distance, the speed of the excitation beam relative to the reference origin is required. To a first approximation, this is just the swarm speed minus the plate pushing speed. For a second scan performed on the same plate, the swarm speed decreased as the dispersed monolayer approached the far end of the pad, so we linearly interpolated the swarm speed from 6 μm/s to the respective swarm expansion rate on agar. The resulting osmolarity versus distance plots are shown in Fig. 4 A.
Figure 4.
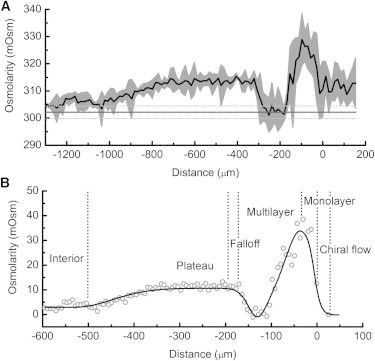
Change of osmolarity inside swarms spreading on liposome pads revealed by scanning. Position 0 corresponds to the monolayer/multilayer interface in (A) or to the leading edge of the swarm in (B). Negative values are toward the center of the swarms. (A) Means (black line) and standard deviations (gray areas) from nine scans on seven different plates. The osmolarity baseline measured on the agar surface is shown by a thick horizontal line and its standard deviation is shown by the thin dotted lines. Note that each scan was completed in less than 2 min with liposomes submerged in swarm fluid, so evaporation was not a problem. (B) Osmolarity profile inside a swarm after subtraction of the baseline value for virgin agar and conversion of the distance on the liposome pad to that on agar. The gray open circles are measured values and the black line is the curve that fits the data. The monolayer was normalized (shrunk to normal size), and the osmolarity values were corrected for dilution that occurred when the swarms spread over the liposome pads. The notations monolayer, multilayer, falloff, and plateau refer to regions of different surface cell density as described in Darnton et al. (11).
The scanning experiment revealed that the multilayer region of the swarm contained two distinct bands: an outer high-osmolarity band of 327.1 ± 6.5 mOsm and an inner low-osmolarity band of 302.5 ± 5.0 mOsm (Fig. 4 A). The osmolarity baseline on the agar surface measured with the fixed liposome spot under identical conditions was 302.1 ± 2.3 mOsm (n = 8) (Fig. 4 A, thick solid and thin dotted lines). The outer band was therefore 25.3 ± 7.3 mOsm higher than the baseline and the inner band was almost isotonic to the agar. Farther inside, there was a stable plateau that was 11.3 ± 3.1 mOsm higher than the baseline. The osmolarity eventually went down to the interior level, which was 3.7 ± 2.9 mOsm above the baseline. The dispersed monolayer, a band ∼150 μm wide to the right of position 0, had an osmolarity of 9.2 ± 5.3 mOsm above the baseline.
The osmolarity profile inside a bacterial swarm
To increase stability, we covered the liposomes with polyethylene glycol, which has certain surface properties (e.g., hydrophilicity) that make swarms expand faster on liposome spots or pads than on virgin agar. In the scanning experiments, different regions of the swarm were fully expanded and well separated from their counterparts on agar (Fig. 3), but on the fixed spot, such expansion was restricted (Fig. S2 B). As a result, we did not see the low-osmolarity band on spots (the low-osmolarity band is obvious in Fig. 4 A, but not in Fig. 2 D). Part of the problem was the low spatial resolution of the fixed-spot method; for example, if the leading edge of a swarm advances 50 μm over a spot of diameter 450 μm, the swarm will cover only 6% of the area of the spot, and 94% of the light reaching the detectors will come from the cell-free region.
The fixed-spot measurements generated an osmolarity increment of 2.8 mOsm when the fluid that flowed in front of the swarms drained into the liposome spots. This is an underestimate because, as noted above, most liposomes in the spots were not influenced by this small amount of fluid. The experimental value was corrected by multiplying by the ratio of the area of the whole spot to the area of the segment that was wetted by the flow. The height of this segment was obtained by assuming that the width of the flow on agar was expanded 4.5 times. The corrected osmolarity was as high as the plateau (12.1 mOsm), as shown in Fig. 4 B.
To find the dimensions of swarms on agar from the dimensions on liposome pads (Fig. 3), we multiplied the dimensions on pads by 2.6/6.0, the ratio of the speeds on agar (2.6 ± 1.0 μm/s; n = 15) to those on pads (6.0 ±1.0 μm/s; n = 5). The monolayer thus converted spanned 66 μm. The width of the monolayer in a normal swarm measures 31 μm on average (10). For comparison with the previous results, we used this smaller width and set the front of the monolayer as the reference origin (Fig. 4 B). The osmolarity of the monolayer was scaled using the dilution factor of 4.5 mentioned above. This assumes that the fluid and the cells were diluted to the same degree, which would be the case if the film of fluid expanded to its original thickness. We note that the cells at the outer edge of the monolayer were transiently stuck to the agar, whereas those in the multilayer were actively swimming (10,11). The expanded multilayer on pads was filled by cells that flowed in from the sides and thus maintained its thickness.
Mapping the fluid-flow pattern inside swarms with micron-sized air bubbles (10) revealed that swarm fluid flows inward from the edge of the swarm toward the center, whereas beginning from ∼300 μm inside the swarm, it flows outward. Fluid balance requires that water moves out of the agar and into the swarm within a region centered ∼30 μm from the edge of the swarm, with a peak at the monolayer/multilayer interface. After we corrected for differences in migration rates on pads versus those on virgin agar, the osmolarity profile agreed well with the model prediction (Fig. 4 B). The osmolarity increased rapidly at the monolayer/multilayer interface and reached the highest value at ∼30 μm inside the swarm. The osmolarity profile for the first ∼130 μm paralleled the surface cell density measured earlier (see Fig. 5 of Wu and Berg (10)).
Fluid balance also requires that the agar absorb water from the swarm in a region centered ∼120 μm from the edge of the swarm. The lowest point on our curve was at −128 μm, and it reached the baseline or slightly lower, within the error range of our measurements (Fig. 4 A, thin dotted lines). The osmolarity reached a stable plateau at ∼200 μm from the edge of the swarm (Fig. 4 B), also in accordance with the model prediction (10). The maximum obtained in the fixed-spot measurements corresponded to the plateau detected by scanning. It was ∼11.3 mOsm higher than the baseline as revealed by both methods. This plateau extended to ∼300 μm inside the swarm. The osmolarity began to decrease beyond that point and reached the level for the swarm interior ∼500 μm from the edge of the swarm. Active fluid flow was not observed beyond 300 μm from edge of the swarm (10), suggesting that this region of the swarm is in equilibrium with the agar.
Properties of osmolytes
The primary osmolyte that has been implicated in the swarming of E. coli or Salmonella is lipopolysaccharide (17,18). Other candidates of high molecular weight include enterobacterial common antigen and colanic acid (18). Mutants of Salmonella that rotate their flagella exclusively clockwise or exclusively counterclockwise fail to swarm, yielding plates that are relatively dry (19). However, revertants that remain nonchemotactic yet frequently switch the direction of flagellar rotation do swarm, leading to the suggestion that erratically moving flagella strip lipopolysaccharide off of the cell surface (20).
We need osmolytes of relatively high molecular weight (with relatively small diffusion coefficients) to explain our results. Substances of low molecular weight, such as salts, acetate, glutamate, proline, glycine betaine, and trehalose, will not do. The swarm fluid is only a few micrometers thick, whereas the underlying agar is ∼1400 μm deep. Both are nearly 100% water. A small molecule with diffusion coefficient D ∼103 μm2/s (10−5 cm2/s) will diffuse (2Dt)1/2 ∼2 μm in ∼0.002 s, 20 μm in ∼0.2 s, and 200 μm in ∼20 s. The timescale of interest (Fig. 4 B) is ∼50 s (a swarm displaced ∼130 μm at the rate of 2.6 μm/s). In that interval, substances of low molecular weight will be diluted by a factor of >100 by diffusion perpendicular to the surface of the plate, and peaks and troughs, such as those apparent in Fig. 4 B, will be washed out by diffusion in a direction parallel to the surface of the plate. However, substances 100 times larger with diffusion coefficients 100 times smaller will fit the bill. A substance of this size can diffuse out of the swarm fluid into the agar (neglecting opposing fluid flow) in ∼0.2 s, or into the agar a distance 10 times as far, in ∼20 s. So once the bulk flow subsides, the osmolyte will move into the agar. Thus, one expects a concentration inversion when the measured value of the osmotic pressure falls to the baseline, as it does in Fig. 4 B. When this happens, some fluid will flow from the swarm back into the agar. Therefore, the general features of our osmolarity measurements are consistent with the predictions of the fluid-flow model. If the osmolytes are polyelectrolytic, counterions will contribute to the osmolarity. We do not know whether the fall in concentration of osmolytes is precipitated simply by a decreased rate of cell growth (10) or the osmolytes are actively resorbed.
If the swarm spreads at 2.6 μm/s, the peak in osmolarity shown in Fig. 4 B extending from 0 to 520 μm can be scanned in 200 s. We let the concentration of osmolyte at the surface of the agar vary in time according to the output of this scan and follow the concentration of the secreted osmolyte as it diffuses into the agar. We ask, as a function of the value of the diffusion coefficient, how long it takes before the gradient normal to the surface of the agar changes sign. This time interval would equal that measured previously (10) before fluid begins to flow from the swarm back into the agar, 41 ± 7 s. The acceptable range of diffusion coefficients proves to be 0.7–6.8 μm2/s, which is in the ballpark predicted by our order-of-magnitude arguments. A diffusion coefficient in this range (1.1 μm2/s) has been measured for aggregates of phenol-extracted lipopolysaccharide of molecular weight ∼2 × 108 (21). Therefore, lipopolysaccharide is an attractive candidate.
Conclusions
We optimized the liposome sensor and used it to monitor the osmolality of bacterial swarms in real time. The results revealed a well-defined osmolality profile inside the bacterial swarm. The previously described flow pattern of E. coli swarm fluid was explained. The diffusion coefficient of the potential osmolyte(s) was predicted based on our model. The chemical structure of the osmolyte(s) remains a subject for future investigation; however, lipopolysaccharide is a reasonable candidate. Our technique is highly reproducible when applied to E. coli, which does not produce surfactants. The method can be applied to surfactant-producing bacteria as well, provided that the surfactants are not strong enough to destroy the liposomes. We look forward to the application of this technique in other swarming bacteria and in other research fields.
Author Contributions
L.P., Y.W., J.T., and H.B. designed the research; H.B. assembled the microscope; Y.W. did preliminary work on the liposome prep; Y.W. and G.H. did preliminary work on the fluorescence measurements; GH perfected the data-acquisition system; J.T. helped with the scanning experiments; L.P. did the final measurements and analyzed the data; and L.P. and H.B. wrote the paper with input from the other authors.
Acknowledgments
This research was supported by NIH grant AI100902.
Supporting Material
References
- 1.Harshey R.M. Bacterial motility on a surface: many ways to a common goal. Annu. Rev. Microbiol. 2003;57:249–273. doi: 10.1146/annurev.micro.57.030502.091014. [DOI] [PubMed] [Google Scholar]
- 2.Kearns D.B. A field guide to bacterial swarming motility. Nat. Rev. Microbiol. 2010;8:634–644. doi: 10.1038/nrmicro2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg H.C. Swarming motility: it better be wet. Curr. Biol. 2005;15:R599–R600. doi: 10.1016/j.cub.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 4.Hola V., Peroutkova T., Ruzicka F. Virulence factors in Proteus bacteria from biofilm communities of catheter-associated urinary tract infections. FEMS Immunol. Med. Microbiol. 2012;65:343–349. doi: 10.1111/j.1574-695X.2012.00976.x. [DOI] [PubMed] [Google Scholar]
- 5.Callegan M.C., Novosad B.D., Senesi S. Role of swarming migration in the pathogenesis of bacillus endophthalmitis. Invest. Ophthalmol. Vis. Sci. 2006;47:4461–4467. doi: 10.1167/iovs.06-0301. [DOI] [PubMed] [Google Scholar]
- 6.Murray T.S., Ledizet M., Kazmierczak B.I. Swarming motility, secretion of type 3 effectors and biofilm formation phenotypes exhibited within a large cohort of Pseudomonas aeruginosa clinical isolates. J. Med. Microbiol. 2010;59:511–520. doi: 10.1099/jmm.0.017715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medina-Ruiz L., Campoy S., Barbé J. Overexpression of the recA gene decreases oral but not intraperitoneal fitness of Salmonella enterica. Infect. Immun. 2010;78:3217–3225. doi: 10.1128/IAI.01321-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harshey R.M., Matsuyama T. Dimorphic transition in Escherichia coli and Salmonella typhimurium: surface-induced differentiation into hyperflagellate swarmer cells. Proc. Natl. Acad. Sci. USA. 1994;91:8631–8635. doi: 10.1073/pnas.91.18.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg H.C. Springer-Verlag Inc.; New York: 2004. E. coli in Motion. [Google Scholar]
- 10.Wu Y., Berg H.C. Water reservoir maintained by cell growth fuels the spreading of a bacterial swarm. Proc. Natl. Acad. Sci. USA. 2012;109:4128–4133. doi: 10.1073/pnas.1118238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darnton N.C., Turner L., Berg H.C. Dynamics of bacterial swarming. Biophys. J. 2010;98:2082–2090. doi: 10.1016/j.bpj.2010.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y., Hosu B.G., Berg H.C. Microbubbles reveal chiral fluid flows in bacterial swarms. Proc. Natl. Acad. Sci. USA. 2011;108:4147–4151. doi: 10.1073/pnas.1016693108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner L., Zhang R., Berg H.C. Visualization of flagella during bacterial swarming. J. Bacteriol. 2010;192:3259–3267. doi: 10.1128/JB.00083-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayaraman S., Song Y., Verkman A.S. Airway surface liquid osmolality measured using fluorophore-encapsulated liposomes. J. Gen. Physiol. 2001;117:423–430. doi: 10.1085/jgp.117.5.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong J.B., Adler J., Dahl M.M. Nonchemotactic mutants of Escherichia coli. J. Bacteriol. 1967;93:390–398. doi: 10.1128/jb.93.1.390-398.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner L., Stern A.S., Berg H.C. Growth of flagellar filaments of Escherichia coli is independent of filament length. J. Bacteriol. 2012;194:2437–2442. doi: 10.1128/JB.06735-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toguchi A., Siano M., Harshey R.M. Genetics of swarming motility in Salmonella enterica serovar typhimurium: critical role for lipopolysaccharide. J. Bacteriol. 2000;182:6308–6321. doi: 10.1128/jb.182.22.6308-6321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Partridge J.D., Harshey R.M. Swarming: flexible roaming plans. J. Bacteriol. 2013;195:909–918. doi: 10.1128/JB.02063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q., Suzuki A., Harshey R.M. Sensing wetness: a new role for the bacterial flagellum. EMBO J. 2005;24:2034–2042. doi: 10.1038/sj.emboj.7600668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariconda S., Wang Q., Harshey R.M. A mechanical role for the chemotaxis system in swarming motility. Mol. Microbiol. 2006;60:1590–1602. doi: 10.1111/j.1365-2958.2006.05208.x. [DOI] [PubMed] [Google Scholar]
- 21.Hartley J.L., Adams G.A., Tornabene T.G. Chemical and physical properties of lipopolysaccharide of Yersinia pestis. J. Bacteriol. 1974;118:848–854. doi: 10.1128/jb.118.3.848-854.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.