

Abstract
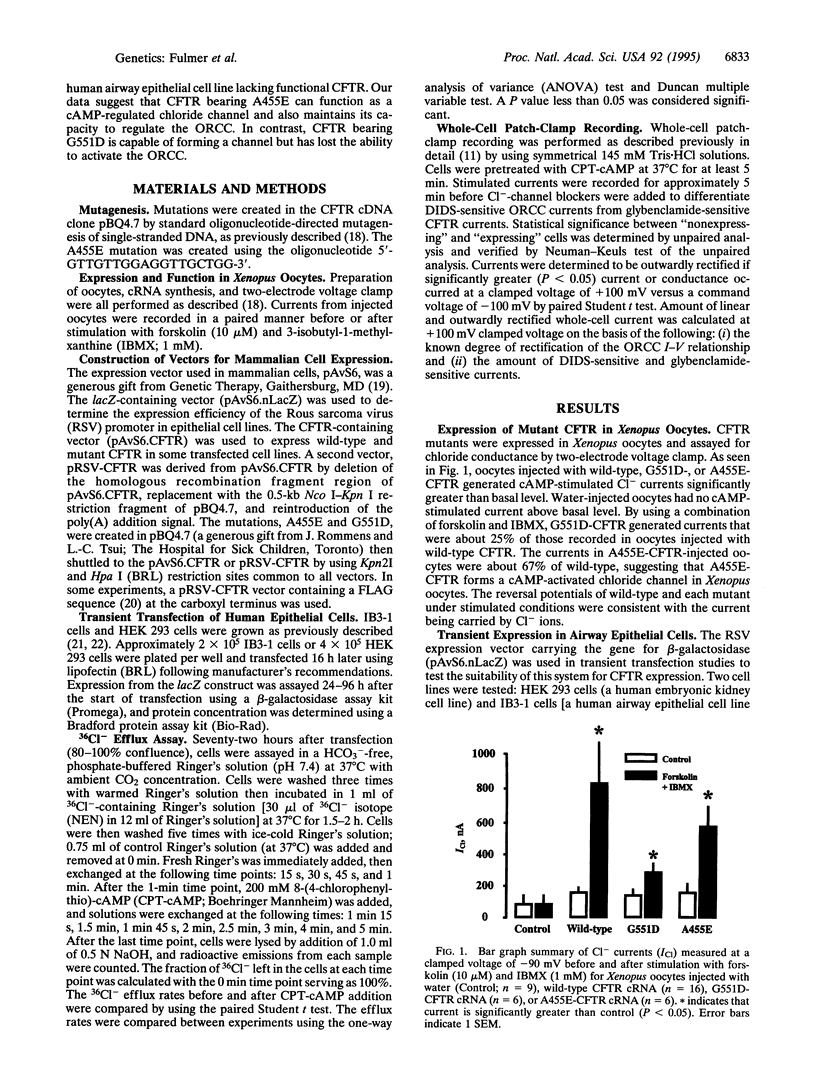
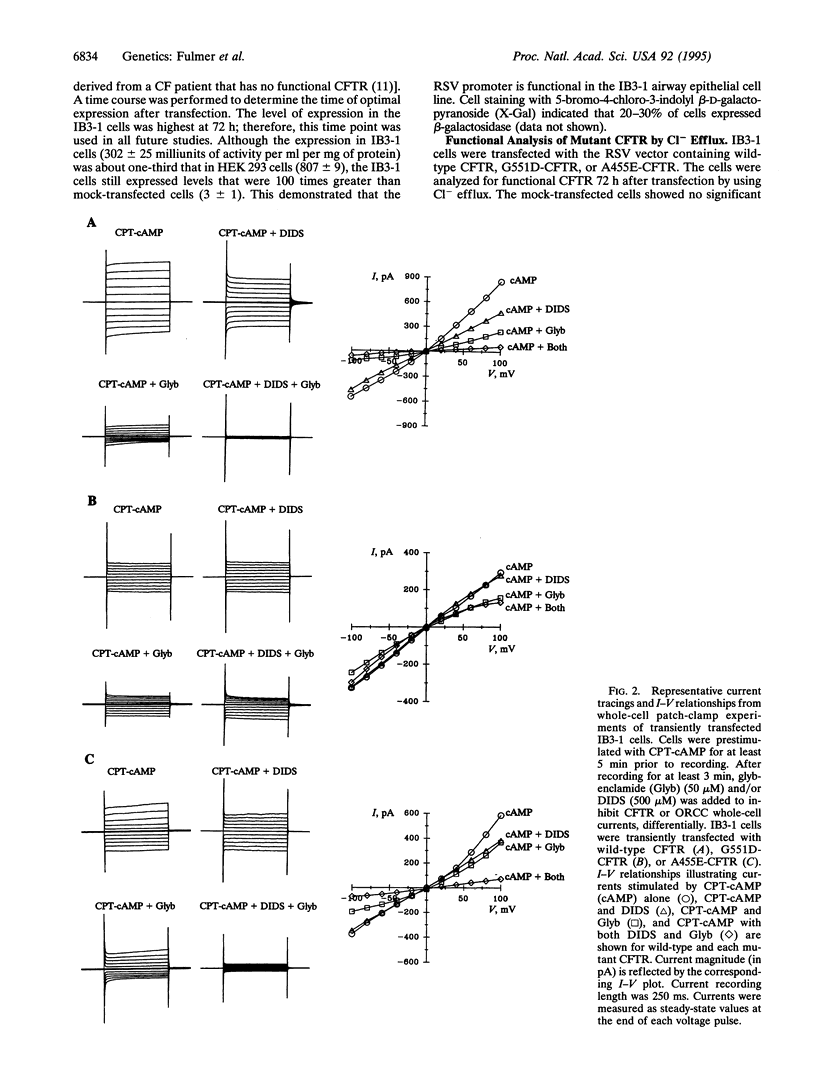
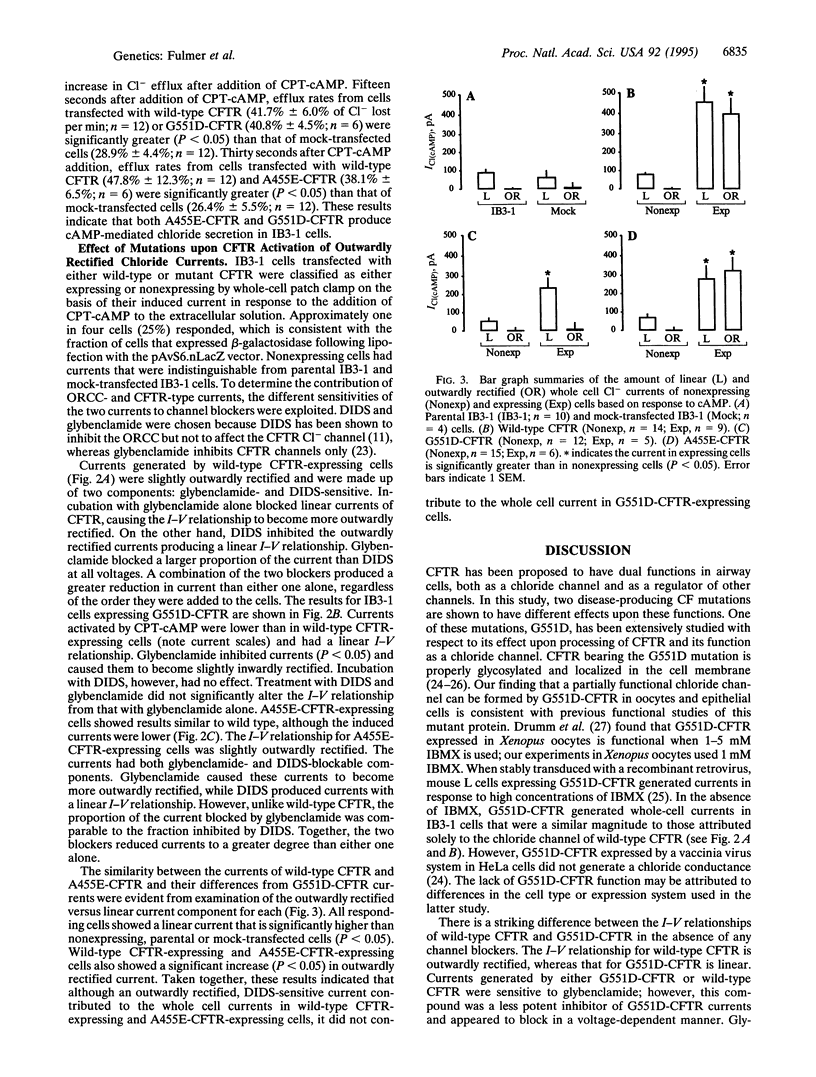
Cystic fibrosis (CF), a disorder of electrolyte transport manifest in the lungs, pancreas, sweat duct, and vas deferens, is caused by mutations in the CF transmembrane conductance regulator (CFTR). The CFTR protein has been shown to function as a cAMP-activated chloride channel and also regulates a separate protein, the outwardly rectifying chloride channel (ORCC). To determine the consequence of disease-producing mutations upon these functions, mutant CFTR was transiently expressed in Xenopus oocytes and in human airway epithelial cells lacking functional CFTR. Both G551D, a mutation that causes severe lung disease, and A455E, a mutation associated with mild lung disease, altered but did not abolish CFTR's function as a chloride channel in Xenopus oocytes. Airway epithelial cells transfected with CFTR bearing either A455E or G551D had levels of chloride conductance significantly greater than those of mock-transfected and lower than those of wild-type CFTR-transfected cells, as measured by chloride efflux. A combination of channel blockers and analysis of current-voltage relationships were used to dissect the contribution of CFTR and the ORCC to whole cell currents of transfected cells. While CFTR bearing either mutation could function as a chloride channel, only CFTR bearing A455E retained the function of regulating the ORCC. These results indicate that CF mutations can affect CFTR functions differently and suggest that severity of pulmonary disease may be more closely associated with the regulatory rather than chloride channel function of CFTR.
Full text
PDF


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991 Jul 12;253(5016):202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Bear C. E., Li C. H., Kartner N., Bridges R. J., Jensen T. J., Ramjeesingh M., Riordan J. R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 1992 Feb 21;68(4):809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., Smith A. E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991 Sep 6;66(5):1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991 Dec 20;254(5039):1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Egan M., Flotte T., Afione S., Solow R., Zeitlin P. L., Carter B. J., Guggino W. B. Defective regulation of outwardly rectifying Cl- channels by protein kinase A corrected by insertion of CFTR. Nature. 1992 Aug 13;358(6387):581–584. doi: 10.1038/358581a0. [DOI] [PubMed] [Google Scholar]
- Flotte T. R., Afione S. A., Solow R., Drumm M. L., Markakis D., Guggino W. B., Zeitlin P. L., Carter B. J. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J Biol Chem. 1993 Feb 15;268(5):3781–3790. [PubMed] [Google Scholar]
- Gabriel S. E., Clarke L. L., Boucher R. C., Stutts M. J. CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature. 1993 May 20;363(6426):263–268. doi: 10.1038/363263a0. [DOI] [PubMed] [Google Scholar]
- Gan K. H., Heijerman H. G., Bakker W. Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med. 1994 Mar 24;330(12):865–867. [PubMed] [Google Scholar]
- Gregory R. J., Rich D. P., Cheng S. H., Souza D. W., Paul S., Manavalan P., Anderson M. P., Welsh M. J., Smith A. E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991 Aug;11(8):3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb B. R., Vick R. N., Boucher R. C. Hyperabsorption of Na+ and raised Ca(2+)-mediated Cl- secretion in nasal epithelia of CF mice. Am J Physiol. 1994 May;266(5 Pt 1):C1478–C1483. doi: 10.1152/ajpcell.1994.266.5.C1478. [DOI] [PubMed] [Google Scholar]
- Hamosh A., King T. M., Rosenstein B. J., Corey M., Levison H., Durie P., Tsui L. C., McIntosh I., Keston M., Brock D. J. Cystic fibrosis patients bearing both the common missense mutation Gly----Asp at codon 551 and the delta F508 mutation are clinically indistinguishable from delta F508 homozygotes, except for decreased risk of meconium ileus. Am J Hum Genet. 1992 Aug;51(2):245–250. [PMC free article] [PubMed] [Google Scholar]
- Hwang T. C., Lu L., Zeitlin P. L., Gruenert D. C., Huganir R., Guggino W. B. Cl- channels in CF: lack of activation by protein kinase C and cAMP-dependent protein kinase. Science. 1989 Jun 16;244(4910):1351–1353. doi: 10.1126/science.2472005. [DOI] [PubMed] [Google Scholar]
- Hyde S. C., Emsley P., Hartshorn M. J., Mimmack M. M., Gileadi U., Pearce S. R., Gallagher M. P., Gill D. R., Hubbard R. E., Higgins C. F. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature. 1990 Jul 26;346(6282):362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- Johnson L. G., Boyles S. E., Wilson J., Boucher R. C. Normalization of raised sodium absorption and raised calcium-mediated chloride secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J Clin Invest. 1995 Mar;95(3):1377–1382. doi: 10.1172/JCI117789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovov B., Ismailov I. I., Benos D. J. Cystic fibrosis transmembrane conductance regulator is required for protein kinase A activation of an outwardly rectified anion channel purified from bovine tracheal epithelia. J Biol Chem. 1995 Jan 27;270(4):1521–1528. doi: 10.1074/jbc.270.4.1521. [DOI] [PubMed] [Google Scholar]
- Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992 Aug;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kunz D., Gerard N. P., Gerard C. The human leukocyte platelet-activating factor receptor. cDNA cloning, cell surface expression, and construction of a novel epitope-bearing analog. J Biol Chem. 1992 May 5;267(13):9101–9106. [PubMed] [Google Scholar]
- Logan J., Hiestand D., Daram P., Huang Z., Muccio D. D., Hartman J., Haley B., Cook W. J., Sorscher E. J. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucleotide binding. J Clin Invest. 1994 Jul;94(1):228–236. doi: 10.1172/JCI117311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittereder N., Yei S., Bachurski C., Cuppoletti J., Whitsett J. A., Tolstoshev P., Trapnell B. C. Evaluation of the efficacy and safety of in vitro, adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator cDNA. Hum Gene Ther. 1994 Jun;5(6):717–729. doi: 10.1089/hum.1994.5.6-717. [DOI] [PubMed] [Google Scholar]
- Rich D. P., Gregory R. J., Anderson M. P., Manavalan P., Smith A. E., Welsh M. J. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 1991 Jul 12;253(5016):205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 Sep 8;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Schwiebert E. M., Flotte T., Cutting G. R., Guggino W. B. Both CFTR and outwardly rectifying chloride channels contribute to cAMP-stimulated whole cell chloride currents. Am J Physiol. 1994 May;266(5 Pt 1):C1464–C1477. doi: 10.1152/ajpcell.1994.266.5.C1464. [DOI] [PubMed] [Google Scholar]
- Sheppard D. N., Ostedgaard L. S., Winter M. C., Welsh M. J. Mechanism of dysfunction of two nucleotide binding domain mutations in cystic fibrosis transmembrane conductance regulator that are associated with pancreatic sufficiency. EMBO J. 1995 Mar 1;14(5):876–883. doi: 10.1002/j.1460-2075.1995.tb07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D. N., Welsh M. J. Inhibition of the cystic fibrosis transmembrane conductance regulator by ATP-sensitive K+ channel regulators. Ann N Y Acad Sci. 1993 Dec 20;707:275–284. doi: 10.1111/j.1749-6632.1993.tb38058.x. [DOI] [PubMed] [Google Scholar]
- Yang Y., Devor D. C., Engelhardt J. F., Ernst S. A., Strong T. V., Collins F. S., Cohn J. A., Frizzell R. A., Wilson J. M. Molecular basis of defective anion transport in L cells expressing recombinant forms of CFTR. Hum Mol Genet. 1993 Aug;2(8):1253–1261. doi: 10.1093/hmg/2.8.1253. [DOI] [PubMed] [Google Scholar]