

Abstract
It is now well established that the immune system can control and eliminate cancer cells. Adoptive T cell transfer has the potential to overcome the significant limitations associated with vaccine-based strategies in patients who are often immune compromised. Application of the emerging discipline of synthetic biology to cancer, which combines elements of genetic engineering and molecular biology to create new biological structures with enhanced functionalities, is the subject of this focused research review.
Keywords: Adoptive cell transfer, Gene transfer, Leukemia, Chimeric antigen receptor, 19th Danish Cancer Society Symposium
Introduction
There are currently two major approaches to overcome tolerance and therefore permit the unleashing of T cells for successful cancer immunotherapy. By using checkpoint blockade, the natural inhibitory signals that limit T cell effector function may be circumvented [1]. The other approach involves the use of synthetic biology to engineer T cells into “cancer hunter cells” [2], to perform tasks that they would not otherwise be able to accomplish. In retrospect, it seems that the concept of synthetic biology in medicine actually originated outside of the field, on athletic fields where athletes discovered that muscular performance could be enhanced by use of performance-enhancing drugs. In a similar manner, the fields of immunology and gene transfer have shown that T cell function can be enhanced by altering receptor specificity and signaling functions that control proliferative capacity and other cellular functions [3]. Currently, there are two forms of engineered T cells being tested in early-phase clinical trials (Fig. 1). In one approach, T cells with enhanced affinity or novel specificity are created by expression of heterodimeric T cell receptors (TCRs) in T cells [4, 5]. The other approach uses chimeric antigen receptor (CAR)-modified T cells, also called CARs T cells or T bodies [6, 7].
Fig. 1.
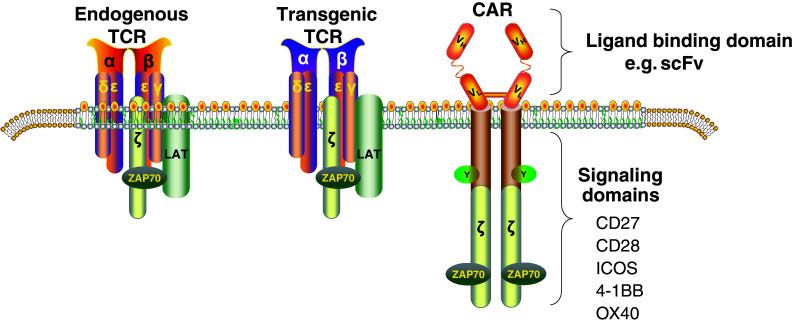
T cells with redirected receptor specificity that is currently under development for cancer and HIV. Dual recognition T cells are created by the introduction of genes that encode T cell receptors (TCR) or chimeric antigen receptors (CARs). The TCR combines TCR-alpha and TCR-beta genes and is MHC-restricted, while CARs are made from antibody-derived scFv, tied to intracellular signaling modules derived from T cell costimulatory proteins, and thus are MHC-independent, but require antigen expression on the surface of target cells
TCR-modified T cells
Affinity-enhanced TCRs are currently being tested at several centers. The rationale for this is that the endogenous repertoire for TCRs is generally of low affinity when targeting self-based cancer antigens [5]. There may be exceptions in the case of so-called neo-antigens, where mutations in the cancer genome create new epitopes that may be targeted by higher affinity TCRs than the TCRs targeting endogenous self-antigens [8]. However, unless the cancer is caused by a virus, most approaches currently rely on targeting non-mutated self-antigens, the most promising of which may be gene products from the cancer testis family of genes [9]. In trials conducted at the University of Pennsylvania and the University of Maryland with colleagues at Adaptimmune Ltd, we have reported the initial results of therapy using two TCRs (clinicaltrials.gov NCT01892293). The first to enter our clinical trials was the NY ESO-1-specific and HLA-A2-restricted TCR [10]. This TCR is being tested in several types of cancer, including melanoma, myeloma, and synovial cell sarcoma [11]. More than 20 patients with myeloma have been treated, and the initial results are quite promising; notably, there has been no serious toxicity. In contrast, the two patients who were treated with a TCR engineered for enhanced affinity to MAGE A3 experienced serious toxicity. This HLA-A1-restricted TCR was found to have off-target recognition of the cardiac and striated muscle-specific protein Titin [12]. This resulted in serious cardiac toxicity that was lethal in both patients that experienced it [13]. As a result, the clinical development of this particular TCR was terminated. In addition, toxicity has been noted with an independently derived MAGE A3 TCR tested in trials at the National Cancer Institute. This TCR was obtained by immunizing transgenic mice with human MAGE A3 peptide, and an HLA-A2-restricted TCR was obtained. In patients with melanoma, this TCR caused severe neurotoxicity [14] due to unforeseen expression of a cross-reactive related antigen in the brain. One lesson from the toxicity that has been observed with these engineered TCRs is that the toxicity was not always manageable with corticosteroid administration. In addition, the onset of the cardiac and neurologic toxicity was rapid, calling into question whether conditional T cell suicide systems would be effective in the management of this sort of acute toxicity observed in non-dispensable organs such as the brain and heart. While these events illustrate the potency that engineered autologous T cells can display, they highlight the urgent need for improved preclinical systems to uncover off-target reactivity before initiation of clinical trials.
CAR-modified T cells
The concept of CARs arose from the cloning of the TCR CD3zeta chain and the observation that the cytoplasmic tail of the zeta chain could activate T cells independently of the rest of the TCR complex [15–17]. The initial studies with CAR-modified T cells having a signaling domain comprised of the CD3zeta chain were conducted in patients with HIV infection [18]. The CAR T cells tested in these studies were found to be safe, and in a retrospective analysis, it was determined that the CAR T cells persisted for more than a decade following infusion [19]. The first CAR T cell trials in cancer patients were conducted by scientists at Cell Genesys targeting the TAG-72 antigen [20]. In unpublished studies led by George Fisher, Alan Venook and Emily Bergsland, patients with metastatic colon cancer were given intravenous or intrahepatic infusions of a first-generation CAR targeted via the anti-TAG-72 cc49 monoclonal antibody. Most patients developed antibodies to the CAR and no antitumor activity was observed (RAC protocols 9707-198 and 9802-239, K. Hege MD, personal communication).
In more recent studies, the field has developed so-called second and third-generation CARs that have additional signaling domains attached to the cytosolic activation domain [21]. In principle, this permits T cells to be redirected toward diverse targets by varying the single chain variable fragments, as well as by reprograming the T cells to survive in various tumor microenvironments that are often toxic for natural lymphocytes (Fig. 2). The first published studies with first-generation CAR T cells in cancer patients were disappointing because they showed limited persistence of the T cells and no antitumor effects [22, 23]. In contrast to these early results, several groups testing second-generation CAR T cells in patients with hematologic malignancies have reported potent antitumor effects [24–28]. CAR T cells endowed with CD28 or CD137 (4-1BB) signaling domains have been found to have potent antitumor effects in patients with B cell malignancies, including chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL). In studies at the University of Pennsylvania, CAR T cells that employ the 4-1BB signaling domain have been found to persist for more than 3 years in patients with CLL, and for more than 2 years now in patients with ALL. The response rate in these patients has been high, with more than 50 % of patients achieving partial or complete remission. The magnitude of proliferation of the CAR T cells following infusion is a biomarker that correlates with response. In general, patients that have high level proliferation of the CAR T cells have complete remissions, while those who have limited T cell engraftment of the CAR T cells have only limited partial or no response. The expansion of the CAR T cells appears to be more rapid in children than in elderly patients, perhaps reflecting a more vigorous immune response potential in children. A corollary of this is that the cell dose required for children with gene-modified T cells may be lower than elderly patients who often have T cells with a more limited replicative capacity [29].
Fig. 2.
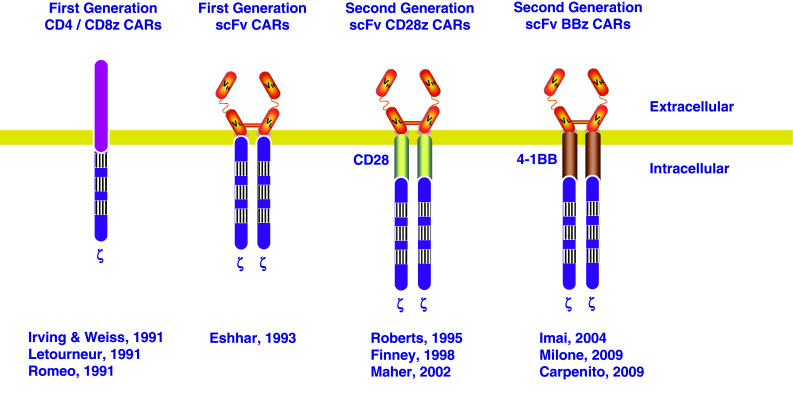
CAR development. Initial CARs were comprised of CD4 or CD8 linked to the CD3zeta molecule. Then, Eshhar attached scFv to the CDzeta molecule. Second-generation CARs using either CD28 or 4-1BB have now been developed by several groups and tested in clinical trials
Studies with CAR T cells targeting CD19 that encode a CD28 signaling domain have also shown potent effects in patients with ALL [28]; however to date, the clinical effects of this particular CAR in patients with CLL have been modest [30]. A potential mechanistic explanation is that CAR T cells with CD28 signaling domains have not displayed long-term engraftment and persistence. Second-generation CD28-based CARs have demonstrated improved survival over first-generation CD3zeta CAR T cells [31]. However, the average survival of CAR T cells in patients with ALL treated at the Memorial Sloan Kettering Cancer Center was approximately 28 days [28]. Thus the strategy employed with CAR T cells that have short duration of engraftment has been to use this therapy as a bridge to transplant, rather than as a definitive therapy. Perhaps the major controversy in the field at this point is whether CAR T cells can replace the need for allogeneic stem cell transplantation, or rather, will they serve as a bridge to transplantation? It is now clear that the duration of B cell aplasia is a biomarker of in vivo activity of CAR T cells; in other words, when normal B cells return, it is an indication that the CAR T cells have been eliminated or otherwise failed to maintain effector function. In some cases, but not all, this heralds a relapse of leukemia. Related to this issue, it is not yet known if in patients with remission, every last tumor cell has been eradicated, or if there are dormant tumor cells that remain. In the latter case, this would suggest that long-term persistence of CAR T cells would be desirable, or in the case of a more limited persistence, that maintenance infusions may be required.
CARtastrophies: adverse events from CAR T cells
As with all therapies, the toxicity from CAR T cells may be classified as on-target or off-target. On-target toxicities have occurred and can be categorized as those toxicities occurring from the targeting of tumor cells, as well as normal cells that have shared expression of the CAR-specific antigen. On-target off tumor toxicity was first reported in the biliary tract with carbonic anhydrase IX CAR T cells [23]. This report, along with others since [32], has disproven the commonly held belief by those mostly outside of the field that clinical safety with antibody studies suggests that the same antibody-targeting moiety in a CAR should also be safe. In the case of CD19- and CD20-directed CARs, B cell aplasia has been reported in many trials [33, 34]. This is due to the fact that in addition to leukemic cells, normal B cells express lineage differentiation antigens CD19 and CD20. One approach to limit the extent of B cell aplasia is being pursued at Baylor College of Medicine, where CAR T cells directed against the kappa light chain subset of the immunoglobulin receptor are being tested [35], thereby potentially sparing a portion of the B cell repertoire. For CD19- and CD20-directed CAR T cells, the major on-target on-tumor toxicity has been tumor lysis syndrome, and related to this, cytokine release syndrome. The tumor lysis syndrome that has been observed is generally related to the bulk of tumor in the patient and is managed as per standard practice by medical oncologists. The unique feature of tumor lysis syndrome following CAR T cell therapy is that it may be delayed for up to 50 days following infusion of the T cells [26], likely reflecting the time required for the CAR T cells to proliferate in those patients.
Cytokine release syndrome occurs in nearly all patients who are responding to B cell-directed CAR therapy and, as was mentioned above, is proportional to the tumor burden in the patient. Cytokine release syndrome is characterized by fever and in more severe cases, renal insufficiency, pulmonary insufficiency and altered mental status. Surprisingly, we found that this can be managed with systemically administered cytokine blockade [36], sparing the use of more broadly immunosuppressive agents such as corticosteroids. To this point, the anti-IL6 receptor antibody tocilizumab has been an effective agent to manage cytokine release syndrome [27]. It is possible that other anti-cytokine reagents such as drugs that would block the IL-1 cascade would also be effective. Similarly, it is possible that inhibitors such as JAK kinase inhibitors may also diminish or prevent the symptoms of cytokine release syndrome. A subset of patients treated with CART19 at the University of Pennsylvania have had macrophage activation syndrome, a clinical syndrome that is related to hemophagocytic lymphohistiocytosis [37, 38]. This is characterized by elevations in serum ferritin, C-reactive protein and biochemical evidence of coagulopathy without overt bleeding diathesis. The cause of this syndrome is not yet known, but it may reflect hyperactivation of the immune system, leading the CART19 T cells to trigger macrophage activation. The most serious toxicity reported to date with anti-CD19 CAR T cells has been neurologic toxicity. This appears to be more prominent with CAR T cells that use a CD28 signaling domain [28, 39, 40], perhaps related to the enhanced TNFalpha secretion associated with CD28 signaling [41]. It is interesting to note that significant neurologic toxicity has also been reported with the CD19-directed bispecific antibody blinatumomab [42]. Whether caused by CAR T cells or blinatumomab, neurologic toxicity is usually reversible [43].
Comparisons of TCR- and CAR-modified T cells
The relative advantages and disadvantages of approaches using T cells engineered with TCRs and CARs can be compared and contrasted. On the one hand, the TCR is a more sensitive signaling receptor, requiring very few ligands (<10 under optimized conditions) in order to trigger sufficient signaling within the T cell to induce in target-specific cytolysis [44]. In contrast, CAR T cells have been shown to require approximately 100 targets at the cell surface in order to trigger killing [45]. The affinity of TCRs for antigen is approximately 1000-fold lower than CAR T cells, so that the signaling intensity triggered by tumor targets in CAR-modified T cells may be higher and potentially non-physiologic. There is potentially a larger set of “druggable targets” in cancer cells targeted with TCR-engineered T cells, as the TCR can in principle sense the entire intracellular proteome that is presented by MHC molecules. In contrast the set of targets for CAR-modified T cells is limited to structures on the tumor surface, notably proteins, glycolipids and carbohydrate antigens. However, a significant limitation of TCR-modified T cells is that they require the tumor target to retain MHC class I expression and normal endogenous processing of antigen. In contrast, the CAR-modified T cells can kill tumor cells that have lost MHC expression, an unfortunately common occurrence in human cancers [46, 47]. In addition, a CAR-modified T cell is more off-the-shelf in that it does not have to be HLA-matched to the patient and the tumor, unlike TCR-modified T cells. In studies to date, it appears that both TCR-modified T cells and CAR-modified T cells can engraft and function in patients; however, to date only CAR-modified T cells have been shown to persist and exhibit sustained effector functions for more than a year in patients [19].
Future directions
There is an increasing excitement in the field that engineered T cell therapies can be a potent new addition to the toolbox for cancer therapy. It is encouraging to note that several pharmaceutical companies and biotechnology companies have now begun development of T cells using TCR and CAR engineering technology. The major question in the field is whether or not engineered T cells will be effective for solid tumors. It is encouraging that in preclinical studies, recent clinical trials show that CAR T cells and TCR T cells are effective for a variety of solid cancers [5, 11, 48–50]. It is likely that CAR T cells will have even more potent effects when combined with checkpoint blockade, as reported in preclinical models [51]. Another question facing the field is what the frequency of target loss will be. At this time, approximately 10 % of patients treated the University of Pennsylvania with CD19-specific CAR T cells have undergone immunoediting of their tumor as revealed by recurrent tumor with antigen loss variants [27]. Finally, the major engineering issue facing the field of CAR T cell therapy is that the manufacturing of CAR T cells is currently accomplished on an “n of 1” basis. This is done using manual cell culture technologies. In order for the therapy to become routinely applied in the practice of medicine, it is necessary that the process be developed by so-called “out-scaling,” using automation and robotic culture technologies so that this promising form of therapy can enter the mainstream practice of oncology therapy [52]. Finally, it is possible that universal donor CAR T cells will have a role [53, 54], in addition to autologous T cells.
Acknowledgments
The authors declare sponsored research support from Novartis.
Conflict of interest
The authors have intellectual property in this field that is owned by the University of Pennsylvania, and licensed by Novartis.
Footnotes
This paper is a Focussed Research Review based on a presentation given at the 19th Danish Cancer Society Symposium in Copenhagen, Denmark, 23rd–25th September 2013, on the topic “Immunotherapy of Cancer—Present Status and Future Promise”. It is part of a CII series of Focussed Research Reviews and meeting report.
References
- 1.Drake CG, Jaffee E, Pardoll DM (2006) Mechanisms of immune evasion by tumors. Adv Immunol 90:51–81 [DOI] [PubMed]
- 2.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437. doi: 10.1016/S1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. Gene therapy with human and mouse T cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. 2001;248:67–76. doi: 10.1016/S0022-1759(00)00343-4. [DOI] [PubMed] [Google Scholar]
- 7.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. 2012;42:3174–3179. doi: 10.1002/eji.201242606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simpson A, Caballero O, Jungbluth A, Chen Y, Old L. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G et al (2013) Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-Directed T cells. Sci Transl Med 5(197):197ra103. doi:10.1126/scitranslmed.3006034 [DOI] [PMC free article] [PubMed]
- 13.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. Cancer regression and neurological toxicity Following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-O. [DOI] [PubMed] [Google Scholar]
- 16.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–1046. doi: 10.1016/0092-8674(91)90327-U. [DOI] [PubMed] [Google Scholar]
- 17.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci USA. 1991;88:8905–8909. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsuyasu RT, Anton P, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4 z gene-modified autologous CD4+ and CD8+ T cells in HIV-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 19.Scholler J, Brady T, Binder-Scholl G, Hwang W-T, Plesa G, Hege K, Vogel A, Kalos M, Riley J, Deeks S et al (2012) Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4(132):132Ra153. doi:10.1126/scitranslmed.3003761 [DOI] [PMC free article] [PubMed]
- 20.McGuinness RP, Ge Y, Patel SD, Kashmiri SV, Lee HS, Hand PH, Schlom J, Finer MH, McArthur JG. Anti-tumor activity of human T cells expressing the CC49-zeta chimeric immune receptor. Hum Gene Ther. 1999;10:165–173. doi: 10.1089/10430349950018968. [DOI] [PubMed] [Google Scholar]
- 21.Finney HM, Lawson ADG, Bebbington CR, Weir ANC. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791–2797. [PubMed] [Google Scholar]
- 22.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 24.Kochenderfer J, Wilson W, Janik J, Dudley M, Stetler-Stevenson M, Feldman S, Maric I, Raffeld M, Nathan D, Lanier B, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically-engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH (2011) T cells expressing chimeric receptors establish memory and potent antitumor effects in patients with advanced leukemia. Sci Transl Med 3(95):95ra73. doi:10.1126/scitranslmed.3002842 [DOI] [PMC free article] [PubMed]
- 26.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold S, Teachey D, Chew A, Hauck B, Wright J, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M et al (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5(177):177ra138. doi:10.1126/scitranslmed.3005930 [DOI] [PMC free article] [PubMed]
- 29.Effros RB, Pawelec G. Replicative senescence of T cells: does the Hayflick Limit lead to immune exhaustion? Immunol Today. 1997;18:450–454. doi: 10.1016/S0167-5699(97)01079-7. [DOI] [PubMed] [Google Scholar]
- 30.Brentjens R, Rivière I, Park J, Davila M, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1825. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan R, Yang J, Kitano M, Dudley M, Laurencot C, Rosenberg S. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jena B, Dotti G, Cooper L. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maus MV, Grupp SA, Porter DL, June CH. Antibody modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–2635. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, Wu J, Heslop HE, Rooney CM, Brenner MK, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr. 2014;26:43–49. doi: 10.1097/MOP.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Xu X, Song H, Yang S, Shi S, Wei J, Pan B, Zhao F, Liao C, Luo C. Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. Br J Haematol. 2008;143:84–91. doi: 10.1111/j.1365-2141.2008.07298.x. [DOI] [PubMed] [Google Scholar]
- 38.Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, Attard L, Bosi A, Pende D, Moretta L, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi: 10.1371/journal.pone.0044649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6(224):224ra225. doi:10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed]
- 41.Saha B, Harlan DM, Lee KP, June CH, Abe R. Protection against lethal toxic shock by targeted disruption of the CD28 gene. J Exp Med. 1996;183:2675–2680. doi: 10.1084/jem.183.6.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gokbuget N, Neumann S, Goebeler M, Viardot A, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 43.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine directed therapy. Blood. 2013;121:5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis MM, Krogsgaard M, Huse M, Huppa J, Lillemeier BF, Li QJ. T cells as a self-referential, sensory organ. Annu Rev Immunol. 2007;25:681–695. doi: 10.1146/annurev.immunol.24.021605.090600. [DOI] [PubMed] [Google Scholar]
- 45.Stone JD, Aggen DH, Schietinger A, Schreiber H, Kranz DM. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell engagers (BiTEs) Oncoimmunology. 2012;1:863–873. doi: 10.4161/onci.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/S0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 47.Vitale M, Pelusi G, Taroni B, Gobbi G, Micheloni C, Rezzani R, Donato F, Wang X, Ferrone S. HLA class I antigen down-regulation in primary ovary carcinoma lesions: association with disease stage. Clin Cancer Res. 2005;11:67–72. [PubMed] [Google Scholar]
- 48.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bendle GM, Linnemann C, Bies L, Song JY, Schumacher TN. Blockade of TGF-beta signaling greatly enhances the efficacy of TCR gene therapy of cancer. J Immunol. 2013;191:3232–3239. doi: 10.4049/jimmunol.1301270. [DOI] [PubMed] [Google Scholar]
- 50.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.John LB, Devaud C, Duong CM, Yong C, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 52.Levine BL, June CH. Perspective: assembly line immunotherapy. Nature. 2013;498:S17. doi: 10.1038/498S17a. [DOI] [PubMed] [Google Scholar]
- 53.Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, Chu V, Paschon DE, Zhang L, Kuball J, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, Crossland DL, Huls H, Littman N, Zhang Z, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–1349. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]