

Abstract
Background. Fine-needle aspiration (FNA) can cause misdiagnosis of cytomorphological findings between parathyroid and thyroid lesions. Case Presentation. A 31-year-old man presented with a palpable neck mass on the right thyroid lobe. FNA cytology was reported as intrathyroidal lymphoid hyperplasia. After 5 years, repeated FNA was done on the enlarged nodule with result of Hürthle cell lesion. Prior to right lobectomy, laboratories revealed elevated serum calcium and parathyroid hormone (PTH). Careful history taking revealed chronic knee pain and ossifying fibroma at the maxilla. Ultrasonography showed a 2.8 cm mass inferior to right thyroid lobe. Pathology from en bloc resection was parathyroid carcinoma and immunohistochemical study revealed positivity for PTH. Genetic analysis found somatic mutation of CDC73 gene in exon1 (c.70delG) which caused premature stop codon in amino acid 26 (p.Glu24Lysfs*2). The final diagnosis was hyperparathyroidism-jaw tumor syndrome. Conclusions. FNA cytology of parathyroid can mimic thyroid lesion. It is important to consider and correlate the entire information from clinical history, laboratory, imaging, and FNA.
1. Introduction
Parathyroid carcinoma is a rare malignancy, affecting less than 1% of patients with primary hyperparathyroidism [1, 2]. It is usually associated with severe clinical presentations with markedly elevated levels of serum calcium >14 mg/dL and parathyroid hormone (PTH) levels from >5 to 10 times the upper normal limit [3]. Therefore, almost all the patients are symptomatic at the time of presentation [4]. However, it can be an indolent tumor with subtle manifestation reported in 2–7% and up to 30% in the previous case series [5–7]. Patients with parathyroid carcinoma frequently present with a palpable neck mass that can masquerade as a thyroid nodule because of the close localization [8]. The fine-needle aspiration (FNA) can cause misdiagnosis of cytomorphological findings because of significant overlap in cytological features of parathyroid and thyroid lesions. Oncocytic parathyroid adenoma can be confused with Hürthle cell thyroid neoplasm, especially in the absence of clinical information [9].
A high prevalence of parathyroid carcinoma has been demonstrated in patients with hyperparathyroidism-jaw tumor syndrome (HPT-JT; OMIM#145001). It is an exceedingly rare cancer syndrome characterized by primary hyperparathyroidism, ossifying fibromas of the maxilla and mandible, and less commonly renal hamartoma, Wilms' tumor, polycystic kidney disease, degenerative cysts, and/or uterine tumors. Primary hyperparathyroidism is usually the presenting manifestation occurring in late adolescence or in young adulthood. Approximately 15% of these cases have parathyroid carcinoma. A germline inactivating mutation of the CDC73 tumor suppressor gene (formerly known as HRPT2 gene; OMIM#607393) can be identified in most patients with HPT-JT and in approximately 20% of patients with sporadic parathyroid carcinoma [10–12]. Also, somatic mutation of CDC73 gene is present in 60–100% of sporadic parathyroid carcinoma [10, 12–14].
We herein reported a case of a parathyroid carcinoma misinterpreted as Hürthle cell thyroid neoplasm on FNA results and subsequently uncovered HPT-JT.
2. Case Report
A 31-year-old man presented with palpable mass on the right side of his neck. Palpation of the thyroid glands showed a 1.5 cm nodule at the lower pole of the right lobe, with no cervical lymphadenopathy. Thyroid function tests were normal. FNA without ultrasound guidance was successfully performed. The cytology was reported as intrathyroidal lymphoid hyperplasia. After observation for 5 years, the size of nodule was slowly enlarged without compressive symptoms. Repeated FNA was done with the result of Hürthle cell lesion (Figure 1). The surgeon decided to perform a right lobectomy. His preoperative laboratory investigations revealed normal thyroid function tests. Serum calcium, phosphorus, and parathyroid hormone (PTH) were 13.5 mg/dL (reference range: 8.5–10.1 mg/dL), 1.9 mg/dL (reference range: 2.5–4.5 mg/dL), and 1,859 pg/mL (reference range: 15–65 pg/mL), respectively. Renal functions were within normal limits. Then, the patient was referred to endocrinologists. On ultrasound examination, a 2.8 cm heterogenous hypervascular mass was located posterior to the right inferior pole of thyroid gland (Figure 2). The technetium-99 m sestamibi single-photon emission computed tomography (Tc-99 m MIBI SPECT) showed increased uptake corresponding to ultrasound findings. Careful systemic history taking revealed that he had been suffering from chronic knee pain for 7 years. Bone radiography showed generalized demineralization, multiple osteolytic lesions at distal femur, proximal tibia, fibula, and patella. Subperiosteal bone resorption along the radial aspect of the middle phalanges and acroosteolysis of digital tufts was shown in X-ray of hands. These radiographic findings were compatible with osteitis fibrosa cystica. Bone mineral density Z-scores of lumbar spine, neck of femur, and one-third of distal radius were −3.8, −3.7, and −7.5, respectively, indicating low bone density. Therefore, parathyroid carcinoma was suspected, and en bloc resection was performed (Figure 3(a)). After the operation, the patient developed hungry bone syndrome. The pathology result was parathyroid carcinoma which microscopically demonstrated vascular invasion in the tumor capsule (Figure 3(b)). The tumor composed mixture of chief cell and oncocytic cell types with occasional nuclear atypia and prominent nucleoli, arranged in solid sheets trabecular and nest pattern (Figure 3(c)). Immunohistochemical (IHC) study revealed positive immunoreactivity for chromogranin A and parathyroid hormone (Figure 3(d)) but negative immunoreactivity for synaptophysin, calcitonin, and thyroglobulin. This confirmed the parathyroid nature of the lesion.
Figure 1.
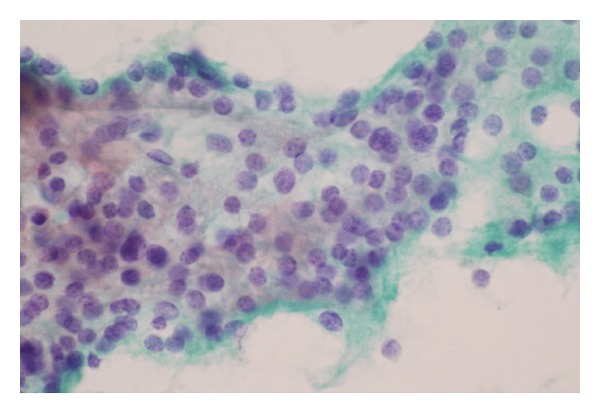
FNA initially reported sheets of follicular cells with oncocytic metaplasia, some naked nuclei, and focal inspissated colloid, compatible with Hürthle cell thyroid lesion. This was difficult to differentiate from parathyroid lesion if lacking of clinical information.
Figure 2.
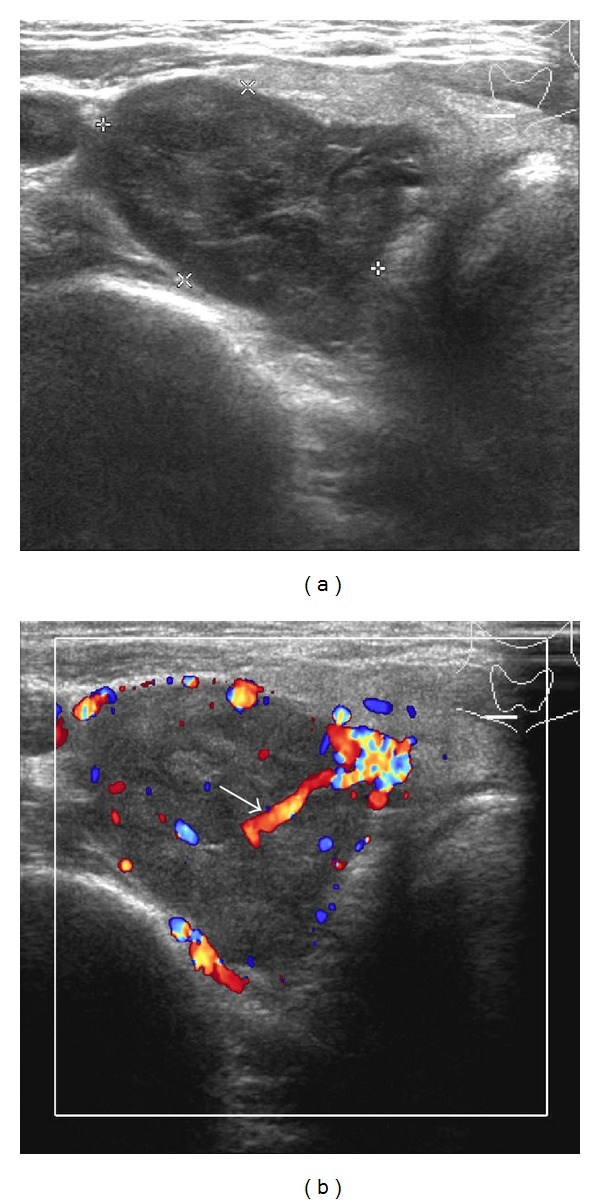
Ultrasonogram of thyroid gland and parathyroid glands. (a) Ultrasonogram and (b) color Doppler flow of right thyroid lobe showed a 2.8 × 1.9 cm heterogenous hypo-to-isoechoic with central hypervascularity solid mass located just inferior to right lobe of thyroid gland. The presence of extrathyroidal feeding artery or polar artery was shown (arrow). This mass was corresponding to palpable neck mass.
Figure 3.
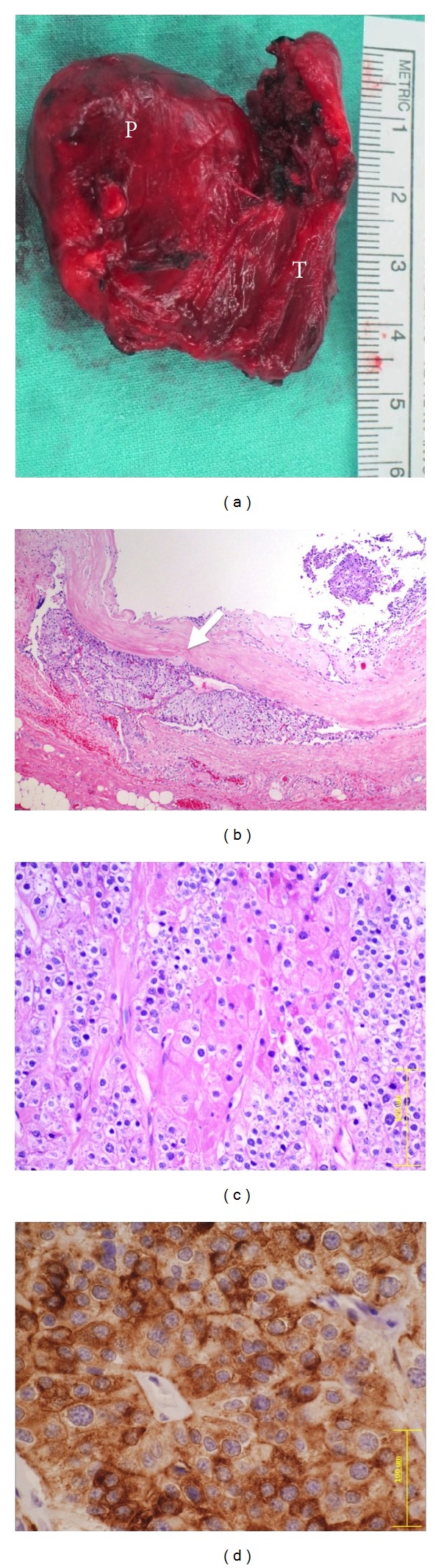
Pathology of parathyroid carcinoma. (a) Gross pathology of parathyroid carcinoma. A large solid tan mass (left, P) was adhered to the right lobe of thyroid gland (right, T). (b) Histopathology demonstrated vascular invasion (arrow) in the tumor capsule (H&E, 40x). (c) The tumor composed mixture of chief cells and oncocytic cells types arranged in solid sheets trabecular and nest pattern (H&E, 200x). (d) Immunohistochemical study for parathyroid hormone (PTH) revealed immunoreactivity in the tumor cells (PTH, 400x).
Additional review of the patient's history revealed the past history of operation for the tumor of the maxilla when he was 10 years old. Pathology showed ossifying fibroma at the right maxilla. Additional computed tomography of chest and abdomen found normal kidney structure and no evidence of metastasis. After informed consent was obtained, we directly sequenced the full coding and flanking splice-junctional regions of the CDC73 gene in the patient's blood and parathyroid tumor and found a somatic frameshift mutation of CDC73 gene in exon1 (c.70delG) which caused premature stop codon in amino acid 26 (p.Glu24Lysfs*2). His mother who had history of endometrial cancer of uterus underwent genetic testing for a CDC73 mutation and was found to be negative. This study was performed with approval from the Ramathibodi Hospital Institutional Review Board. The final diagnosis was parathyroid carcinoma with HPT-JT.
3. Discussion
FNA cytology of the parathyroid might be misdiagnosed as Hürthle cell associated lesions of thyroid such as Hürthle cell thyroid neoplasm [9, 15–17]; adenomatous thyroid nodules with Hürthle cell change; or chronic lymphocytic thyroiditis [18] because they might share some cytomorphologic similarities such as follicular structures, colloid-like material in the background [9]. Moreover, the presence of oncocytic cells and naked nuclei of chief cells in parathyroid cytologic specimen can be mimicking Hürthle cells and lymphocytes, respectively [19, 20]. This can be more challenging when the parathyroid lesion is mainly composed of oncocytic cells.
There are some cytomorphologic features that are helpful to differentiate between oncocytic parathyroid and Hürthle cell thyroid neoplasm. Parathyroid cells are smaller and have pale scant cytoplasm in combination with the highly eosinophilic cytoplasm. The cell borders are poorly defined. The nuclei of the parathyroid cells are frequently round to oval with stippled nuclear chromatin or salt-and-pepper appearance and sometimes contain nucleoli. Numerous naked nuclei, approximately the size of erythrocytes, in the background of the smear favor a parathyroid lesion. Hürthle cell thyroid neoplasms have much larger and more prominent nucleoli, and the cells tend to be more dyscohesive [16]. There is no single definite diagnostic criterion that helps to differentiate reliably parathyroid lesions from those of the thyroid, but rather a combination of the cytomorphological features should be applied (Table 1) [21].
Table 1.
Comparison features between oncocytic parathyroid lesions and Hürthle cell thyroid neoplasm.
| Cytomorphologic features | Oncocytic parathyroid lesions | Hürthle cell thyroid neoplasm |
|---|---|---|
| Patterns | Cells isolated and in loose aggregates or syncytial fragments; nuclear overlapping molding; and anisokaryosis | Cells in syncytial fragments; more dyscohesive |
|
| ||
| Cells | Small 6–9 μm in diameter; round to cuboidal | Larger than parathyroid; variable in size |
|
| ||
| Naked nuclei | Numerous | Few |
|
| ||
| Nuclei | Small; coarsely granular chromatin | Variably enlarged; fine to coarsely chromatin |
|
| ||
| Nucleoli | Inconspicuous | Prominent |
|
| ||
| Cytoplasm | Scant; clear, granular, or eosinophilic | Scant |
|
| ||
| Colloid | Absent but colloid-like material | Scant/− |
|
| ||
| IHC studies | ||
| PTH | + | − |
| TTF-1 | − | + |
| Thyroglobulin | − | + |
| Chromogranin | + | − |
PTH: parathyroid hormone; TTF-1: thyroid transcription factor-1; IHC: immunohistochemical.
In addition to cytomorphology, the clinical information of hyperparathyroidism including pathological fractures, joint and bone pain, muscular asthenia, fatigue, nausea, vomiting, loss of appetite, polyuria, polydipsia, nephrolithiasis, constipation, and weight loss is important. Despite this, these presentations are nonspecific and subtle. High-resolution ultrasonography of neck provides a noninvasive technique to facilitate the differential diagnosis of neck mass. American Thyroid Association recommends that a thyroid ultrasonography should be performed on all patients with suspected thyroid nodules [22]. Furthermore, ultrasound may reveal an unsuspected parathyroid lesion. Normal parathyroid glands are seldom seen on ultrasound. Enlarged parathyroids appear as ovoid mass with homogeneously hypoechoic echogenicity in relation to the thyroid gland and mostly locates adjacent the posterior aspect of the thyroid separated by a fibro-fatty capsule seen as a hyperechoic line between the thyroid gland and the parathyroid mass. Intrathyroidal parathyroid adenoma is about 2–3.4% of cases [23]. Doppler images help in distinguishing suspected parathyroid glands from other structures because parathyroid adenomas typically have a peripheral rim of vascularity and asymmetrically increased blood flow compared to the thyroid. Identification of an extrathyroidal artery (polar artery) feeding a parathyroid mass can discriminate parathyroid glands from lymph nodes, which usually have a hilar blood supply [24]. A panel of IHC study for the PTH, thyroglobulin, thyroid transcription factor-1 (TTF-1), and chromogranin and analyses of the PTH level of the washouts of FNA might be helpful [25, 26]. However, false negative case of PTH staining can occur due to poor cellularity of smears or previously stained smears [21].
Cytologic features can be useful in identifying a parathyroid origin of cells, but it is difficult to make a distinction between normal parathyroid gland and hyperplasia, adenoma, or carcinoma [27]. The appropriate treatment of parathyroid carcinoma is en bloc resection which includes resection of the ipsilateral thyroid lobe, together with the isthmus and lymphadenopathy of central compartment of the neck. Thus, it is important to preoperatively identify patients with parathyroid carcinoma to raise the chance of cure by performing complete surgical resection at the initial operation. Several preoperative factors can predict parathyroid carcinoma such as tumor size larger than 3 cm, alkaline phosphatase more than 285 IU/L [28], younger age, hard consistency, high serum calcium, and PTH levels [8, 29].
In addition to thyroid follicular cells, medullary thyroid carcinoma is also included in differential diagnosis for parathyroid cells [30]. Medullary thyroid carcinoma and parathyroid lesions have shared the same features on stippled nuclear chromatin, round to oval cells, loose clusters, and single cells. But the concurrent presence of spindle and oval cells with a granular cytoplasm and an absence of naked nuclei usually are usually considered a diagnosis of medullary thyroid carcinoma. Use of IHC studies for calcitonin in difficult cases has diagnostic value in the differentiation between medullary thyroid carcinoma and parathyroid lesions.
This patient presented with parathyroid carcinoma and later was found to have a history of ossifying fibroma of the maxilla that is compatible with rare endocrine syndrome, HPT-JT. These ossifying tumors are histologically different from the osteitis fibrosa cystica seen in primary hyperparathyroidism. Less commonly (15% of cases), patients suffer from renal complications, for example, various cystic and neoplastic renal abnormalities [31]. Moreover, up to 75% of women with HPT-JT present with uterine tumors [32]. It is an autosomal dominant familial cancer syndrome with variable expression and incomplete penetrance, with 10% of gene carriers showing no clinical manifestations [31]. In the majority of cases, primary hyperparathyroidism is the presenting symptom. A majority of cases with HPT-JT have a single-gland parathyroid involvement and a relatively high risk of parathyroid carcinoma up to 15% [33]. Untreated, HPT-JT may result in significant morbidity and mortality due to severe hypercalcemia. The HRPT2 gene, also known as CDC73, is located at chromosome 1q31.2 underlining the HPT-JT syndrome [11]. CDC73 consists of 17 exons and encodes 531 amino acids. It codes for a protein known as parafibromin that acts as a tumor suppressor involving the regulation of transcriptional events and histone modification; however, the exact mechanism by which parafibromin serves as a tumor suppressor remains unknown. Mutations in CDC73 are scattered throughout the coding region and splice sites and most mutations are predicted to cause inactivation of the protein product. About 80% of germline mutations have been reported in exons 1, 2, and 7 [34]. The majority of mutations are frameshift mutations, and a smaller proportion of mutations are nonsense and missense. Most mutations (more than 80%) result in premature truncation of parafibromin. Additionally, some patients with whole gene deletions have also been reported [35]. Somatic mutation of CDC73 is uncommon in sporadic benign parathyroid adenomas [36]. In contrast, mutations of CDC73 are frequently seen in apparently sporadic cases of parathyroid carcinoma [10, 12]. Our patient was found to have a frameshift somatic mutation in exon 1 causing premature stop codon. This mutation has been reported in ossifying fibroma of the jaw [37]. The identification of a mutation in the coding region would provide a definitive diagnosis, although a negative result could not exclude the possibility of a mutation in the promoter or other noncoding regions of CDC73. Carpten et al. reported that CDC73 sequencing fails to identify a germline mutation in about half of the index patients from families with classic HPT-JT and with proven genetic linkage to 1q24-q32 [11]. Unidentified germline mutation may locate in the 5′-regulatory region of CDC73. Gross deletions or rearrangements in CDC73 may not be readily identified using direct sequencing. Also it is possible that other tumor suppressor genes may contribute to the development of parathyroid carcinoma.
Accordingly, all patients with newly diagnosed parathyroid carcinoma should have a meticulous review of family history and should be considered for CDC73 mutation screening whether or not other features of HPT-JT are present. Genetic tests of this recognized mutation in at-risk asymptomatic family members would emphasize regular clinical and biochemical surveillance of carriers of the mutation and reassure to family members who have no mutation. Children with the gene mutation are recommended to undergo baseline screening of calcium and PTH levels, jaw X-rays, and renal ultrasound as early as age 5 [38]. Monitoring serum calcium concentrations in all subjects at risk provides an alternative to definitive genetic diagnosis.
4. Conclusions
It is difficult to distinguish parathyroid lesions from thyroid lesions based on solely FNA cytomorphologic diagnosis because of their morphologic similarities. Therefore, it is important to consider and relate the whole information from careful clinical history taking, laboratory, imaging studies, and FNA. PTH assay in FNA specimen and immunohistochemical study can definitely distinguish between parathyroid and thyroid lesions. HPT-JT is a rare condition that physicians should recognize and genetically counsel the patient and family members for regular surveillance for early detection of complications.
Acknowledgments
This study was supported byresearch funds from Ramathi- bodi Foundation and the Division of Endocrinology and Metabolism, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University.
Consent
Written informed consent was obtained from the patient for publication of this case report and the accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contributions
Chutintorn Sriphrapradang carried out the clinical work and drafted the manuscript. Pattana Sornmayura, Niramol Chanplakorn, and Rangsima Aroonroch expatiated on the pathology and provided the pictures. Objoon Trachoo and Pattarana Sae-Chew performed genetic analysis. All authors read, revised, and approved the final paper.
References
- 1.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. Two hundred eighty-six cases of parathyroid carcinoma treated in the U.S. between 1985–1995: a National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1999;86(3):538–544. doi: 10.1002/(sici)1097-0142(19990801)86:3<538::aid-cncr25>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 2.Schaapveld M, Jorna FH, Aben KKH, Haak HR, Plukker JTM, Links TP. Incidence and prognosis of parathyroid gland carcinoma: a population-based study in the Netherlands estimating the preoperative diagnosis. The American Journal of Surgery. 2011;202(5):590–597. doi: 10.1016/j.amjsurg.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 3.Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. Journal of Bone and Mineral Research. 2008;23(12):1869–1880. doi: 10.1359/jbmr.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao L, Liu J, He X, et al. The changing clinical patterns of primary hyperparathyroidism in chinese patients: data from 2000 to 2010 in a single clinical center. The Journal of Clinical Endocrinology and Metabolism. 2013;98(2):721–728. doi: 10.1210/jc.2012-2914. [DOI] [PubMed] [Google Scholar]
- 5.Wynne AG, Van Heerden J, Carney JA, Fitzpatrick LA. Parathyroid carcinoma: clinical and pathologic features in 43 patients. Medicine. 1992;71(4):197–205. [PubMed] [Google Scholar]
- 6.Shane E, Bilezikian JP. Parathyroid carcinoma: a review of 62 patients. Endocrine Reviews. 1982;3(2):218–226. doi: 10.1210/edrv-3-2-218. [DOI] [PubMed] [Google Scholar]
- 7.Busaidy NL, Jimenez C, Habra MA, et al. Parathyroid carcinoma: a 22-year experience. Head and Neck. 2004;26(8):716–726. doi: 10.1002/hed.20049. [DOI] [PubMed] [Google Scholar]
- 8.Shane E. Clinical review 122: parathyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 2001;86(2):485–493. doi: 10.1210/jcem.86.2.7207. [DOI] [PubMed] [Google Scholar]
- 9.Paker I, Yilmazer D, Yandakci K, Arikok AT, Alper M. Intrathyroidal oncocytic parathyroid adenoma: a diagnostic pitfall on fine-needle aspiration. Diagnostic Cytopathology. 2010;38(11):833–836. doi: 10.1002/dc.21338. [DOI] [PubMed] [Google Scholar]
- 10.Shattuck TM, Välimäki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. The New England Journal of Medicine. 2003;349(18):1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- 11.Carpten JD, Robbins CM, Villablanca A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nature Genetics. 2002;32(4):676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 12.Cetani F, Pardi E, Borsari S, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5583–5591. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 13.Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. Journal of Medical Genetics. 2003;40(9):657–663. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haven CJ, Van Puijenbroek M, Tan MH, et al. Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clinical Endocrinology. 2007;67(3):370–376. doi: 10.1111/j.1365-2265.2007.02894.x. [DOI] [PubMed] [Google Scholar]
- 15.Weymouth MD, Serpell JW, Chambers D. Palpable parathyroid adenomas presenting as clinical solitarythyroid nodules and cytologically as follicular thyroid neoplasms. ANZ Journal of Surgery. 2003;73(1-2):36–39. doi: 10.1046/j.1445-2197.2003.02636.x. [DOI] [PubMed] [Google Scholar]
- 16.Giorgadze T, Stratton B, Baloch ZW, LiVolsi VA. Oncocytic parathyroid adenoma: problem in cytological diagnosis. Diagnostic Cytopathology. 2004;31(4):276–280. doi: 10.1002/dc.20115. [DOI] [PubMed] [Google Scholar]
- 17.Lieu D. Cytopathologist-performed ultrasound-guided fine-needle aspiration of parathyroid lesions. Diagnostic Cytopathology. 2010;38(5):327–332. doi: 10.1002/dc.21203. [DOI] [PubMed] [Google Scholar]
- 18.Auger M, Charbonneau M, Huttner I. Unsuspected intrathyroidal parathyroid adenoma: mimic of lymphocytic thyroiditis in fine-needle aspiration specimens-a case report. Diagnostic Cytopathology. 1999;21(4):276–279. doi: 10.1002/(sici)1097-0339(199910)21:4<276::aid-dc9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Bondeson L, Bondeson AG, Nissborg A, Thompson NW. Cytopathological variables in parathyroid lesions: a study based on 1,600 cases of hyperparathyroidism. Diagnostic Cytopathology. 1997;16(6):476–482. doi: 10.1002/(sici)1097-0339(199706)16:6<476::aid-dc2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Abati A, Skarulis MC, Shawker T, Solomon D. Ultrasound-guided fine-needle aspiration of parathyroid lesions: a morphological and immunocytochemical approach. Human Pathology. 1995;26(3):338–343. doi: 10.1016/0046-8177(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 21.Absher KJ, Truong LD, Khurana KK, Ramzy I. Parathyroid cytology: avoiding diagnostic pitfalls. Head and Neck. 2002;24(2):157–164. doi: 10.1002/hed.10003. [DOI] [PubMed] [Google Scholar]
- 22.Cooper DS, Doherty GM, Haugen BR, et al. Revised American thyroid association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- 23.Yabuta T, Tsushima Y, Masuoka H, et al. Ultrasonographic features of intrathyroidal parathyroid adenoma causing primary hyperparathyroidism. Endocrine Journal. 2011;58(11):989–994. doi: 10.1507/endocrj.ej11-0069. [DOI] [PubMed] [Google Scholar]
- 24.Lane MJ, Desser TS, Weigel RJ, Jeffrey RB., Jr. Use of color and power doppler sonography to identify feeding arteries associated with parathyroid adenomas. American Journal of Roentgenology. 1998;171(3):819–823. doi: 10.2214/ajr.171.3.9725323. [DOI] [PubMed] [Google Scholar]
- 25.Owens CL, Rekhtman N, Sokoll L, Ali SZ. Parathyroid hormone assay in fine-needle aspirate is useful in differentiating inadvertently sampled parathyroid tissue from thyroid lesions. Diagnostic Cytopathology. 2008;36(4):227–231. doi: 10.1002/dc.20783. [DOI] [PubMed] [Google Scholar]
- 26.Kwak JY, Kim E, Moon HJ, et al. Parathyroid incidentalomas detected on routine ultrasound-directed fine-needle aspiration biopsy in patients referred for thyroid nodules and the role of parathyroid hormone analysis in the samples. Thyroid. 2009;19(7):743–748. doi: 10.1089/thy.2008.0263. [DOI] [PubMed] [Google Scholar]
- 27.Dimashkieh H, Krishnamurthy S. Ultrasound guided fine needle aspiration biopsy of parathyroid gland and lesions. CytoJournal. 2006;3, article 6 doi: 10.1186/1742-6413-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae JH, Choi HJ, Lee Y, et al. Preoperative predictive factors for parathyroid carcinoma in patients with primary hyperparathyroidism. Journal of Korean Medical Science. 2012;27(8):890–895. doi: 10.3346/jkms.2012.27.8.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karakas E, Müller H-H, Lyadov VK, et al. Development of a formula to predict parathyroid carcinoma in patients with primary hyperparathyroidism. World Journal of Surgery. 2012;36(11):2605–2611. doi: 10.1007/s00268-012-1707-9. [DOI] [PubMed] [Google Scholar]
- 30.Kruljac I, Pavić I, Mateša N, et al. Intrathyroid parathyroid carcinoma with intrathyroidal metastasis to the contralateral lobe: source of diagnostic and treatment pitfalls. Japanese Journal of Clinical Oncology. 2011;41(9):1142–1146. doi: 10.1093/jjco/hyr094.hyr094 [DOI] [PubMed] [Google Scholar]
- 31.Teh BT, Farnebo F, Kristoffersson U, et al. Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21- q32 and loss of the wild type allele in renal hamartomas. The Journal of Clinical Endocrinology & Metabolism. 1996;81(12):4204–4211. doi: 10.1210/jcem.81.12.8954016. [DOI] [PubMed] [Google Scholar]
- 32.Bradley KJ, Hobbs MR, Buley ID, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism- jaw tumour syndrome. Journal of Internal Medicine. 2005;257(1):18–26. doi: 10.1111/j.1365-2796.2004.01421.x. [DOI] [PubMed] [Google Scholar]
- 33.Iacobone M, Masi G, Barzon L, et al. Hyperparathyroidism-jaw tumor syndrome: a report of three large kindred. Langenbeck's Archives of Surgery. 2009;394(5):817–825. doi: 10.1007/s00423-009-0511-y. [DOI] [PubMed] [Google Scholar]
- 34.Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Human Mutation. 2010;31(3):295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- 35.Bricaire L, Odou MF, Cardot-Bauters C, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. The Journal of Clinical Endocrinology and Metabolism. 2013;98(2):E403–E408. doi: 10.1210/jc.2012-2789. [DOI] [PubMed] [Google Scholar]
- 36.Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism. 2005;90(9):5015–5017. doi: 10.1210/jc.2005-0717. [DOI] [PubMed] [Google Scholar]
- 37.Pimenta FJ, Gontijo Silveira LF, Tavares GC, et al. HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncology. 2006;42(7):735–739. doi: 10.1016/j.oraloncology.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Kelly TG, Shattuck TM, Reyes-Mugica M, et al. Surveillance for early detection of aggressive parathyroid disease: carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. Journal of Bone and Mineral Research. 2006;21(10):1666–1671. doi: 10.1359/jbmr.060702. [DOI] [PubMed] [Google Scholar]