

Abstract
Currently, there is particular interest in the molecular mechanisms of adaptive evolution in bacteria. Neisseria is a genus of gram negative bacteria, and there has recently been considerable focus on its two human pathogenic species N. meningitidis and N. gonorrhoeae. Until now, no genome-wide studies have attempted to scan for the genes related to adaptive evolution. For this reason, we selected 18 Neisseria genomes (14 N. meningitidis, 3 N. gonorrhoeae and 1 commensal N. lactamics) to conduct a comparative genome analysis to obtain a comprehensive understanding of the roles of natural selection and homologous recombination throughout the history of adaptive evolution. Among the 1012 core orthologous genes, we identified 635 genes with recombination signals and 10 genes that showed significant evidence of positive selection. Further functional analyses revealed that no functional bias was found in the recombined genes. Positively selected genes are prone to DNA processing and iron uptake, which are essential for the fundamental life cycle. Overall, the results indicate that both recombination and positive selection play crucial roles in the adaptive evolution of Neisseria genomes. The positively selected genes and the corresponding amino acid sites provide us with valuable targets for further research into the detailed mechanisms of adaptive evolution in Neisseria.
1. Introduction
Homologous recombination and positive selection are two indispensable sources of genetic variation and play central roles in the adaptive evolution of many bacteria species [1, 2]. Of the two mechanisms, homologous recombination occurs frequently in some bacteria, such as Streptomyces [3], Helicobacter pylori [4], and Neisseria [5], and could possibly speed adaptation by reducing competition between beneficial mutations [6]. There is also evidence for positive selection in specific genes in certain pathogens, such as Listeria monocytogenes [7], Salmonella [8], Streptococcus [9], Campylobacter [10], and Actinobacilus pleuropneumoniae [11]. These positively selected genes are usually involved in the dynamic interaction between host and pathogen [12, 13].
At present, there are well-developed methods for detecting genes undergoing recombination and selection. Phi [14] and GENECONV [15] are two common methods used to detect recombination based on different statistical tests. The dN/dS-based method is typically used to estimate the ratio of the rate of nonsynonymous nucleotide substitutions to that of synonymous substitutions [16, 17]. This ratio indicates whether a gene has been under positive selection (ω > 1), neutral selection (ω = 1), or purifying selection (ω < 1). Combined with the codon models developed by Nielsen and Yang [16, 18], which allow variation in ω among sites, this method can identify positive selection signals when there are only few positive sites. All these methods will be employed in this study to detect the genes with the history of recombination or positive selection.
Neisseria is a genus of bacteria that colonizes the mucosal surfaces of many animals. Of the known 14 species, only 2 species, Neisseria meningitides and Neisseria gonorrhoeae, are human pathogens; and the remainders are all commensal or nonpathogenic. Until now, there have been many comparative genomic studies on the genomic evolution of these two pathogenic species [5, 19–27]. Homologous recombination has been found to play a key role in the adaptive evolution of Neisseria; however, few studies have characterised the effect of positive selection on the Neisseria genome. Only two genes, porB [28] and pilE [29], have received attention, and both have undergone strong positive selection pressure. In this study, we used the genome sequences available for the strains of N. meningitidis, N. gonorrhoeae, and nonpathogenic N. lactamica to investigate the contributions of recombination and positive selection to the evolution of Neisseria genomes. Considering the high sequence diversity and open pan-genome, we focused on the core genome genes during our scan for recombined genes and positively selected genes. Statistical tests and a literature review were conducted to determine the association between genes and the properties of this genus.
2. Materials and Methods
2.1. Data Preparation
Eighteen genome sequences of Neisseria, including complete proteomes and the corresponding coding genes, were retrieved from the NCBI Genome database (http://www.ncbi.nlm.nih.gov/genome/bacteria/). Detailed information, such as Genbank ID and genome size, is listed in Table 1. The COGs (clusters of orthologous groups of proteins) functional classification for each proteome was conducted with ID mapping from the Uniprot database [30]. Then, using Neisseria gonorrhoeae FA 1090 as the reference genome, stand-alone BLAST was performed against the proteomes of the remaining 17 strains for homologs (sequence identity > 80% and alignment coverage > 80%) of each of the FA_1090 proteins. For each of the core genes from FA_1090, BLAST was performed against all 18 genomes (including the reference genome) with the same thresholds, and multiple copies in any genome were reported and removed from further analysis. The remaining core proteins were defined as the core orthologs of Neisseria.
Table 1.
Genome sequences used in this study.
| Strain name | GenBank accession no. | Genome size (Mbp) | No. of CDS | CC ID |
|---|---|---|---|---|
| Neisseria_meningitidis_FAM18 | NC_008767 | 2.19 | 1917 | CC11 |
| Neisseria_meningitidis_G2136 | NC_017513 | 2.18 | 1928 | CC8 |
| Neisseria_meningitidis_WUE_2594 | NC_017512 | 2.23 | 1941 | CC5 |
| Neisseria_meningitidis_Z2491 | NC_003116 | 2.18 | 1909 | CC4 |
| Neisseria_meningitidis_8013 | NC_017501 | 2.28 | 1913 | CC18 |
| Neisseria_meningitidis_053442 | NC_010120 | 2.15 | 2020 | CC4821 |
| Neisseria_meningitidis_alpha14 | NC_013016 | 2.14 | 1872 | CC53 |
| Neisseria_meningitidis_M04_240196 | NC_017515 | 2.25 | 1947 | CC269 |
| Neisseria_meningitidis_H44_76 | NC_017516 | 2.24 | 1961 | CC32 |
| Neisseria_meningitidis_MC58 | NC_003112 | 2.27 | 2063 | CC32 |
| Neisseria_meningitidis_alpha710 | NC_017505 | 2.24 | 2017 | CC41/44 |
| Neisseria_meningitidis_M01_240149 | NC_017514 | 2.22 | 1936 | CC41/44 |
| Neisseria_meningitidis_NZ_05_33 | NC_017518 | 2.24 | 1948 | CC41/44 |
| Neisseria_meningitidis_M01_240355 | NC_017517 | 2.29 | 1971 | CC213 |
| Neisseria_gonorrhoeae_TCDC_NG08107 | NC_017511 | 2.15 | 2196 | |
| Neisseria_gonorrhoeae_FA_1090 | NC_002946 | 2.15 | 2002 | |
| Neisseria_gonorrhoeae_NCCP11945 | NC_011035 | 2.23 | 2680 | |
| Neisseria_lactamica_020_06 | NC_014752 | 2.22 | 1972 |
2.2. Alignment and Calculation of Nucleotide Diversity, Informative Sites, Codon Bias, dN, and dS
The orthologous protein sequences were aligned using the method implemented in muscle [31]. Then, multiple codon alignments of genes corresponding to protein sequence alignments were obtained using PAL2NAL [32]. Using the resulting gene alignments, the gene-by-gene number of informative sites and the nucleotide diversity were obtained from the output of the PhiPack program [14].
In this study, the effective number of codons (Nc) was used to measure the codon bias. The Nc value ranges from 20 for the strongest bias to 61 for no bias [33], and the program CodonW (http://sourceforge.net/projects/codonw/) was used to calculate the values of Nc for each gene. The number of synonymous nucleotide substitutions per synonymous site (dS) and the number of nonsynonymous nucleotide substitutions per nonsynonymous site (dN) were estimated from the gene alignments using the program SNAP [34].
2.3. Detection of Recombination
Four statistical procedures GENECONV [15], pairwise homoplasy index (Phi) [14], maximum χ 2 [35], and neighbor similarity score (NSS) [36] were run on the aligned genes to discover the homologous recombination signals. For the analyses of GENECONV, the parameter g-scale was set to 1, which allows mismatches within a recombining fragment. The P values were calculated from 10000 random permutations of the data. The remaining three programs were implemented in the PhiPack package and were run with default parameters.
2.4. Detection of Selection
FastTree [37] was used to construct maximum likelihood phylogenetic trees with a general time-reversible (GTR) model of nucleotide substitution for each gene alignment. The resulting topologies of ML trees were applied to subsequent selection analysis.
The codeml program from PAML [38] was used to detect the genes under positive selection. Two site-specific models were applied: the null model M1a (nearly neutral) and the alternative model M2a (positive selection); the two models differ by the statistical distribution assumed for the ω ratio. The latter model allows sites with ω > 1, whereas the former only allows sites with ω varying between 0 and 1. To ensure convergence to the best likelihood, all calculations were performed three times. A likelihood ratio test (LRT) was then carried out to infer the occurrence of sites under positive selection pressure through comparing M1a against M2a. P values were determined from the LRT scores calculated by the module χ 2 of the PAML package.
2.5. Statistical Analysis
Correction for multiple testing was performed using the method presented by Benjamini and Hochberg [39]. For all genes tested for recombination and positive selection, q-values were calculated for each P value using the R package [40, 41] (q-value with the proportion of true null hypothesis set to 1). According to the conservation of tests, false discovery rates of 10% and 20% were used for the recombination analyses and positive selection detection, respectively.
The significance level for differences among the properties, including nucleotide diversity, codon bias, dS, and dN, between a COG and other COGs was determined using the nonparametric Mann-Whitney U-test. Correlation between each COG and evolutionary forces (homologous recombination and positive selection) was estimated using a binomial test. Then, Bonferroni corrections for multiple comparisons were performed according to the number of one-sided tests. The significance level was set to 5%. All statistical tests were carried out using Python scripts and R.
3. Results and Discussion
3.1. Characterization of the Orthologous Genes in 18 Neisseria Genomes
Previous studies [42–45] showed that both intraspecies and interspecies recombination could act as the important genetic mechanism in generating new clones and alleles in Neisseria. The genus Neisseria consists of two important pathogenic species and a dozen species that are never or rarely pathogenic. At present, there are only 18 completely sequenced genomes of genus Neisseria available, including 14 N. meningitidis, 3 N. gonorrhoeae, and 1 N. lactamics genomes. Thus, we selected all 18 genomes to conduct a genome-wide scan for the identification of genes exhibiting recombination or positively selected signals.
The phylogenetic relationships of the 18 strains were first established based on the 7 housekeeping genes frequently used for multilocus sequence typing (MLST) analysis of Neisseria: abcZ, adk, aroE, fumC, gdh, pdhC, and pgm [46]. The 7 genes were concatenated to construct a maximum likelihood tree with high bootstrap values as shown in Figure 1. In the tree, the three species were divided into three clades and formed a monophyly, respectively.
Figure 1.
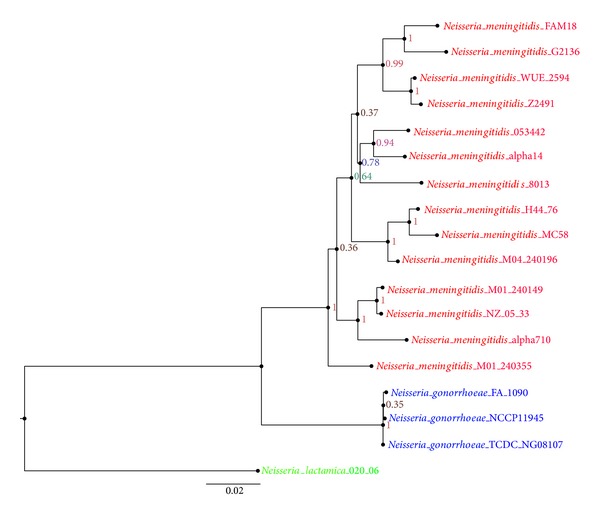
Phylogram of concatenated sequences of 7 housekeeping genes (abcZ, adk, aroE, fumC, gdh, pdhC, and pgm) for the 18 Neisseria genomes analyzed. The genomes in different species are marked with different colors: red for Neisseria meningitides, blue for Neisseria gonorrhoeae, and green for Neisseria lactamics. The numbers labeled on each internal node are the boostrap values.
In the next step, N. gonorrhoeae FA 1090 was used as the reference genome to perform a BLAST search against the other 17 Neisseria genomes for orthologs. Finally, 1034 genes were identified as present in all 18 genomes, containing the initial definition of the core genome for these Neisseria strains and accounting for 38.73% to 55.45% of the coding genes in each genome. This proportion is similar to that in previous analysis of Neisseria meningitides genomes [5, 27]. Of the 1034 core genes, 22 genes occurred as two or more copies in some genomes and were excluded from further analysis. The remaining 1012 genes with a single copy per genome were then defined as the core orthologous genes for subsequent analysis of homologous recombination and natural selection.
Among these genes, genes in COGs “Replication, recombination, and repair” were found to show higher nucleotide diversity than genes in other COGs (Table 2). For the association between the number of informative sites and COGs, the same result was obtained, which means genes in category “Replication, recombination, and repair” also had more informative sites than genes in other COGs (Table 2).
Table 2.
Association between COGs and descriptive variables.
| Functional category | Number of genes analyzed | Bonferroni-corrected P value for one-sided U−test for association between genes in a given COG and(1) | |||||
|---|---|---|---|---|---|---|---|
| >nt diversity | >Number of Informative sites | >Codon bias(2) | <Codon bias(2) | >dS | >dN | ||
| Energy production and conversion | 85 | <0.001 | |||||
| Nucleotide metabolism and transport | 37 | 0.03 | |||||
| Translation, ribosomal structure, and biogenesis | 110 | <0.001 | 0.03 | ||||
| Replication, recombination, and repair | 70 | <0.001 | <0.001 | 0.03 | |||
| Cell wall/membrane/envelope biogenesis | 75 | 0.002 | |||||
| Function unknown | 93 | 0.023 | 0.03 | ||||
| Intracellular trafficking, secretion and vesicular transport | 29 | 0.020 | 0.039 | ||||
| Not in COGs | 47 | 0.040 | <0.001 | ||||
(1)“>” or “<” indicates the direction of the one-sided tests (i.e. “>Codon bias” shows Bonferroni-corrected P-values for associations between genes in a given COG and higher codon bias as compared to the genes in other COGs, and “<Codon bias” represents a contrast tendency).
(2)Tests for codon bias were performed using Nc values (a lower Nc means increased codon bias).
The effective number of codons, abbreviated as Nc, was used to measure the codon bias for each orthologous gene. Genes categorised into the COG “Translation, ribosomal structure and biogenesis” were evident to have a significant higher codon bias compared with genes in other COG categories (Table 2). It is well known that genes with a lower Nc can have a strong bias and are more likely to be highly expressed [47–49]. So, the genes in the two COGs might present housekeeping features in the fundamental life cycle and essential physiological activities of Neisseria.
In the same way, an association between COGs and dN or dS was also observed. There were 4 COGs in which genes were found to have higher rates of synonymous nucleotide substitutions in comparison with other categories. On the other hand, genes in the other 4 COGs also showed a tendency to have higher rates of nonsynonymous substitutions in comparison with genes in other COGs (Table 2). It is worth noting that all the genes in the core genome in Neisseria had higher dS and dN rates than the genes in other bacteria, for example, E. coli [50] and A. pleuropneumoniae [11], indicating that strong natural selection might act on Neisseria.
3.2. A Considerable Number of Genes Showing Evidence of Recombination
Until now, there were several different strategies for identifying the homologous recombination regions in sequences. In this study, four common statistical test methods, including NSS, Max-χ2, Phi, and GENECONV, were employed to detect the recombination signals among the 1012 orthologous genes. As a result, a total of 996 genes (98.4% of all 1012 core genome genes) were found to show significant evidence (FDR < 10%) of recombination by at least one of the four tests. Overall, 951, 968, 842, and 727 genes were identified to show significant evidence of recombination by NSS, Max-χ2, Phi, and GENECONV, respectively. Additionally, a total of 635 genes (62.7% of 1012 core genome genes) were showed recombination signals in all four tests. The proportion of genes undergoing recombination ranged from 62.7% to 98.4%, which is higher than those typically observed in other bacteria, such as E. coli. The result suggests that homologous recombination plays an important role in the evolution of Neisseria genomes.
In a previous work [5], Joseph et al. identified 459 ortholog genes with signs of recombination in Neisseria meningitidis genomes, which accounts for 39.6% of all core genome genes. In this work, only Neisseria meningitidis genomes were for recombination test, the abovementioned 459 orthologous genes with signs of recombination could be considered intraspecies recombinations. In our present work, in addition to the N. meningitidis genomes, the genomes of Neisseria gonorrhoeae, and Neisseria lactamica were also selected for the recombination analyses and several interspecies recombination genes were identified. The interspecies recombination events in the genus Neisseria have been reported many times [44–46]. It is not surprising that the proportion of genes with recombination signals in the present work is markedly higher than the value observed by Joseph et al. It can be deduced that both intraspecies and interspecies recombination could act as important genetic mechanisms for generating new clones and alleles [47] in Neisseria.
To test whether the high percentage of core genome genes with a recombination signal is caused by the choice of genomes, we carried out the same analysis on the 14 N. meningitidis genome sequences with the same parameters. We first obtained 1211 orthologous genes with a single copy per genome. Among these orthologous genes, 634 (52.4%) genes were identified to show significant evidence of recombination by all the four tests. In this case, a lower percentage of genes with recombination signals were identified, confirming that the choice of genomes really has an impact on the percentage of recombined genes in the core genome. It also indicated that interspecies recombination indeed has a role in the evolution of Neisseria genomes. Additionally, a higher proportion of genes with recombination signals were observed in these 14 N. meningitidis genomes compared with the results in Joseph's work. The reason could lie in the differences in the specific genomes in both analyses, suggesting that intraspecies recombination plays an unexpected role in the evolution of the N. meningitidis genome. In a word, recombination acts as an important and irreplaceable genetic mechanism in shaping the genomes of genus Neisseria.
Moreover, it is worth noting that the core genes identified as recombinants have high rates of dS and dN, nucleotide diversity and the number of information sites (P < 0.001, P < 0.001, P < 0.001 and P < 0.001, respectively, one-sided U-test). The association between COG categories and the number of recombined genes was also estimated (Figure 2). Only two COGs “general function prediction only” and “function unknown” were significantly overrepresented with recombined genes. However, after Bonferroni correction, all the genes exhibiting evidence of recombination were distributed with no significance in all COGs. This unbiasedness of recombined genes in function further confirmed the role of recombination in shaping genomes during the evolution of Neisseria.
Figure 2.
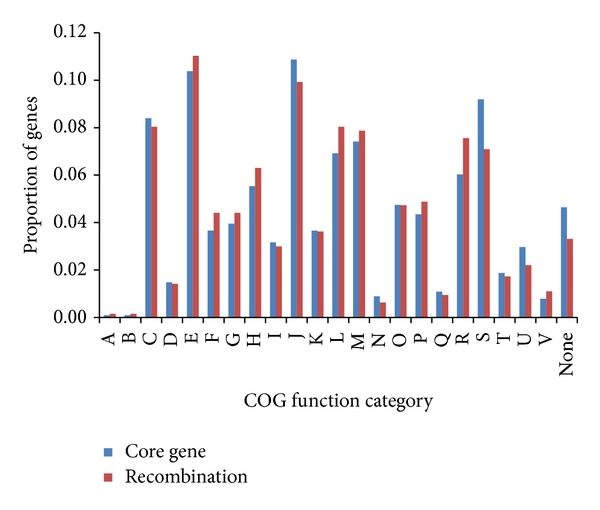
Genes with recombination signals are distributed with no significance in all COGs. The x axis represents different COG categories. The y axis represents the proportion of genes in each COG category. The proportion of genes with evidence for recombination and core genes for each COG are represented by red and blue bars, respectively. The COG categories are coded as follows: A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control, cell division, and chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, and chaperone; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms; None, not in COGs.
3.3. 10 Genes Showing Evidence of Positive Selection
The detection of positive selection for the 1012 orthologs was conducted in PAML, and models M1a and M2a of variable selective pressure across codon sites were used to estimate selective pressure and test for positive selection. Based on LRT statistics for comparing the null model and alternative model with χ 2 distribution and correction for multiple testing (FDR < 20%), a total of 10 genes were identified to be under strong selected pressure. Of the 10 genes, 4 belonged to the COG “Replication, recombination, and repair”, and 3 were in the COG “Inorganic ion transport and metabolism.” The remaining three genes were classified into the “cell wall/membrane/envelope biogenesis,” “nucleotide transport and metabolism,” and “function unknown”, respectively (Table 3).
Table 3.
Genes under positive selection.
| Gene | Cluster ID | COG | Function | 2ΔL | q-value | ω | Positively selected sites |
|---|---|---|---|---|---|---|---|
| dnaE | N35 | L | DNA polymerase III alpha subunit | 48.459 | 0.016 | 13.082 | 413, 968, 971, 972 |
| N139 | P | Ammonium transporter | 42.474 | 0.096 | 63.631 | 12, 14, 18, 19, 20, 21, 67 | |
| recB | N245 | L | DNA helicase | 67.811 | 0.000 | 4.287 | 4, 251, 865, 869, 882, 1036, 1137, 1184 |
| hup | N352 | P | TonB-dependent receptor | 129.195 | 0.000 | 7.064 | 263, 265, 282, 287, 288, 290, 291, 293, 304, 378, 380, 535, 538, 553, 557, 561, 646, 810, 884, 889, 891 |
| N380 | M | Hypothetical protein | 46.418 | 0.029 | 152.674 | 18, 19, 20, 23, 24, 25, 26, 28, 30 | |
| dnaX | N436 | L | DNA polymerase III gamma and tau subunit | 57.997 | 0.001 | 6.032 | 228, 294, 329, 512, 559 |
| uraA | N514 | F | Uracil permease | 55.222 | 0.002 | 16.737 | 2, 9, 10, 17, 24, 25, 29, 31, 395, 455 |
| N832 | S | Hypothetical protein | 51.544 | 0.006 | 6.580 | 190, 207, 212, 228, 232, 276, 314, 368, 401, 514, 729 | |
| frpB | N966 | P | Iron-regulated outer membrane protein | 125.098 | 0.000 | 4.333 | 341, 342, 343, 348, 394, 409, 415, 451, 459, 466, 467, 471, 472, 473, 476, 483, 674, 688, 718, 730, 739 |
| polA | N973 | L | DNA polymerase I | 54.283 | 0.003 | 6.489 | 212, 866, 867, 881, 882, 898 |
In the same way, two obvious discrepancies were observed, respectively, for values of dS and the number of informative sites between genes under positive selection and the remaining genes (P = 0.024 and P = 0.005, one-sided U-test). Furthermore, all 10 positively selected genes were found to show significant evidence of recombination detected by at least one recombination test. Only one gene was not in the genes identified by all four tests. The probable reason for this is that recombination could form phylogenetic incongruence [51, 52].
Compared to the high proportion of recombined genes, few positively selected genes (10) were identified, accounting for approximately 1% of the core genome. Similar proportion was also obtained in E. coli [12], but is smaller than those of other pathogenic bacteria, such as A. pleuropneumoniae [11].
Among the protein products encoded by the 10 positively selected genes, only 8 proteins were annotated with definite functions. We found that these proteins were either involved in DNA processing or inorganic transport and metabolism.
Of the 10 genes, recB, encoding the DNA helicase, is an integral part of recBCD homologous recombined enzyme. Mutations in recB are required for double-strand break repair [53] and can also reduce the frequency of many types of recombination events [54]. dnaE, dnaX and polA are all DNA polymerase genes. The first two encode the polymerase iii subunits, and the last encodes polymerase I. All three play fundamental roles in DNA metabolism, including DNA replication, recombination, and repair. In a word, positive selection on the four genes might ensure the strain to adapt to frequent recombination in the genomes.
AmtB encodes an ammonium transporter and is involved in ammonium transmembrane transporter activity. uraA encodes a uracil permease involved in transmembrane transport as well and acts as a membrane-bound facilitator for the transport of uracil across the cell membrane into the cytoplasm [55]; it is therefore necessary for uracil uptake, especially at low exogenous uracil concentrations and even under conditions with high UPRTase activity.
Hup encodes a TonB-dependent receptor that utilizes heme as an iron source [56]. It has been reported that mutations in the hemoglobin receptor gene have profound effects on the survival of N. meningitidis in an infant rat, indicating that this gene is important for the virulence of Neisseria [57].
FrpB is clearly a virulence gene, encoding an iron-regulated outer membrane protein. It is a member of the TonB-dependent transporter family and is responsible for iron uptake into the periplasm. FrpB is subject to a high degree of antigenic variation, principally through a region of hypervariable sequence exposed on the cell surface [58, 59].
In a word, the four genes play important roles in the uptake of nutrition. So the adaptive changes in these proteins might be beneficial for Neisseria to survive in the host.
4. Conclusion
Our analysis reported here indicates that both homologous recombination and positive selection play important roles in the evolution of the core genome in Neisseria. Additionally, homologous recombination has a greater contribution to the genetic variation of a large number of genes with recombination signals. Only 10 genes were identified to be under positive selection, which also showed significant evidence of recombination. However, the positively selected genes were found to be involved in DNA processing or located on the cell membrane. The former reduce the frequency of recombination and enables a stable genetic environment, while the latter maintain a dynamic interaction with the external environment, as well as with the host. Overall, the changes in these positively selected genes result in an improvement in bacterial fitness in response to a variety of environmental signals. These genes can be regarded as a screened gene set for further analysis of the mechanisms of adaptive evolution in Neisseria.
Acknowledgments
This work was supported by the National Key Program for Infectious Diseases of China (2011ZX10004-001), the National Basic Research Program of China (2013CB910804), and the Innovation Foundation of AMMS (No. 2012CXJJ023).
Conflict of Interests
The authors declare that they have no conflict of interests.
Authors' Contribution
Junjie Yue and Long Liang formulated the study. Dong Yu performed the research. Yuan Jin and Zhiqiu Yin analysed the data. Hongguang Ren and Wei Zhou participated in analysis and discussion. Dong Yu wrote the paper. All authors read and approved the final paper.
References
- 1.Bell G. Selection: The Mechanism of Evolution. Chapman & Hall; 1997. [Google Scholar]
- 2.Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. Garland Science; 2002. DNA replication, repair, and recombination; p. p. 845. [Google Scholar]
- 3.Doroghazi JR, Buckley DH. Widespread homologous recombination within and between Streptomyces species. ISME Journal. 2010;4(9):1136–1143. doi: 10.1038/ismej.2010.45. [DOI] [PubMed] [Google Scholar]
- 4.Suerbaum S, Maynard Smith J, Bapumia K, et al. Free recombination within Helicobacter pylori. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(21):12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joseph B, Schwarz RF, Linke B, et al. Virulence evolution of the human pathogen neisseria meningitidis by recombination in the core and accessory genome. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0018441.e18441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper TF. Recombination speeds adaptation by reducing competition between beneficial mutations in populations of Escherichia coli. PLoS Biology. 2007;5(9) article e225 doi: 10.1371/journal.pbio.0050225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai YL, Maron SB, McGann P, Nightingale KK, Wiedmann M, Orsi RH. Recombination and positive selection contributed to the evolution of Listeria monocytogenes lineages III and IV, two distinct and well supported uncommon L. monocytogenes lineages. Infection, Genetics and Evolution. 2011;11(8):1881–1890. doi: 10.1016/j.meegid.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soyer Y, Orsi RH, Rodriguez-Rivera LD, Sun Q, Wiedmann M. Genome wide evolutionary analyses reveal serotype specific patterns of positive selection in selected Salmonella serotypes. BMC Evolutionary Biology. 2009;9(1, article 264) doi: 10.1186/1471-2148-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lefébure T, Stanhope MJ. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biology. 2007;8(5, article R71) doi: 10.1186/gb-2007-8-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefébure T, Stanhope MJ. Pervasive, genome-wide positive selection leading to functional divergence in the bacterial genus Campylobacter. Genome Research. 2009;19(7):1224–1232. doi: 10.1101/gr.089250.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Chen H, Zhou R. Genome-wide evidence for positive selection and recombination in Actinobacillus pleuropneumoniae. BMC Evolutionary Biology. 2011;11(1, article 203) doi: 10.1186/1471-2148-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen L, Bollback JP, Dimmic M, Hubisz M, Nielsen R. Genes under positive selection in Escherichia coli. Genome Research. 2007;17(9):1336–1343. doi: 10.1101/gr.6254707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunham RC, Plummer FA, Stephens RS. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infection and Immunity. 1993;61(6):2273–2276. doi: 10.1128/iai.61.6.2273-2276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172(4):2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawyer S. Statistical tests for detecting gene conversion. Molecular Biology and Evolution. 1989;6(5):526–538. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z, Nielsen R, Goldman N, Pedersen AK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155(1):431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z, Bielawski JR. Statistical methods for detecting molecular adaptation. Trends in Ecology and Evolution. 2000;15(12):496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148(3):929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunning Hotopp JC, Grifantini R, Kumar N, et al. Comparative genomics of Neisseria meningitidis: core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology. 2006;152, part 12:3733–3749. doi: 10.1099/mic.0.29261-0. [DOI] [PubMed] [Google Scholar]
- 20.Joseph B, Schneiker-Bekel S, Schramm-Glück A, et al. Comparative genome biology of a serogroup B carriage and disease strain supports a polygenic nature of meningococcal virulence. Journal of Bacteriology. 2010;192(20):5363–5377. doi: 10.1128/JB.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unemo M, Shafer WM. Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Annals of the New York Academy of Sciences. 2011;1230:E19–E28. doi: 10.1111/j.1749-6632.2011.06215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caugant DA. Genetics and evolution of Neisseria meningitidis: importance for the epidemiology of meningococcal disease. Infection, Genetics and Evolution. 2008;8(5):558–565. doi: 10.1016/j.meegid.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Bennett JS, Bentley SD, Vernikos GS, et al. Independent evolution of the core and accessory gene sets in the genus Neisseria: insights gained from the genome of Neisseria lactamica isolate 020-06. BMC Genomics. 2010;11(1, article 652) doi: 10.1186/1471-2164-11-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckee CO, Jolley KA, Recker M, et al. Role of selection in the emergence of lineages and the evolution of virulence in Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(39):15082–15087. doi: 10.1073/pnas.0712019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett JS, Jolley KA, Sparling PF, et al. Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from multilocus sequence typing. BMC Biology. 2007;5(article 35) doi: 10.1186/1741-7007-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolley KA, Wilson DJ, Kriz P, McVean G, Maiden MCJ. The influence of mutation, recombination, population history, and selection on patterns of genetic diversity in Neisseria meningitidis. Molecular Biology and Evolution. 2005;22(3):562–569. doi: 10.1093/molbev/msi041. [DOI] [PubMed] [Google Scholar]
- 27.Schoen C, Blom J, Claus H, et al. Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3473–3478. doi: 10.1073/pnas.0800151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith NH, Smith JM, Spratt BG. Sequence evolution of the porB gene of Neisseria gonorrhoeae and Neisseria meningitidis: evidence of positive Darwinian selection. Molecular Biology and Evolution. 1995;12(3):363–370. doi: 10.1093/oxfordjournals.molbev.a040212. [DOI] [PubMed] [Google Scholar]
- 29.Andrews TD, Gojobori T. Strong positive selection and recombination drive the antigenic variation of the PilE protein of the human pathogen Neisseria meningitidis. Genetics. 2004;166(1):25–32. doi: 10.1534/genetics.166.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Activities at the Universal Protein Resource (UniProt) Nucleic Acids Research. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright F. The “effective number of codons” used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 34.Ota T, Nei M. Variance and covariances of the numbers of synonymous and nonsynonymous substitutions per site. Molecular Biology and Evolution. 1994;11(4):613–619. doi: 10.1093/oxfordjournals.molbev.a040140. [DOI] [PubMed] [Google Scholar]
- 35.Smith JM. Analyzing the mosaic structure of genes. Journal of Molecular Evolution. 1992;34(2):126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 36.Jakobsen IB, Easteal S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Computer Applications in the Biosciences. 1996;12(4):291–295. doi: 10.1093/bioinformatics/12.4.291. [DOI] [PubMed] [Google Scholar]
- 37.Price MN, Dehal PS, Arkin AP. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B. 1995;57(1):289–300. [Google Scholar]
- 40.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2014. [Google Scholar]
- 41.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowler LD, Zhang QY, Riou JY, Spratt BG. Interspecies recombination between the penA genes of Neisseria meningitidis and commensal Neisseria species during the emergence of penicillin resistance in N. meningitidis: natural events and laboratory simulation. Journal of Bacteriology. 1994;176(2):333–337. doi: 10.1128/jb.176.2.333-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou J, Bowler LD, Spratt BG. Interspecies recombination, and phylogenetic distortions, within the glutamine synthetase and shikimate dehydrogenase genes of Neisseria meningitidis and commensal Neisseria species. Molecular Microbiology. 1997;23(4):799–812. doi: 10.1046/j.1365-2958.1997.2681633.x. [DOI] [PubMed] [Google Scholar]
- 44.Feil E, Zhou J, Smith JM, Spratt BG. A comparison of the nucleotide sequences of the adk and recA genes of pathogenic and commensal Neisseria species: evidence for extensive interspecies recombination within adk. Journal of Molecular Evolution. 1996;43(6):631–640. doi: 10.1007/BF02202111. [DOI] [PubMed] [Google Scholar]
- 45.Holmes EC, Urwin R, Maiden MCJ. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Molecular Biology and Evolution. 1999;16(6):741–749. doi: 10.1093/oxfordjournals.molbev.a026159. [DOI] [PubMed] [Google Scholar]
- 46.Maiden MCJ, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Research. 1982;10(22):7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carbone A, Képès F, Zinovyev A. Codon bias signatures, organization of microorganisms in codon space, and lifestyle. Molecular Biology and Evolution. 2005;22(3):547–561. doi: 10.1093/molbev/msi040. [DOI] [PubMed] [Google Scholar]
- 49.Willenbrock H, Ussery DW. Prediction of highly expressed genes in microbes based on chromatin accessibility. BMC Molecular Biology. 2007;8, article 11 doi: 10.1186/1471-2199-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Research. 2002;12(6):962–968. doi: 10.1101/gr.87702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164(3):1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orsi RH, Sun Q, Wiedmann M. Genome-wide analyses reveal lineage specific contributions of positive selection and recombination to the evolution of Listeria monocytogenes. BMC Evolutionary Biology. 2008;8(1, article 233) doi: 10.1186/1471-2148-8-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saveson CJ, Lovett ST. Tandem repeat recombination induced by replication fork defects in Escherichia coli requires a novel factor, RadC. Genetics. 1999;152(1):5–13. doi: 10.1093/genetics/152.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovett ST, Luisi-DeLuca C, Kolodner RD. The genetic dependence of recombination in recD mutants of Escherichia coli . Genetics. 1988;120(1):37–45. doi: 10.1093/genetics/120.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersen PS, Frees D, Fast R, Mygind B. Uracil uptake in Escherichia coli K-12: isolation of uraA mutants and cloning of the gene. Journal of Bacteriology. 1995;177(8):2008–2013. doi: 10.1128/jb.177.8.2008-2013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perkins-Balding D, Ratliff-Griffin M, Stojiljkovic I. Iron transport systems in Neisseria meningitidis. Microbiology and Molecular Biology Reviews. 2004;68(1):154–171. doi: 10.1128/MMBR.68.1.154-171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stojiljkovic I, Hwa V, de Saint Martin L, et al. The Neisseria meningitidis haemoglobin receptor: Its role in iron utilization and virulence. Molecular Microbiology. 1995;15(3):531–541. doi: 10.1111/j.1365-2958.1995.tb02266.x. [DOI] [PubMed] [Google Scholar]
- 58.van der Ley P, van der Biezen J, Sutmuller R, Hoogerhout P, Poolman JT. Sequence variability of FrpB, a major iron-regulated outer-membrane protein in the pathogenic neisseriae. Microbiology. 1996;142(11, part 1):3269–3274. doi: 10.1099/13500872-142-11-3269. [DOI] [PubMed] [Google Scholar]
- 59.Beucher M, Sparling PF. Cloning, sequencing, and characterization of the gene encoding FrpB, a major iron-regulated, outer membrane protein of Neisseria gonorrhoeae. Journal of Bacteriology. 1995;177(8):2041–2049. doi: 10.1128/jb.177.8.2041-2049.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]