

Abstract
Defective control of lipid metabolism leading to lipotoxicity causes insulin resistance in skeletal muscle, a major factor leading to diabetes. Here, we demonstrate that perilipin (PLIN) 5 is required to couple intramyocellular triacylglycerol lipolysis with the metabolic demand for fatty acids. PLIN5 ablation depleted triacylglycerol stores but increased sphingolipids including ceramide, hydroxylceramides and sphingomyelin. We generated perilipin 5 (Plin5)−/− mice to determine the functional significance of PLIN5 in metabolic control and insulin action. Loss of PLIN5 had no effect on body weight, feeding or adiposity but increased whole-body carbohydrate oxidation. Plin5−/− mice developed skeletal muscle insulin resistance, which was associated with ceramide accumulation. Liver insulin sensitivity was improved in Plin5−/− mice, indicating tissue-specific effects of PLIN5 on insulin action. We conclude that PLIN5 plays a critical role in coordinating skeletal muscle triacylglycerol metabolism, which impacts sphingolipid metabolism, and is requisite for the maintenance of skeletal muscle insulin action.
Keywords: Lipid metabolism, Perilipin, Lipid droplet, Insulin resistance, Skeletal muscle
Graphical Abstract
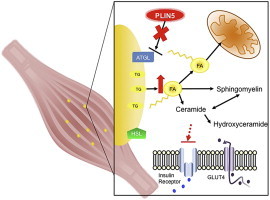
1. Introduction
The close association between the growing epidemics of obesity and type 2 diabetes continues to fuel interest in understanding how alterations in lipid metabolism contribute to the development of insulin resistance. An excess of fatty acids beyond the cells energy requirements results in the accumulation of triacylglycerol within lipid droplets, and the deposition of lipids in tissues other than adipose tissue is a feature of obesity and type 2 diabetes. Skeletal muscle serves as an important site for glucose disposal after a meal [1] and it has been known for many years that the accumulation of intramyocellular triacylglycerol predicts insulin resistance in muscle, independent of adiposity [2–5]. However, the association is not clear cut because highly trained endurance athletes store as much triacylglycerol in their muscles as patients with type 2 diabetes, yet display remarkable insulin sensitivity [6]. Thus, the matching of fatty acid flux from the lipid droplet with mitochondrial demand for fatty acid substrate may be an important regulatory process to avoid defects in insulin action.
Both skeletal and cardiac muscle store significant amounts of triacylglycerol within intracellular lipid droplets, which are located in close proximity to the mitochondria and endoplasmic reticulum [7]. Intracellular triacylglycerol is an important energy substrate in skeletal [8] and cardiac muscle [9] and triacylglycerol turnover is relatively high in muscles [9,10], which ensures a continuous substrate supply to buffer changes in systemic free fatty acid levels. However, an oversupply of fatty acids, beyond the energetic needs of the myocyte, eventually causes insulin resistance via a diverse range of mechanisms including the accumulation of lipid metabolites, maladaptive proinflammatory signaling and alterations in membrane physical properties [11]. In this context, an inability to mobilize fatty acids from triacylglycerol [12,13] and/or increase triacylglycerol breakdown in skeletal muscle [14], without an increase in energy demand, can result in excessive intracellular lipid accumulation and insulin resistance.
The understanding of lipid droplet biology and the regulation of intracellular lipid fluxes have expanded in recent years with the discovery of perilipin (PLIN) family members [15]. PLIN1 was first discovered as a lipid droplet-associated protein in white adipocytes and is a critical regulator of lipid metabolism [16,17], and more recently PLIN2 and PLIN5 have been shown to play major roles in liver and cardiac metabolism, respectively [18–21]. While PLIN5 is highly expressed in skeletal muscle [22], its role in the control of intramyocellular lipid trafficking, substrate utilization and insulin action is not well understood. Mild overexpression of PLIN5 via DNA electrotransfer into the glycolytic muscle of rats increased triacylglycerol accumulation [23], which is consistent with the general consensus that PLIN5 plays an important role in lipid droplet accumulation [22,24,25]. While PLIN5 increased triacylglycerol accumulation, this appeared to have no effect on skeletal muscle insulin action [26]. Intriguingly, and perhaps paradoxically, the same authors have shown that PLIN5 may also localize to the mitochondria, suggesting that PLIN5 might direct fatty acids from the lipid droplet to the mitochondria for oxidation [23]. While these short-term overexpression models have provided useful insights into PLIN5 function in muscle, the physiological significance of PLIN5 deletion on skeletal muscle and systemic metabolic control remains unresolved.
In the present study, we tested the hypothesis that PLIN5 plays an important role in regulating skeletal muscle fatty acid metabolism, and in turn, in regulating insulin action. We have generated Plin5 deficient mice and show that PLIN5 regulates skeletal muscle triacylglycerol flux, whole-body substrate metabolism and insulin action in a tissue-specific context.
2. Methods
2.1. Generation of PLIN5 null mice and mouse breeding
A targeted vector containing Plin5 (NM_001013706.2; Figure 1A) interrupted in exon 5, 6, 7 was generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (Project ID CSD40589, ES clone EPD0301_4_E04). Blastocysts were collected from the mating of Balb/c mice. Cells were microinjected into the blastocyl cavity and injected blastocysts were transferred to the uteri of pseudopregnant CD1 female mice. To test for germline transmission, male chimeras were bred to C57BL/6N wild type female mice. Mice were confirmed to contain the PLIN5 disrupted allele by Southern blot analysis of KpnI digested genomic tail DNA (Figure 1B). Genomic DNA was used for PCR. Mice were backcrossed onto a C57Bl/6 background for 2 generations. All studies were approved by the Monash University Animal Ethics Committee.
Figure 1.
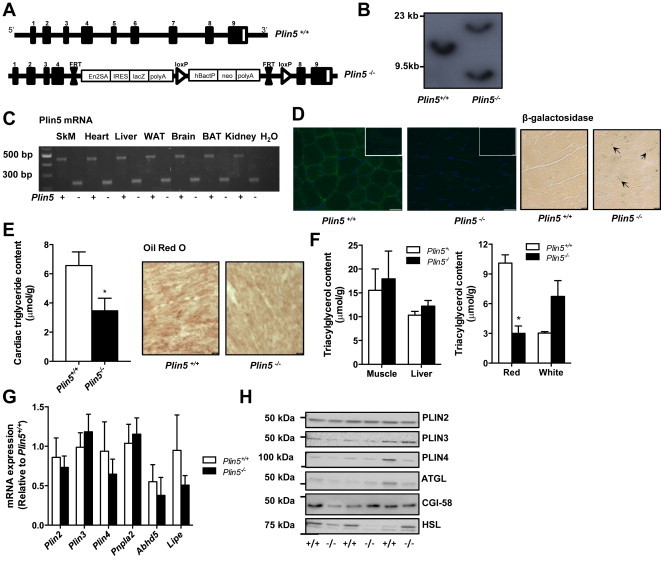
Generation of Plin5−/− mice. (A) Targeting vector used for the generation of Plin5−/− mice. (B) Confirmation of Plin5 disruption by Southern blotting of genomic tail DNA and (C) RT-PCR of RNA from Plin5+/+ and Plin5−/− mice. SkM, skeletal muscle, WAT, white adipose tissue, BAT, brown adipose tissue. Upper band (Plin5+/+) 466 bp; lower band (Plin5−/−) 254 bp. (D) Left: Representative images of skeletal muscle sections stained with PLIN5 antibody from Plin5+/+ and Plin5−/− mice, with concentration matched serum controls inset. Scale bar = 50 μm. Right: Confirmation of lacZ expression in Plin5−/− hearts with β-galactosidase staining. Arrows pointing to regions of staining. Scale bar = 10 μm. (E) Cardiac triacylglycerol content (♂, n = 13 Plin5+/+, n = 15 Plin5−/−, Age = 12–17 weeks). Representative image of lipid droplet staining with ORO in heart. Scale bar = 10 μm. (F) Left: Skeletal muscle (mixed quadriceps) and liver triacylglycerol content (♂, n = 13 Plin5+/+, n = 15 Plin5−/−, Age = 12–17 weeks). Right: Triacylglycerol content in red and white portions of the quadriceps (♂, n = 4 Plin5+/+, n = 4 Plin5−/−, Age = 12–17 weeks). *P < 0.05 vs. Plin5+/+ mice. (G) Skeletal muscle expression of PLIN genes Plin2, Plin3 and Plin4 and of lipase and lipase coactivator genes Pnp1a2, Abhd5 and Lipe (♂, n = 4–5 Plin5+/+, n = 6–7 Plin5−/−). (H) Representative immunoblots of PLIN proteins and lipases in skeletal muscle (♂, n = 3 Plin5+/+, n = 3 Plin5−/−).
2.2. Mice
Heterozygous mice were mated, with male Plin5−/− and Plin5+/+ littermates used for experiments. Mice were maintained at 22 °C on a 12:12-h light–dark cycle. Mice were fed a low fat (5% energy from fat) or high-fat diet (43% energy from fat, Specialty Feeds, WA, Australia) and had free access to water. All experiments were performed in 4 h fasted mice commencing at 1100 h unless otherwise stated.
2.3. Assessment of whole body metabolism, body composition and physical activity
Whole body metabolism was measured in metabolic cages (Columbus Instruments, Columbus, OH). Mice were housed individually for 48 h for assessment of oxygen uptake, carbon dioxide production and physical activity. Whole body fat and lean mass were measured by Dual Energy X-ray absorptiometry (Lunar Pixi #51042, PIXImus, WI, USA). Running experiments were performed on an Eco 3/6 treadmill (Columbus Instruments, Columbus, OH) as described in Ref. [27]. To determine maximal running speed, mice ran at 10 m/min on a 5% grade for 2 min and the speed was increased by 2 m/min until exhaustion.
2.4. Fatty acid and glucose metabolism ex vivo
Mice were anaesthetized with isoflurane and kept under sedation while the soleus muscle was excised. Fatty acid [27] and glucose metabolism were assessed by radiometric methods as described in Ref. [28].
2.5. Tissue lipids
Lipids were extracted [29] and triacylglycerol content determined using the TG-GPO-PAP reagent (Cat. no. 04657594190, Roche Diagnostics, Basel, Switzerland). Diacylglycerol and ceramide content was determined by an [32P]ATP-linked enzymatic method as previously described in Ref. [30]. Lipidomics was performed on cell lysates as described in Ref. [31].
2.6. Glucose tolerance tests
Mice were intraperitoneally injected with D-glucose (2 g/kg mass). Tail blood was collected and blood glucose was determined using a glucometer (Accu-chek, Roche, Mannheim, Germany). Plasma insulin was determined by an in-house ELISA.
2.7. Hyperinsulinemic-euglycemic clamp studies
Mice were anaesthetized and a catheter was inserted into the jugular vein as previously described [32]. Following five days recovery, mice were fasted for 4 h prior to beginning the clamp. Mice were conscious but restrained for the duration of the procedure. Blood glucose was equilibrated with a 90 min continuous infusion of [3-3H]-glucose (0.05 μCi/min). Hyperinsulinemic-euglycemic clamp commenced with the increase of [3-3H]-glucose tracer to 2 μCi/min and insulin infusion (4 mU/kg/min). Exogenous glucose (25% w/v) was infused to maintain basal blood glucose levels, which averaged 8.1 ± 0.3 mM. Tail blood samples were collected and blood glucose was measured every 5 min. After 120 min, a 10 μCi bolus of 2-deoxy-d-[1-14C] glucose (2-[14C] DG; Perkin Elmer) was administered to assess tissue-specific glucose uptake (Rg′). At the conclusion of the clamp, mice were anaesthetized with an intravenous injection of sodium pentobarbitone (100 mg/kg iv) and tissues rapidly dissected and snap frozen in liquid nitrogen for analysis of 2-[14C] DG uptake.
2.8. Intravenous insulin tolerance tests
Whole body and tissue specific insulin sensitivity was assessed in anaesthetized (2%, isoflurane) mice as described in Ref. [33]. Briefly, 2-[1,2-3H] deoxyglucose (10 μCi) was injected intravenously with 0.5 U/kg insulin. Blood was taken from the tip of the tail at 2, 5, 10, 15 and 20 min after injection. Samples were deproteinized, spun and the supernatant used to determine radioactivity. Blood glucose was also determined at these time points. Animals were sacrificed by exsanguination with tissue removed quickly and tissue specific glucose clearance was determined from total and phosphorylated 2-[1,2-3H] deoxyglucose [33].
2.9. Blood biochemistry
Whole blood was collected into EDTA tubes then centrifuged for 3 min at 8000 × g. Plasma free fatty acids and triacylglycerol were determined by enzymatic colorimetric assays (Wako Chemicals, Wako, VA, USA). High density lipoproteins (HDL) and very (VLDL) and low density lipoproteins (LDL) were assessed by ELISA (Ab65390, Abcam, Cambridge, UK). Plasma β-hydroxybutyrate was analyzed by a colorimetric assay (700190, Cayman Chemicals, Ann Arbor, MI, USA).
2.10. Immunohistochemistry
Tissues were rapidly frozen in melting isopentane after being coated in OCT Tissue-Tek (Sakura Finetek, The Netherlands). Serial 10 μm sections were cut at −20 °C on to SuperFrost Ultra Plus glass slides. Slides were fixed in Bouin's solution with 0.1% Triton X-100 (Sigma–Aldrich) for 30 min. Sections were stained for lipid droplets with freshly made, filtered oil red O (Sigma–Aldrich) made in 60% triethylphosphate (Sigma–Aldrich). β-galactosidase expression was identified using commercial reagents (MIR 2600, Mirus Bio, Madison, WI, USA). Sections were blocked in PBS with 1% BSA for 1 h then incubated overnight with PLIN5 antibody (Cat. no. GP31, Progen Biotechnik, Germany, ∼0.02 mg/ml) while mitochondrial staining was performed using the mitochondrial antibody directed against the oxidative phosphorylation complexes I–V (Total OXPHOS Cat. no. ab110413, Abcam, Cambridge, UK). For negative controls, primary antibodies were substituted for concentration matched guinea pig serum for PLIN5 (Antibodies Australia), or OXPHOS cocktail pre-absorbed 4:1 overnight with mouse IgG1 and IgG2a (Dako, Denmark). Immunohistochemistry images were analyzed for colocalization with ImageJ (NIH) by using Manders' colocalization coefficient, M2, which provides a fraction of the colocalizing objects in the images. Electron microscopy was performed as described previously in Ref. [34].
2.11. qRT-PCR
Total RNA was extracted, reverse transcribed and cDNA used for qRT-PCR as described in Ref. [31]. TaqMan Gene Expression Assays are listed in Supplementary Table 1.
2.12. Immunoblot analysis
Immunoblot analysis was performed as described previously in Ref. [35]. Stain-free images were collected after transfer for loading control (ChemiDoc MP and ImageLab software Version 4.1, Bio-rad Laboratories, NSW, Australia). Membranes were probed with commercial antibodies (Supplementary Table 2).
2.13. Measurement of oxidative stress
Thiobarbituric acid reactive substances (TBARS) was measured in skeletal muscle lysates by using a colorimetric assay for the formation of malondialdehyde (10009055, Cayman Chemicals, Ann Arbor, MI, USA).
2.14. In vitro phosphorylation
Recombinant HA-tagged murine PLIN5 was phosphorylated by cAMP-dependent protein kinase (PKA) as described in Ref. [35] and the location of the HA-PLIN5 was confirmed by immunoblot.
2.15. Cell culture and mitochondrial metabolism
Primary myoblasts were isolated using the explant culture method [36]. Myoblasts were differentiated to myotubes by switching to 3% horse serum medium and were left to differentiate for five days prior to experiments. Fatty acid metabolism (radiometric methodology) was assessed as described in Ref. [35] and mitochondrial function using the Seahorse XF analyzer.
2.16. Statistics
Data are expressed as means ± SEM. Statistical analysis was performed by one-way or two-way analysis of variance (ANOVA) with repeated measures, and specific differences were located using a Bonferroni post hoc test. Unpaired t-tests were used where appropriate (GraphPad Prism Version 5.02). Statistical significance was set a priori at P ≤ 0.05.
3. Results
3.1. Generation of PLIN5 deficient mice
Interbreeding of heterozygous mice for the Plin5 null allele yielded mice that were homozygous for each of the two alleles. The absence of the Plin5 allele was confirmed by Southern blotting (Figure 1B) and the absence of PLIN5 mRNA was confirmed by PCR (Figure 1C). The absence of PLIN5 protein was confirmed with immunohistochemistry of skeletal muscle (Figure 1D). A secondary confirmation was the presence of the lacZ in the hearts of Plin5−/− mice, as detected by β-galactosidase staining (Figure 1D). Triacylglycerol content was reduced by 52% in the hearts of Plin5−/− mice (Figure 1E) and coincided with a marked reduction of lipid droplets stained by ORO (Figure 1F). This is consistent with the prominent reduction in cardiac lipid content reported in another line of Plin5−/− mice [20]. There was no such reduction in triacylglycerol content in the liver or mixed skeletal muscle (Figure 1F). Interestingly, triacylglycerol content was lower in red quadriceps (oxidative) muscle and tended (122%, P < 0.08) to be higher in white quadriceps (glycolytic) muscle of Plin5−/− compared with Plin5−/− mice (Figure 1F). This may reflect the greater capacity of oxidative fibers to oxidize triglyceride-derived fatty acids compared with glycolytic fibers [37]. While the deletion of one Plin gene can induce compensatory increases in the other Plin genes [20,38], the expression of Plin 2, 3 and 4 were not altered in Plin5−/− mice (Figure 1G), nor were there changes in the expression of prominent lipases or co-activator proteins associated with triacylglycerol lipolysis (Figure 1G, H). Plin1 was not detected in skeletal muscle.
3.2. Whole body fatty acid oxidation is decreased in the absence of PLIN5
Plin5+/+ and Plin5−/− mice had similar body weights during development (Figure 2A) and DEXA examination revealed no difference in lean and fat mass (Figure 2B). This was supported by post mortem examination showing no difference in epididymal fat, heart and liver mass, indicating that the absence of PLIN5 had no effect on adiposity and gross morphology (Table 1). Neither food intake (Figure 2C) nor energy expenditure (VO2) was different between genotypes (Figure 2D). However, the respiratory exchange ratio was increased in Plin5−/− mice (Figure 2E), demonstrating an ∼11% decrease in whole-body fatty acid oxidation and a reciprocal increase in carbohydrate oxidation. These changes cannot be attributed to increased physical activity (Figure 2F). Fasting muscle and liver glycogen (Figure 2G), plasma glucose and insulin concentrations were not different between groups; neither were plasma free fatty acids, triacylglycerols and HDL or VLDL/LDL cholesterol levels (Table 1). Plasma ß-hydroxybutyrate, a marker of liver fatty acid oxidation, was not altered by genotype (Table 1).
Figure 2.
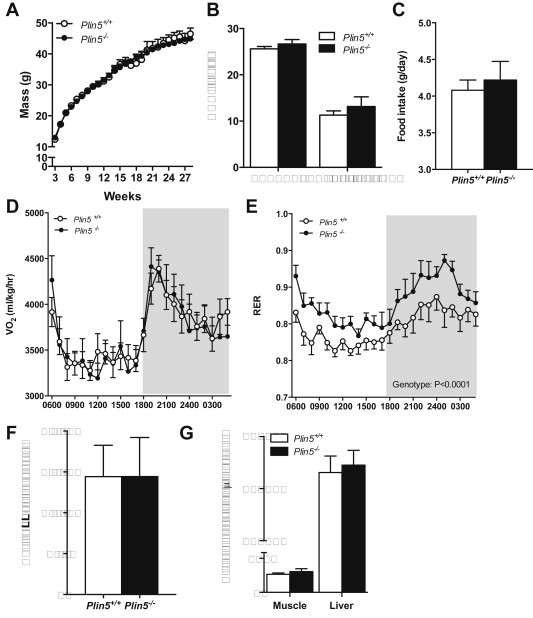
Plin5 deletion does not alter growth, adiposity, or energy expenditure but does impact substrate partitioning. (A) Body mass (♂, n = 6–16 Plin5+/+, n = 10–16 Plin5−/−). (B) Lean mass and fat mass assessed by DEXA (♂, n = 10 Plin5+/+, n = 6 Plin5−/−, Age = 14–16 weeks). (C) Food intake (♂, n = 5 Plin5+/+, n = 5 Plin5−/−, Age = 14–16 weeks). (D) Oxygen consumption (VO2) and (E) respiratory exchange ratio (RER) were assessed by indirect calorimetry (♂, n = 10 Plin5+/+, n = 6 Plin5−/−, Age = 14–16 weeks). Shaded area represents dark phase. (F) Total daily activity (♂, n = 9 Plin5+/+, n = 5 Plin5−/−, Age = 14–16 weeks). (G) Muscle (n = 7 Plin5+/+, n = 8 Plin5−/−) and liver (n = 8 Plin5+/+, n = 9 Plin5−/−) glycogen content in 4 h fasted mice (♂, Age = 14–16 weeks).
Table 1.
Tissue weights and blood chemistry in Plin5−/− and Plin5+/+ mice fed a low-fat diet.
| Plin5+/+ | Plin5−/− | P value | |
|---|---|---|---|
| Heart mass (mg) | 135.5 ± 4.9 | 138.8 ± 5.6 | 0.66 |
| Liver mass (g) | 1.3 ± 0.1 | 1.2 ± 0.1 | 0.82 |
| Epidydimal fat mass (g) | 0.78 ± 0.1 | 0.77 ± 0.1 | 0.98 |
| Blood glucose (mmol/l) | 8.6 ± 0.3 | 8.9 ± 0.3 | 0.57 |
| Plasma insulin (pmol/l) | 133 ± 34 | 140 ± 12 | 0.86 |
| Plasma FFA (mmol/l) | 0.66 ± 0.07 | 0.72 ± 0.06 | 0.56 |
| Plasma TG (mmol/l) | 1.03 ± 0.17 | 1.28 ± 0.12 | 0.24 |
| Plasma HDL (mmol/l) | 0.98 ± 0.02 | 1.00 ± 0.04 | 0.69 |
| Plasma VLDL/LDL (mmol/l) | 0.24 ± 0.02 | 0.26 ± 0.03 | 0.59 |
| Plasma ß-hydroxybutyrate (μmol/l) | 133 ± 17 | 135 ± 26 | 0.95 |
Tissue masses were from mice aged 14–16 weeks. Heart (♂, n = 17 Plin5+/+, n = 18 Plin5−/−), liver (♂, n = 5 Plin5+/+, n = 3 Plin5−/−) and epididymal fat pad (♂, n = 17 Plin5+/+, n = 18 Plin5−/−).
All mice were fasted for 4 h before blood sampling. n = 8 for each genotype. Data are mean ± SEM.
3.3. Effects of PLIN5 deletion on skeletal muscle substrate metabolism
Based on the high abundance of PLIN5 in skeletal muscle [22] and the quantitative importance of skeletal muscle in regulating whole-body substrate metabolism [39], we next examined the effects of PLIN5 deletion on skeletal muscle fatty acid metabolism in isolated soleus muscle. Total fatty acid uptake, fatty acid oxidation and the incorporation of fatty acids into tri- and diacylglycerol were not different between the two genotypes (Figure 3A–D). ß-adrenergic activation of PKA is a major regulator of triglyceride metabolism in adipocytes and most likely oxidative skeletal muscle [37,40,41], although PKA action in muscle requires further confirmation. We first performed in vitro kinase assays to demonstrate that PLIN5 is a substrate for protein kinase A (Figure 3E), then we examined fatty acid metabolism in response to forskolin, an activator of cAMP/PKA signaling. Forskolin reduced fatty acid uptake, oxidation and storage; however, these effects were not impacted by Plin5 deletion. Thus, PLIN5 deletion does not affect the uptake, oxidation or esterification of extracellular-derived fatty acids in muscle under basal and PKA-stimulated conditions. Further studies in isolated soleus muscle revealed no difference in glucose oxidation between Plin5+/+ and Plin5−/− mice (104 ± 5 vs. 105 ± 5 nmol/mg tissue/h, respectively. P = 0.92, n = 8 per genotype).
Figure 3.
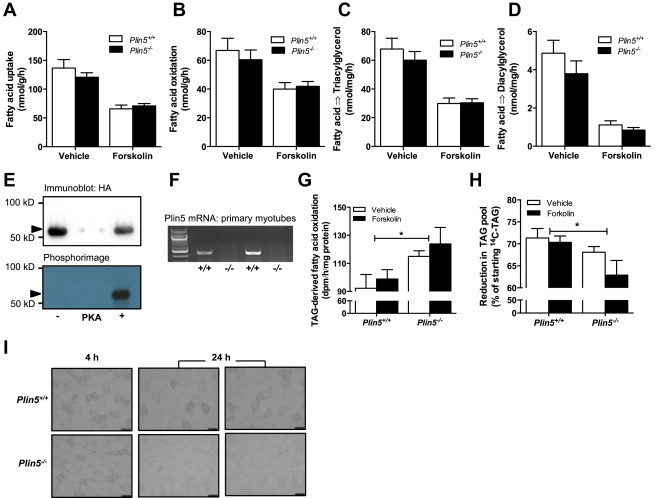
Effects of PLIN5 on skeletal muscle fatty acid metabolism. Fatty acid metabolism was examined in isolated soleus muscle. (A) Fatty acid uptake, (B) fatty acid oxidation, (C) fatty acid incorporation into triacylglycerol and (D) fatty acid incorporation into diacylglycerol (♂, Vehicle: n = 12–16 Plin5+/+, n = 15–18 Plin5−/−; Forskolin, n = 7–12 Plin5+/+, n = 9–12 Plin5−/−, Age = 14–16 weeks). (E) Recombinant murine PLIN5 was phosphorylated by cAMP-dependent protein kinase (PKA) catalytic subunit in kinase assay buffer containing [γ-32P]ATP. The protein mixture was separated by SDS-PAGE and PLIN5 identified by immunoblotting against HA (upper band). The 32P-labeled PLIN5 was detected by phosphorimaging (lower band). Arrow denotes PLIN5. Representative of 2 independent experiments. (F) Plin5 mRNA expression in primary myotubes of Plin5−/− and Plin5+/+ mice. (G) Oxidation of triacylglycerol (TAG)-derived fatty acids in myotubes and (H) the percentage reduction of the radiolabeled TAG lipid pool (n = 8 for each group from 2 independent donor mice and two independent experiments). *Main effect for genotype, P < 0.05. (I) Representative image of lipid droplets stained with oil red O in skeletal muscle of Plin5−/− and Plin5+/+ mice following a 4 h and 24 h fast. Scale bar = 50 μm.
PLIN5 interacts with ATGL to inhibit spontaneous and ß-adrenergic stimulated lipolysis [42], suggesting that PLIN5 is more likely to regulate fatty acid flux from endogenous triacylglycerol stored in lipid droplets rather than modulate the oxidation of extracellular-derived fatty acids [20,42]. We performed pulse-chase experiments on primary myotubes generated from the skeletal muscle of wild-type and Plin5−/− mice to evaluate triacylglycerol metabolism. Plin5 mRNA expression was retained in myotubes generated from Plin5+/+ mice and was absent in Plin5−/− myotubes (Figure 3F). Intracellular triacylglycerol was pre-labeled with [1-14C] oleate for 4 h, then incubated in medium without radiolabeled fatty acids for 4 h, with or without isoproterenol. The oxidation of triacylglycerol-derived fatty acids was increased in Plin5−/− myotubes (genotype effect, P = 0.009, Figure 3G) and this coincided with increased depletion of the 14C labeled triacylglycerol in Plin5−/− myotubes (genotype effect, P = 0.02, Figure 3H). Hence, PLIN5 deletion causes degradation of triacylglycerol in skeletal muscle. Consistent with this notion, 24 h fasted wild-type mice stored significant amounts of lipid in their skeletal muscle whereas the skeletal muscle of Plin5−/− mice were devoid of marked lipid droplet staining (Figure 3I), indicating increased intramyocelluar lipolysis and demonstrating an inability to expand the intramyocellular triacylglycerol pool.
3.4. Mitochondrial function is normal in the skeletal muscle of Plin5−/− mice
Intramyocellular lipid droplet biology and mitochondrial biogenesis/metabolism are linked through shared molecular signals [43] and increasing intramyocellular triacylglycerol flux is proposed to enhance mitochondrial biogenesis, most likely via peroxisome proliferator-activated receptor (PPAR) mediated transcriptional reprogramming [44,45]. We first assessed mitochondrial function in primary myotubes. Oxygen consumption rate was not different under basal or maximally stimulated conditions in Plin5−/− vs. Plin5+/+ myotubes (Figure 4A–D). In line with these in vitro observations, markers of mitochondrial content and function were conserved in skeletal muscle in vivo. The expression of genes related to fatty acid uptake and storage (Cd36, Dgat1), fatty acid oxidation (Cpt1b, Mcad, Pdk4) and oxidative phosphorylation (Acox, ATP synthase, Nrf1, Ppargc1a, Tfam, UCP2) were similar in the mixed quadriceps muscle between genotypes (Figure 4E). In support of these observations, the maximal activities of the mitochondrial enzymes citrate synthase (CS) and ß-hydroxyacyl CoA dehydrogenase (β-HAD) were not affected in Plin5−/− mice (Figure 4F) and the maximal running capacity of Plin5−/− mice was not different from Plin5+/+ littermates during the graded exercise test (Figure 4G). Others have suggested that PLIN5 regulates intracellular lipid fluxes by physically recruiting mitochondria to the LD surface [46]. We detected no difference in the mitochondria to lipid droplet contact within skeletal muscle of Plin5−/− and Plin5+/+ mice using confocal microscopy (Figure 4H, I). Electron microscopy images supported these findings (Figure 4J).
Figure 4.
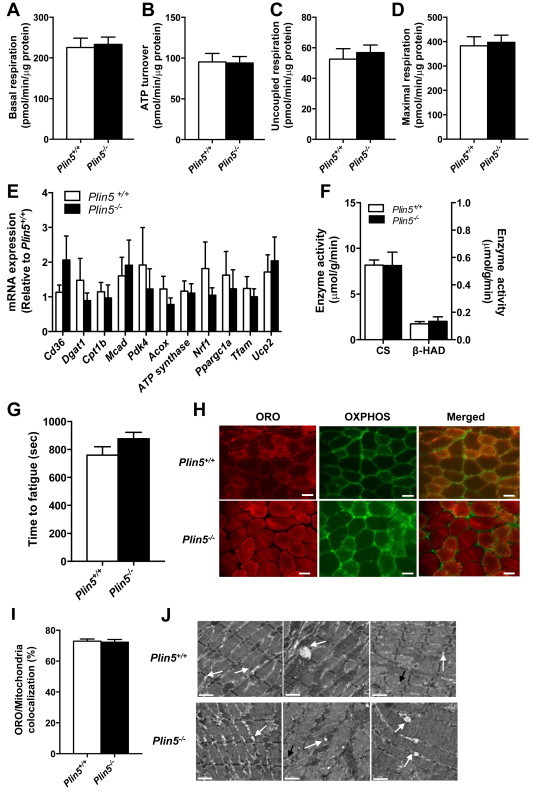
Mitochondrial capacity is not altered by PLIN5 deletion. Measures of mitochondrial capacity in Plin5−/− vs. Plin5+/+ myotubes: (A) basal mitochondrial oxygen consumption rate, (B) ATP turnover, (C) uncoupled respiration and (D) maximal respiration (n = 10 for each group obtained from three independent donor mice and three independent experiments). (E) Expression of fatty acid uptake, storage, and oxidation genes and oxidative phosphorylation genes (♂, n = 4–6 Plin5+/+, n = 5–7 Plin5−/−). (F) Skeletal muscle citrate synthase and ß-hydroxyacyl CoA dehydrogenase (β-HAD) maximal enzyme activity (♂, n = 8 Plin5+/+, n = 8 Plin5−/−). (G) Maximal running capacity assessed on a treadmill (♂, n = 7 Plin5+/+, n = 12 Plin5−/−). The exercise intensity was increased every minute until mice reached exhaustion. (H) Confocal images showing oil red O (lipid, red), OXPHOS (mitochondria, green) staining and the merged images in mixed quadriceps muscle. Scale bar = 50 μm. (I) The mitochondria/oil Red O colocation was not different between genotypes (♂, n = 3 Plin5+/+, n = 3 Plin5−/−. Nine independent sections were counted per animal with an average total of 27 ± 2 fibers per animal). (J) Electron microscopy imaging of mixed quadriceps muscle. The white arrows highlight lipid droplets and mitochondria in contact, black arrows highlight lipid droplets not in contact with mitochondria. Scale bar = 1 μm.
3.5. Glucose tolerance is improved in PLIN5 deficient mice
Skeletal muscle triacylglycerol accumulation is associated with insulin resistance [3,47] yet, paradoxically; decreasing intramyocellular triacylglycerol turnover can improve insulin action [27,48,49]. Therefore, we next asked whether Plin5 deletion would impact glucose tolerance and insulin action. Glucose tolerance was improved in Plin5−/− mice compared to wild-type mice as indicated by an attenuated blood glucose excursion after glucose administration (Figure 5A). Plasma insulin increased (P = 0.07, main effect) mildly after glucose administration and was not different between genotypes (P = 0.26) (Figure 5B). This suggests improved glucose effectiveness or enhanced insulin sensitivity in Plin5−/− mice.
Figure 5.
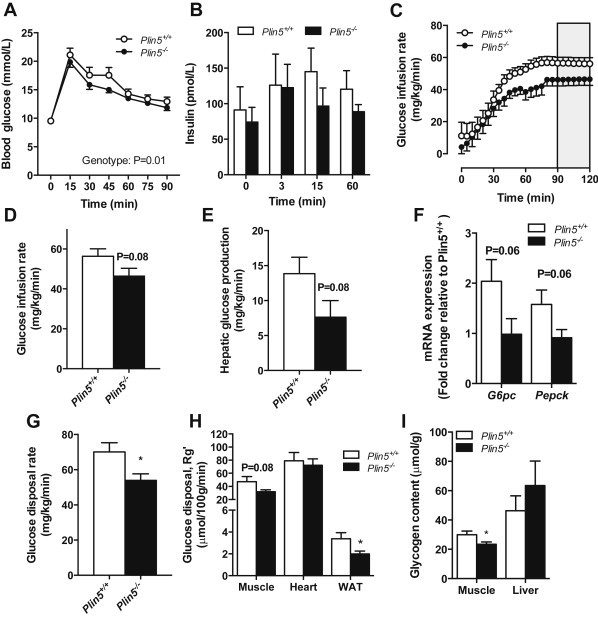
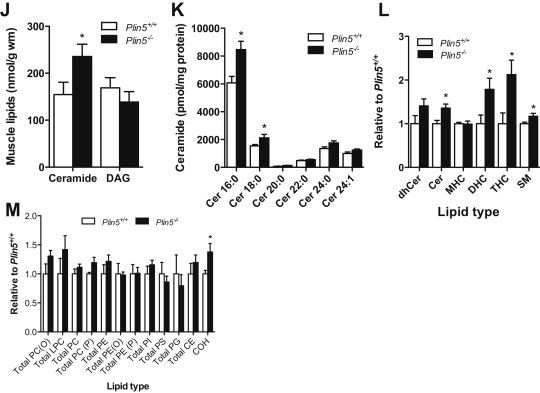
Glucose tolerance is maintained, but insulin sensitivity is reduced in association with muscle ceramide accumulation in Plin5−/− mice fed a low-fat diet. (A) Glucose tolerance of Plin5−/− mice and Plin5+/+ littermates (♂, n = 23 Plin5+/+, n = 22 Plin5−/−, Age = 10–17 weeks). (B) Plasma insulin in response to glucose tolerance test (♂, n = 10 Plin5+/+, n = 7 Plin5−/−, Age = 10–17 weeks). (C) Glucose infusion rate during the hyperinsulinemic euglycemic clamp with the steady-state period highlighted in grey shading. (D) Average glucose infusion rate during steady state (♂, n = 10 Plin5+/+, n = 12 Plin5−/−, Age = 13–16 weeks). (E) Hepatic glucose production during hyperinsulinemic euglycemic clamp (♂, n = 10 Plin5+/+, n = 12 Plin5−/−, Age = 13–16 weeks). (F) Expression of gluconeogenic genes G6pc and Pepck (♂, n = 9 Plin5+/+, n = 10 Plin5−/−, Age = 13–16 weeks). (G) Glucose disposal rate during the hyperinsulinemic euglycemic clamp (♂, n = 10 Plin5+/+, n = 12 Plin5−/−, Age = 13–16 weeks). (H) Uptake of 2-deoxyglucose by skeletal muscle, heart and epididymal white adipose tissue (WAT) (♂, n = 7 Plin5+/+, n = 8 Plin5−/−, Age = 13–16 weeks). (I) Muscle (n = 9 Plin5+/+, n = 9 Plin5−/−) and liver (n = 8 Plin5+/+, n = 8 Plin5−/−) glycogen content following hyperinsulinemic euglycemic clamp (♂, Age = 13–16 weeks). *P < 0.05 vs. Plin5+/+. (J) Ceramide and diacylglycerol content in skeletal muscle (♂, n = 8 Plin5+/+, n = 8 Plin5−/−, Age = 13–16 weeks). (K) Ceramide species in Plin5−/− vs. Plin5+/+ myotubes (n = 6 for each group) *P < 0.05 vs. Plin5+/+. (L) Sphingolipids in Plin5−/− vs. Plin5+/+ myotubes (n = 6 for each group) *P < 0.05 vs. Plin5+/+. dhCer, dihydroceramide; Cer, ceramide; MHC, monohexosylceramide; DHC, dihexosylceramide; THC, trihexosylceramide; SM, sphingomyelin. (M) Lipids were assessed by electrospray ionization-tandem mass spectrometry in Plin5−/− vs. Plin5+/+ myotubes (n = 6 for each group). Alkylphosphatidylcholine (PC(O)), lysophosphatidylcholine (LPC), phosphatidylcholine (PC), alkenylphosphatidylcholine (PC(P)), phosphatidylethanolamine (PE), alkylphosphatidylethanolamine (PE(O)), alkenylphosphatidylethanolamine (plasmalogen) (PE(P)), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylglycerol (PG), cholesterol ester (CE) and cholesterol (COH). *P < 0.05 vs. Plin5+/+.
3.6. PLIN5 deletion exerts tissue-specific effects on insulin action
To assess whole body and tissue-specific insulin action, we performed hyperinsulinemic euglycemic clamps on Plin5−/− and wild-type mice. Mice were clamped at 8.1 ± 0.3 mM glucose, 398 ± 38 pM insulin (no differences between genotypes) and steady-state GIR was achieved in both groups (Figure 5C). The glucose infusion rate required to maintain euglycemia was 15% lower in the Plin5−/− mice (P = 0.08), indicating a strong trend towards impaired whole-body insulin action (Figure 5D). Hepatic glucose production was 41% lower (P = 0.08) in Plin5−/− vs. Plin5+/+ mice during the clamp (Figure 5E). This was associated with reduced expression of the gluconeogenic genes G6pc and Pepck in livers from Plin5−/− mice (Figure 5F), suggestive of enhanced hepatic insulin sensitivity. The glucose disposal rate (GDR) during the clamp was reduced by 22% in Plin5−/− mice (Figure 5G) and this was associated with reduced 2-deoxyglucose uptake into mixed skeletal muscle (34%, P = 0.08, Figure 5H) and reduced muscle glycogen content after the clamp (Figure 5I). Glucose uptake into the heart (Figure 5H) was not different between genotypes and glucose uptake was reduced in white adipose tissue of Plin5−/− mice (40%, Figure 5H), although this is a quantitatively small contribution to systemic glucose disposal. Taken together, these data show that Plin5−/− mice become insulin resistant due to reduced insulin-stimulated glucose disposal in skeletal muscle and adipose tissue, but maintain insulin sensitivity in the liver.
3.7. Evidence of altered lipid partitioning but not ER stress, inflammation or oxidative stress in the skeletal muscle of PLIN5−/− mice
Ceramide and diacylglycerol are widely acknowledged as key drivers of insulin resistance [11]. Ceramide was increased by 53% in the skeletal muscle of Plin5−/− compared with Plin5+/+ mice, whereas diacylglycerol was not different (Figure 5J). Lipidomic analysis of cultured myotubes supported this observation: ceramide (Figure 5K, L), dihydro- and hexosyl-ceramides and sphingomyelin (Figure 5L) were increased in Plin5−/− myotubes, whereas other lipid types such as phospholipids and sterols were mostly unchanged (Figure 5M). There was no evidence for inflammation (JNK phosphorylation, degradation of IΚBα, TNFα and IL-6 mRNA expression), ER stress (phosphorylated GADD34 and PERK) or oxidative stress (thiobarbituric acid reactive substances) in the skeletal muscle of Plin5−/− mice compared with wild-type mice (data not shown).
3.8. Effects of diet-induced obesity on lipid metabolism and insulin action in Plin5−/− mice
Because obesity impacts lipid metabolism and insulin action, we next examined the metabolic phenotype of Plin5−/− mice that were fed a high-fat diet for eight weeks. High-fat feeding increased body mass (Figure 6A) and fat mass (Figure 6B) in mice of both genotypes. Whole-body energy expenditure (Figure 6C), RER (Figure 6D) and daily physical activity (Figure 6E) were not different between Plin5−/− and Plin5+/+ mice. There was no difference in skeletal muscle fatty acid oxidation (Figure 6F) or incorporation into triacylglycerol (Figure 6G) between genotypes and lipid droplet density, diacylglycerol and ceramide levels were similarly unaffected (data not shown). Fasting blood glucose (Figure 6H, P = 0.07) and plasma insulin (Figure 6I) were not different between phenotypes. Glucose tolerance was markedly better in high-fat fed Plin5−/− compared with Plin5+/+ mice (Figure 6J). Indeed, high-fat fed Plin5−/− mice had comparable glucose tolerance to low-fat fed Plin5+/+ mice (Figure 6K). Differences in plasma insulin levels did not explain the improved glucose tolerance (Figure 6L). Insulin sensitivity was assessed by intravenous administration of insulin and 2-[1,2-3H] deoxyglucose. Neither whole body deoxyglucose clearance (P = 0.30) nor tissue-specific clearance into the key sites of glucose disposal was different between genotypes (Figure 6M, N), suggesting insulin-independent mechanisms may be responsible for the improved glucose tolerance i.e. glucose effectiveness (mass action of glucose).
Figure 6.
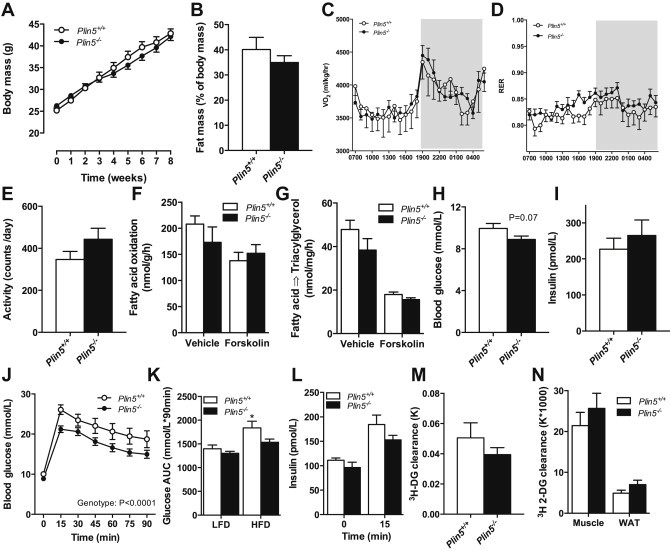
Effects of high-fat feeding on substrate metabolism and insulin action in Plin5−/− mice. (A) Body mass (♂, n = 14 Plin5+/+, n = 12 Plin5−/−). (B) Percent body fat mass assessed by DEXA (♂, n = 6 Plin5+/+, n = 10 Plin5−/−, Age = 14–16 weeks). (C) Oxygen consumption (VO2) and (D) respiratory exchange ratio (RER) were assessed by indirect calorimetry (♂, n = 6 Plin5+/+, n = 10 Plin5−/−, Age = 14–16 weeks). (E) Total daily activity (♂, n = 6 Plin5+/+, n = 9 Plin5−/−, Age = 14–16 weeks). (F) Fatty acid oxidation and (G) fatty acid incorporation into triacylglycerol (TAG) (♂, n = 8 Plin5+/+, n = 5 Plin5−/−; Age = 14–16 weeks). (H) Blood glucose in 4 h fasted mice (♂, n = 14 Plin5+/+, n = 16 Plin5−/−; Age = 14–16 weeks). (I) Plasma insulin in 4 h fasted mice (♂, n = 7 Plin5+/+, n = 8 Plin5−/−; Age = 14–16 weeks). (J) Glucose tolerance (♂, n = 13 Plin5+/+, n = 16 Plin5−/−, Age = 14–16 weeks) (K) Total glucose area under the curve (AUC) for low fat diet (LFD) and high fat diet (HFD) from the glucose tolerance test (LFD, ♂, n = 23 Plin5+/+, n = 22 Plin5−/−, Age = 10–17 weeks; HFD, ♂, n = 14 Plin5+/+, n = 16 Plin5−/−, Age = 14–16 weeks). *P < 0.05 vs. all other groups. (L) Plasma insulin in response to glucose tolerance test (♂, n = 7 Plin5+/+, n = 7 Plin5−/−, Age = 14–16 weeks). (M) Rate of insulin-stimulated 3H 2-deoxyglucose disappearance from the blood and (N) rate of insulin-stimulated 3H 2-deoxyglucose (DG) clearance into mixed quadriceps skeletal muscle and white (WAT) adipose tissue (♂, n = 6 Plin5+/+, n = 8 Plin5−/−, Age = 14–16 weeks).
4. Discussion
Lipid metabolism and insulin action are intimately linked [50] and lipid droplets stored within tissues are critical for controlling intracellular lipid flux [51]. In this regard, the highly conserved perilipin family of proteins reside on the surface of lipid droplets and are important for lipid droplet formation, lipolytic regulation during times of energetic demand and protection against lipotoxicity when cells are exposed to an excess of fatty acids. PLIN5 is unique among this family because it is highly expressed in oxidative, glucoregulatory tissues such as skeletal muscle [22], is localized to multiple intracellular locations including the lipid droplet, endoplasmic reticulum, mitochondria and the cytosol [52] and may regulate a dynamic interaction between the lipid droplet and mitochondria that is proposed to be relevant to the etiology and treatment of insulin resistance [43,46]. Uncertainty surrounds the metabolic roles of PLIN5 (discussed below), partly because most cell systems used to date poorly reflect the metabolic requirements of the major oxidative tissues in vivo. To address this question, we generated Plin5−/− mice and examined lipid metabolism and insulin action in skeletal muscle, a major site for fatty acid and glucose disposal.
The controversy surrounding PLIN5's involvement in metabolism arises from the apparently conflicting observations in immortalized cell lines that PLIN5 promotes both fatty acid oxidation and storage [22] and either increases or decreases lipolysis [24,42,53,54]. The prevailing views with respect to lipolysis are: (1) ATGL and CGI-58 interact separately with PLIN5 on lipid droplets to facilitate their interaction and drive lipolysis [53,54]; or (2) PLIN5 and CGI-58 independently recruit ATGL to lipid droplets with opposite effects, CGI-58-ATGL interactions drive lipolysis while PLIN5 binding to ATGL inhibits lipolysis [24,42]. In the current study, we show that PLIN5 does not influence fatty acid transport into myotubes, fatty acid oxidation or fatty acid esterification into glycerolipids. However, PLIN5 ablation increases intramyocellular lipolysis and invokes a remarkable decrease in muscle lipid droplet content in fasted, but not fed mice. Hence, our data agree with the interpretation that PLIN5 provides a barrier function on lipid droplets to prevent excessive lipolysis. These novel findings in skeletal muscle mostly agree with the previously described role of PLIN5 in the heart. Plin5 ablation reduced lipid droplet size, which was due to increased ATGL-mediated TAG lipolysis and, independently, an increased capacity for FA oxidation in cardiomyocytes [20]. On the other hand, cardiac-specific overexpression of PLIN5 inhibits ATGL-mediated lipolysis resulting in increased lipid droplet size, myocardial steatosis, mild impairments in oxidative metabolism and modest cardiac dysfunction [21,55]. Together, these studies support the view that PLIN5 is a highly abundant protein in muscle that is required to repress intramyocellular lipolysis. This effect is most prominent during prolonged nutrient deprivation, where fatty acid substrate from both intramyocellular and adipose tissue triacylglycerol is enhanced. Hence, PLIN5 may protect myocytes from unnecessary depletion of intramyocellular fuels and/or control intracellular fatty acid flux to prevent lipotoxic outcomes.
Mitochondrial dysfunction in skeletal muscle is proposed to contribute to reduced fatty acid oxidation, lipid deposition and the development of insulin resistance in obese individuals [56]. Indeed, such observations have provided the impetus for developing therapeutics aimed at enhancing mitochondrial function and fatty acid oxidation to treat obesity (e.g. acetyl CoA carboxylase inhibitors, AMPK activators). While this rationale is probably oversimplified [57], emerging evidence indicates that increasing fatty acid flux from lipid droplets can induce mitochondrial biogenesis, most likely via a PPAR-mediated program, and in turn improve cell function [43–45,58]. While we demonstrated that fatty acid oxidation from intramyocellular lipid was increased with Plin5 deletion, mitochondrial content and function were not enhanced in either isolated myotubes or intact skeletal muscle; exogenous fatty acid and glucose oxidation were not enhanced in vitro and whole-body fatty acid oxidation was actually reduced by ∼11% in Plin5−/− mice. The brain and skeletal muscle account for ∼45% of the daily energy expenditure [59], therefore in the absence of increased glucose oxidation in these tissues, other tissues are most likely accounting for the decrease in systemic fatty acid oxidation and reciprocal increase in glucose oxidation. While the effects of PLIN5 overexpression in skeletal muscle are equivocal [23,26], overexpression of PLIN5 in cardiac muscle reduces intracellular lipolysis and is associated with reduced mRNA contents of oxidative phosphorylation proteins, decreased mitochondrial enzyme activities and mildly impaired mitochondrial respiration [21,55]. Aside from tissue specific actions, the reasons for this apparent discrepancy are unclear. It is possible that the increase in intramyocellular lipolysis with Plin5 deletion was insufficient to drive a PPAR transcriptional program. This possibility is supported by the similarity in PGC1α mRNA and PGC1α target genes between Plin5−/− and control mice shown here, and further supported by a recent report in muscle-specific ATGL null and overexpressing mice showing a dissociation of intramyocellular lipolysis, mitochondrial biogenesis and fatty acid oxidation [60]. Alternatively, PLIN5 may modulate the cells molecular program via direct transcription/co-activator functions, which may be functionally redundant in Plin5−/− mice.
While intramyocellular triacylglycerol accumulation is linked to insulin resistance [2,3], emerging data now link the etiology of insulin resistance to the uncoupling of intramyocellular lipolysis from the cellular demand for fatty acids [14,61]. In this context, we proposed that the increase in muscle lipolysis combined with no additional demand for fatty acid substrate would reduce insulin sensitivity in Plin5−/− mice. Indeed, our results show that Plin5−/− mice have reduced skeletal muscle insulin sensitivity, as demonstrated during hyperinsulinemic euglycemic clamps. This was associated with ceramide accumulation, which is a negative regulator of insulin signal transduction [50]. Other complex ceramides and sphingomyelin were increased, while phospholipids, sterol lipids and diacylglycerol were unchanged with Plin5 deletion. Hence, fatty acids derived from accelerated intracellular lipolysis appear to be preferentially directed into sphingolipids, which in turn cause insulin resistance. This interpretation is consistent with a recent human study demonstrating the inverse: increased PLIN5 content, decreased muscle ceramide (not diacylglycerol) and improved insulin sensitivity in the skeletal muscle of endurance trained compared with obese men [62]. The reduction in skeletal muscle insulin sensitivity in Plin5−/− mice is unlikely to be due to impaired blood flow and insulin delivery, secondary to impaired cardiac function, because impairments in the cardiac function of Plin5−/− mice is not apparent until 30–38 weeks of age [20], and we assessed insulin sensitivity in mice aged 16 weeks when cardiac function is normal.
In contrast to muscle, both hepatic glucose production and the insulin sensitive gluconeogenic gene expression were lower in Plin5−/− mice during the insulin clamp conditions, suggestive of improved hepatic insulin sensitivity. Moreover, Plin5−/− mice have normal fasting glucose and insulin and improved glucose tolerance compared with wild-type mice following glucose administration. This highlights the important role of liver in maintaining glycemia under basal and post-prandial conditions (i.e. GTT) [63,64] and suggests that the improved hepatic insulin action compensates for the reduced peripheral insulin sensitivity in Plin5−/− mice. It is important to note that progressive declines in muscle insulin sensitivity have no impact on glucose tolerance in C57Bl/6 mice [32] and that glucose effectiveness is quantitatively more important than insulin sensitivity in the determination of glucose tolerance [65]. The contribution of glucose effectiveness over insulin sensitivity is more pronounced with intraperitoneal glucose administration, which bypasses the gut and precludes an incretin effect, thus resulting in minimal insulin secretion. Hence, our glucose tolerance and insulin clamp data are concordant. The uncoupling of muscle and liver insulin sensitivity is not without precedence either; Plin1−/− mice present with a very similar phenotype [17], although the factors governing these responses in each genotype are unlikely to be conserved given the unique tissue expression patterns of PLIN1 (adipose) and PLIN5 (muscle, liver). Understanding the cell-specific mechanisms that mediate the dichotomy in insulin action is beyond the scope of these studies and future work will address this intriguing question.
PLIN5 plays a protective role in regulating lipid homeostasis in several tissues [20–22], including skeletal muscle, and PLIN5 abundance is a predictor of insulin sensitivity in humans [43]. We tested whether Plin5 deletion would exacerbate the insulin resistance induced by high-fat feeding, through an inability to appropriately sequester lipids into lipid droplets. Energy balance, substrate partitioning and muscle lipid metabolism and storage were mostly unperturbed in Plin5−/− compared with Plin5+/+ mice with high fat feeding. Despite these similarities, Plin5−/− mice had lower blood glucose and enhanced glucose tolerance, which was not explained by hyperinsulinemia or enhanced insulin sensitivity in peripheral tissues. Thus, while PLIN5 acts to sequester triacylglycerol in the lipid droplet and prevent excessive lipolysis under standard dietary conditions, the substantial increase in lipid flux associated with high-fat feeding overwhelms the capacity to store incoming fatty acid as triacylglycerol, resulting in an increase in lipotoxic intermediates. Under these conditions, the contribution of PLIN5 deletion and intramyocellular lipolysis to sphingolipid accumulation is likely to be negligible, and insulin resistance ensues irrespective of PLIN5 deletion. Interestingly, Plin5−/− mice are protected from high-fat feeding induced glucose intolerance, suggesting that blocking PLIN5 may be a mechanism to improve hepatic glucose metabolism. Though at this point we cannot define the precise mechanisms mediating this improvement, future studies in mice with liver-specific ablation of PLIN5 should help delineate how this occurs.
Taken together, our studies show that PLIN5 suppresses skeletal muscle lipolysis, particularly during prolonged nutrient deprivation, does not influence mitochondrial reprogramming and is required for the maintenance of insulin sensitivity in skeletal muscle by preventing ceramide production. Others have shown that PLIN5 expression is increased by PPARα agonists, fasting, fatty acids and endurance exercise training [22,24,25,43]; situations characterized by increased lipid flux in skeletal muscle. Hence, our data support the hypothesis that PLIN5 is required to match lipolysis of intramyocellular triacylglycerol to metabolic demands, which helps to maintain insulin sensitivity in skeletal muscle.
Author contributions
R.R.M. performed experiments, analyzed data, wrote the manuscript. R.M., M.M., A.S., G.M.K. and N.M. performed experiments and edited the manuscript. P.J.M. provided lipidomic support and edited the manuscript. C.R.B. performed experiments, analyzed data, provided discussion and edited the manuscript. M.J.W. performed experiments, analyzed data and wrote the manuscript.
Acknowledgments
We thank Dirk Truman and Leanne Cotton (Monash University) for valuable advice on mouse generation and Adam Costin (Monash Micro Imaging, Monash University) for performing the electron microscopy analysis. These studies were supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (to MJW APP1047138). RRM is supported by Paul McNamee Postgraduate Scholarship and MJW and PJM are supported by research fellowships from the National Health and Medical Research Council (NHMRC, APP 606460). NIH grants to Velocigene at Regeneron Inc (U01HG004085) and the CSD Consortium (U01HG004080) funded the generation of gene-targeted ES cells for 8500 genes in the KOMP Program and archived and distributed by the KOMP Repository at UC Davis and CHORI (U42RR024244).
MJW takes full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Kelley D., Mitrakou A., Marsh H., Schwenk F., Benn J., Sonnenberg G. The Journal of Clinical Investigation. 1988;81:1563–1571. doi: 10.1172/JCI113489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krssak M., Falk P.K., Dresner A., DiPietro L., Vogel S.M., Rothman D.L. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 3.Pan D.A., Lillioja S., Kriketos A.D., Milner M.R., Baur L.A., Bogardus C. Diabetes. 1997;46:983–988. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 4.Perseghin G., Scifo P., De Cobelli F., Pagliato E., Battezzati A., Arcelloni C. Diabetes. 1999;48:1600–1606. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- 5.Jacob S., Machann J., Rett K., Brechtel K., Volk A., Renn W. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 6.Goodpaster B.H., He J., Watkins S., Kelley D.E. The Journal of Clinical Endocrinology and Metabolism. 2001;86:5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 7.Fujimoto T., Parton R.G. Cold Spring Harbor Perspectives in Biology. 2011;3:1–17. doi: 10.1101/cshperspect.a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watt M.J., Heigenhauser G.J.F., Spriet L.L. Journal of Applied Physiology. 2002;93:1185–1195. doi: 10.1152/japplphysiol.00197.2002. [DOI] [PubMed] [Google Scholar]
- 9.Banke N.H., Wende A.R., Leone T.C., O'Donnell J.M., Abel E.D., Kelly D.P. Circulation Research. 2010;107:233–241. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sacchetti M., Saltin B., Olsen D.B., van Hall G. The Journal of Physiology. 2004;561:883–891. doi: 10.1113/jphysiol.2004.075135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watt M.J., Hoy A.J. American Journal of Physiology. Endocrinology and Metabolism. 2012;302:E1315–1328. doi: 10.1152/ajpendo.00561.2011. [DOI] [PubMed] [Google Scholar]
- 12.Levin M.C., Monetti M., Watt M.J., Sajan M.P., Stevens R.D., Bain J.R. American Journal of Physiology. Endocrinology and Metabolism. 2007;293:E1772–1781. doi: 10.1152/ajpendo.00158.2007. [DOI] [PubMed] [Google Scholar]
- 13.Nagle C.A., An J., Shiota M., Torres T.P., Cline G.W., Liu Z.X. The Journal of Biological Chemistry. 2007;282:14807–14815. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badin P.M., Louche K., Mairal A., Liebisch G., Schmitz G., Rustan A.C. Diabetes. 2011;60:1734–1742. doi: 10.2337/db10-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bickel P.E., Tansey J.T., Welte M.A. Biochimica et Biophysica Acta. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg A.S., Egan J.J., Wek S.A., Garty N.B., Blanchettemackie E.J., Londos C. Journal of Biological Chemistry. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 17.Tansey J.T., Sztalryd C., Gruia-Gray J., Roush D.L., Zee J.V., Gavrilova O. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McManaman J.L., Bales E.S., Orlicky D.J., Jackman M., Maclean P.S., Cain S. Journal of Lipid Research. 2013;54:1346–1359. doi: 10.1194/jlr.M035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai Y., Boyle S., Varela G.M., Caron E., Yin X., Dhir R. Physiological Genomics. 2012;44:1125–1131. doi: 10.1152/physiolgenomics.00045.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuramoto K., Okamura T., Yamaguchi T., Nakamura T.Y., Wakabayashi S., Morinaga H. The Journal of Biological Chemistry. 2012;287:23852–23863. doi: 10.1074/jbc.M111.328708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., Sreenivasan U., Gong D.W., O'Connell K.A., Dabkowski E.R., Hecker P.A. Journal of Lipid Research. 2013;54:953–965. doi: 10.1194/jlr.M032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolins N.E., Quaynor B.K., Skinner J.R., Tzekov A., Croce M.A., Gropler M.C. Diabetes. 2006;55:3418–3428. doi: 10.2337/db06-0399. [DOI] [PubMed] [Google Scholar]
- 23.Bosma M., Minnaard R., Sparks L.M., Schaart G., Losen M., de Baets M.H. Histochemistry and Cell Biology. 2012;137:205–216. doi: 10.1007/s00418-011-0888-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalen K.T., Dahl T., Holter E., Arntsen B., Londos C., Sztalryd C. Biochimica et Biophysica Acta. 2007;1771:210–227. doi: 10.1016/j.bbalip.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi T., Matsushita S., Motojima K., Hirose F., Osumi T. The Journal of Biological Chemistry. 2006;281:14232–14240. doi: 10.1074/jbc.M601682200. [DOI] [PubMed] [Google Scholar]
- 26.Bosma M., Sparks L.M., Hooiveld G.J., Jorgensen J.A., Houten S.M., Schrauwen P. Biochimica et Biophysica Acta. 2013;1831:844–852. doi: 10.1016/j.bbalip.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Huijsman E., van de Par C., Economou C., van der Poel C., Lynch G.S., Schoiswohl G. American Journal of Physiology. Endocrinology and Metabolism. 2009;297:E505–513. doi: 10.1152/ajpendo.00190.2009. [DOI] [PubMed] [Google Scholar]
- 28.Shortreed K.E., Krause M.P., Huang J.H., Dhanani D., Moradi J., Ceddia R.B. PLoS One. 2009;4:e7293. doi: 10.1371/journal.pone.0007293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Folch J., Lees M., Sloane Stanley G.H. The Journal of Biological Chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- 30.Watt M.J., Hevener A., Lancaster G.I., Febbraio M.A. Endocrinology. 2006;147:2077–2085. doi: 10.1210/en.2005-1074. [DOI] [PubMed] [Google Scholar]
- 31.Borg M.L., Andrews Z.B., Duh E.J., Zechner R., Meikle P.J., Watt M.J. Diabetes. 2011;60:1458–1466. doi: 10.2337/db10-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner N., Kowalski G.M., Leslie S.J., Risis S., Yang C., Lee-Young R.S. Diabetologia. 2013;56:1638–1648. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 33.Cooney G.J., Caterson I.D., Newsholme E.A. FEBS Letters. 1985;188:257–261. doi: 10.1016/0014-5793(85)80383-5. [DOI] [PubMed] [Google Scholar]
- 34.Crowe S., Turpin S.M., Ke F., Kemp B.E., Watt M.J. Endocrinology. 2008;149:2546–2556. doi: 10.1210/en.2007-1447. [DOI] [PubMed] [Google Scholar]
- 35.Pagnon J., Matzaris M., Stark R., Meex R.C., Macaulay S.L., Brown W. Endocrinology. 2012;153:4278–4289. doi: 10.1210/en.2012-1127. [DOI] [PubMed] [Google Scholar]
- 36.Mokbel N., Ilkovski B., Kreissl M., Memo M., Jeffries C.M., Marttila M. Brain – A Journal of Neurology. 2013;136:494–507. doi: 10.1093/brain/aws348. [DOI] [PubMed] [Google Scholar]
- 37.Peters S.J., Dyck D.J., Bonen A., Spriet L.L. American Journal of Physiology – Endocrinology and Metabolism. 1998;275:E300–E309. doi: 10.1152/ajpendo.1998.275.2.E300. [DOI] [PubMed] [Google Scholar]
- 38.Chen W., Chang B., Wu X., Li L., Sleeman M., Chan L. American Journal of Physiology. Endocrinology and Metabolism. 2013;304:E770–779. doi: 10.1152/ajpendo.00523.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rolfe D.F., Brown G.C. Physiological Reviews. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 40.Watt M.J., Stellingwerff T., Heigenhauser G.J.F., Spriet L.L. Journal of Physiology – London. 2003;550:325–332. doi: 10.1113/jphysiol.2003.043133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jocken J.W., Blaak E.E. Physiology & Behavior. 2008;94:219–230. doi: 10.1016/j.physbeh.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Wang H., Bell M., Sreenevasan U., Hu H., Liu J., Dalen K. The Journal of Biological Chemistry. 2011;286:15707–15715. doi: 10.1074/jbc.M110.207779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koves T.R., Sparks L.M., Kovalik J.P., Mosedale M., Arumugam R., DeBalsi K.L. Journal of Lipid Research. 2013;54:522–534. doi: 10.1194/jlr.P028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haemmerle G., Moustafa T., Woelkart G., Buttner S., Schmidt A., van de Weijer T. Nature Medicine. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmadian M., Duncan R.E., Varady K.A., Frasson D., Hellerstein M.K., Birkenfeld A.L. Diabetes. 2009;58:855–866. doi: 10.2337/db08-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H., Sreenevasan U., Hu H., Saladino A., Polster B.M., Lund L.M. Journal of Lipid Research. 2011;52:2159–2168. doi: 10.1194/jlr.M017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bjorbaek C., El-Haschimi K., Frantz J.D., Flier J.S. The Journal of Biological Chemistry. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 48.Hoy A.J., Bruce C.R., Turpin S.M., Morris A.J., Febbraio M.A., Watt M.J. Endocrinology. 2011;152:48–58. doi: 10.1210/en.2010-0661. [DOI] [PubMed] [Google Scholar]
- 49.Kienesberger P.C., Lee D., Pulinilkunnil T., Brenner D.S., Cai L., Magnes C. The Journal of Biological Chemistry. 2009;284:30218–30229. doi: 10.1074/jbc.M109.047787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samuel V.T., Shulman G.I. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greenberg A.S., Coleman R.A., Kraemer F.B., McManaman J.L., Obin M.S., Puri V. The Journal of Clinical Investigation. 2011;121:2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartholomew S.R., Bell E.H., Summerfield T., Newman L.C., Miller E.L., Patterson B. Biochimica et Biophysica Acta. 2011;1821:268–278. doi: 10.1016/j.bbalip.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Granneman J.G., Moore H.P., Mottillo E.P., Zhu Z. The Journal of Biological Chemistry. 2009;284:3049–3057. doi: 10.1074/jbc.M808251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Granneman J.G., Moore H.P., Mottillo E.P., Zhu Z., Zhou L. The Journal of Biological Chemistry. 2011;286:5126–5135. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pollak N.M., Schweiger M., Jaeger D., Kolb D., Kumari M., Schreiber R. Journal of Lipid Research. 2013;54:1092–1102. doi: 10.1194/jlr.M034710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodpaster B.H. Diabetes. 2013;62:1032–1035. doi: 10.2337/db12-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoehn K.L., Turner N., Swarbrick M.M., Wilks D., Preston E., Phua Y. Cell Metabolism. 2010;11:70–76. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sapiro J.M., Mashek M.T., Greenberg A.S., Mashek D.G. Journal of Lipid Research. 2009;50:1621–1629. doi: 10.1194/jlr.M800614-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frayn K.N., Humphreys S.M., Coppack S.W. Proceedings of the Nutrition Society. 1995;54:177–189. doi: 10.1079/pns19950047. [DOI] [PubMed] [Google Scholar]
- 60.Sitnick M.T., Basantani M.K., Cai L., Schoiswohl G., Yazbeck C.F., Distefano G. Diabetes. 2013;62:3350–3361. doi: 10.2337/db13-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moro C., Galgani J.E., Luu L., Pasarica M., Mairal A., Bajpeyi S. The Journal of Clinical Endocrinology and Metabolism. 2009;94:3440–3447. doi: 10.1210/jc.2009-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amati F., Dube J.J., Alvarez-Carnero E., Edreira M.M., Chomentowski P., Coen P.M. Diabetes. 2011;60:2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rohner-Jeanrenaud F., Proietto J., Ionescu E., Jeanrenaud B. Diabetes. 1986;35:1350–1355. doi: 10.2337/diab.35.12.1350. [DOI] [PubMed] [Google Scholar]
- 64.Mitrakou A., Kelley D., Veneman T., Jenssen T., Pangburn T., Reilly J. Diabetes. 1990;39:1381–1390. doi: 10.2337/diab.39.11.1381. [DOI] [PubMed] [Google Scholar]
- 65.Best J.D., Kahn S.E., Ader M., Watanabe R.M., Ni T.C., Bergman R.N. Diabetes Care. 1996;19:1018–1030. doi: 10.2337/diacare.19.9.1018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.