

Abstract
Overfeeding causes rapid synaptic remodeling in hypothalamus feeding circuits. Polysialylation of cell surface molecules is a key step in this neuronal rewiring and allows normalization of food intake. Here we examined the role of hypothalamic polysialylation in the long-term maintenance of body weight, and deciphered the molecular sequence underlying its nutritional regulation. We found that upon high fat diet (HFD), reduced hypothalamic polysialylation exacerbated the diet-induced obese phenotype in mice. Upon HFD, the histone acetyltransferase MOF was rapidly recruited on the St8sia4 polysialyltransferase-encoding gene. Mof silencing in the mediobasal hypothalamus of adult mice prevented activation of the St8sia4 gene transcription, reduced polysialylation, altered the acute homeostatic feeding response to HFD and increased the body weight gain. These findings indicate that impaired hypothalamic polysialylation contribute to the development of obesity, and establish a role for MOF in the brain control of energy balance.
Keywords: Hypothalamus, Polysialylation, Synaptic plasticity, Obesity, Food intake, Chromatin
1. Introduction
Homeostatic maintenance of energy balance is crucial for health. In the brain, specialized neuronal circuits integrate metabolic signals related to energy stores and needs and consequently adjust energy intake, utilization and expenditure [1]. The melanocortin system is one of the most well described circuits which control body weight and food intake [2,3]. The two main antagonistic components of this system are the anorexigenic POMC and orexigenic NPY neurons located in the arcuate nucleus of the hypothalamus. It has been recently shown that density and input type on POMC and NPY neurons vary in adult mice, depending on the metabolic state and the associated fluctuation of circulating hormones [4–6]. Such synaptic plasticity modifies the activity of these neurons and thereby the responsiveness of the melanocortin system. It has been proposed that the synaptic plasticity of this system in response to acute changes in fuel availability was essential to the accurate control of food intake and the maintenance of body weight in adulthood [7–9]. Moreover, altered synaptic plasticity of POMC and NPY neurons was found in diet-induced obese rodents suggesting that inability to dynamically rewire feeding circuits could contribute to obesity [10].
Polysialic acid (PSA) is a cell-surface glycan that modulates cell-to-cell interactions [11]. Polysialylation of cell adhesion proteins is involved in various synaptic plasticity-dependent processes in the central nervous system including learning [12,13], pain modulation [14,15] and the control of neuroendocrine functions [16,17]. We previously reported that polysialylation was also required for the adaptive synaptic plasticity of feeding circuits during acute positive energy balance [8]. Precisely, high-fat diet (HFD) given for three days caused a PSA-dependent increase in the excitatory inputs connecting POMC neurons in mice. The polysialyltransferase PST-1, encoded by the St8sia4 gene was identified as the enzyme involved in the PSA-dependent control of energy intake [8]. However the biological mediators and the intracellular cascade of events leading to the dietary fat-induced PSA-dependent synaptic plasticity in the hypothalamus have not been characterized yet.
It is now clearly established that modifications in gene expression patterns are required to elicit structural and functional synaptic plasticity in response to experience and environmental cues [18–20]. The molecular mechanisms supporting these changes in gene expression were explored in several synaptic plasticity-dependent processes such as learning, memory formation, addiction and stress [21–23]. In these models, alterations in expression, localization or activity of transcription regulatory factors and in chromatin structure are instrumental in the signaling pathway leading to synapse remodeling [23,24]. In particular, changes in the acetylation of histone tails and in the activity of histone acetyltransferases or histone deacetylases are associated with changes in plasticity-related gene expression in the brain areas supporting these synaptic plasticity-dependent processes, i.e. the hippocampus, the prefrontal cortex and the nucleus accumbens [25–28]. Such mechanisms are still unexplored regarding the synaptic plasticity of the feeding circuits.
Here we explored the role of hypothalamic polysialylation in the long-term maintenance of body weight. We also deciphered the molecular sequence underlying the activation of polysialylation in the hypothalamus upon overfeeding. Hypothalamic PSA removal and St8sia4 silencing increased body weight gain on HFD, indicating that St8sia4 is essential to maintain energy homeostasis. We showed that dietary fat rapidly incited post-translational histone modifications on the St8sia4 gene in the mediobasal hypothalamus (MBH). The histone acetyltransferase MOF was required for the HFD-induced acetylation of histone H4 on lysine 16 and the subsequent activation of St8sia4 gene transcription. Moreover, Mof silencing in the MBH of adult mice fed a HFD reduced hypothalamic PSA levels, caused overfeeding and exacerbated diet-induced obesity.
2. Materials and methods
2.1. Animals and animal procedures
Experiments were carried out on 8-week-old male C57Bl/6JOla mice (Harlan Laboratories). Animal care and experimental procedures were performed with approval from the Animal Care and Use Committee of the University of Burgundy. Mice were housed in individual cages and maintained on a standard light/dark cycle. They were fed either a standard diet (A04; Safe Laboratories) or a high fat diet (HFD) (Safe Laboratories as previously described [8]). All animals had ad libitum access to food and water. Body weight and food consumption were recorded every day.
2.2. Repeated bilateral injection of endoneuraminidase N into the mediobasal hypothalamus
Mice were anesthetized with isoflurane (Abbott) and placed in a stereotaxic apparatus (David Kopf Instruments) in flat skull position. After dermal disinfection with betadine solution, the skin and cranial muscles were incised and the skull was exposed. Small holes were drilled and a double cannula with a cap (C235G-0.8/SP 4.6 mm; Plastic One) was inserted to target above the mediobasal hypothalamus (MBH) using the following coordinates: −1.4 mm posterior to the Bregma, ±0.4 mm lateral to the sagittal suture, and −4.6 mm below the skull surface (Supplementary Figure 1A). Cannula was fixed with screws and dental cement. After cannulation, animals were kept under controlled temperature and rehydrated with intraperitoneal injections of physiological fluid. Mice were then housed individually and were allowed 1 week for recovery before experiment. For injection, mice were anesthetized with isoflurane, and cannula cap was removed. Mice were injected with either vehicle or Endoneuraminidase N (EndoN, 0.28 units/side, 400 nL; Eurobio) at a rate of 100 nL/min, weekly during one month, with a 5.6 mm injector (C235I/SP 5.6 mm; Plastic One) inserted into the cannula targetting the MBH. The injector was maintained for an additional 3 min to avoid back leakage. After injection, mice were replaced in their individual cages.
2.3. Bilateral injection of lentiviral vectors into the hypothalamus
Mice were anesthetized with isoflurane (Abbott) and placed in a stereotactic apparatus (David Kopf Instruments). Mice received a bilateral injection of lentiviral transduction particles (400 nL per side, 2–3 × 109 TU/mL), using the following stereotactic coordinates: −1.4 mm posterior to the Bregma, ±0.4 mm lateral to the sagittal suture, and −5.6 mm below the skull surface (Supplementary Figure 1B). The lentiviral particles contained either a non-target control shRNA (Non-mammalian Control; Sigma–Aldrich), or a shRNA targeting Mof (TRCN0000039323; Sigma–Aldrich) or St8sia4 (TRCN0000093936). The viral suspension was delivered at a rate of 100 nL/min through a 34G blunt needle mounted on a 10 μL syringe (NanoFil device from WPI) controlled by a micropump (UMP2 from WPI). After surgery, animals were rehydrated with intraperitoneal injection of saline. Mice were also injected with analgesic (buprenorphine; 0.1 mg/kg i.p.; Buprecare, Animalcare) and ibuprofen (20 mg/kg; Advil) was added to the drinking water for 3 days. All experiments were initiated after a 3-week recovery period.
2.4. EchoMRI
Nuclear magnetic resonance was used to determine the body composition of the mice (QNMR system EchoMRI-500T, Echo Medical Systems).
2.5. Chromatin immunoprecipitation
Brains were promptly removed and the mediobasal hypothalamic regions dissected using brain matrices. Explants were immediately cross-linked in 1.5% formaldehyde for 10 min at room temperature and quenched by adding glycine to a final concentration of 0.125 M. Explants were then washed 3 times in ice-cold PBS. Tissues were first homogenized in a cell lysis buffer containing 50 mM Hepes pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40 and 0.25% Triton X-100, 1× Complete protease inhibitors cocktail (Roche). The pellets were finally resuspended in a nuclear lysis buffer (Tris–HCl 10 mM, NaCl 140 mM, EDTA 1 mM, EGTA 0.5 mM, 0.1% SDS, 0.1% Na Deoxycholate), 1× Complete protease inhibitors cocktail (Roche) and sonicated for 20 min in a Bioruptor (Diagenode; settings: High; 30 s ON/30 s OFF). This chromatin-shearing protocol produces fragments ranging from 150 to 400 bp in length. For ChIP, antibodies (listed in Table 1) were conjugated to magnetic Dynabeads (Invitrogen). Chromatin was incubated with beads-bound antibodies overnight at 4 °C in a buffer containing Tris–HCl 10 mM, NaCl 140 mM, EDTA 1 mM, EGTA 0.5 mM, 0.1% SDS, 0.5% Na Deoxycholate, 1% NP-40 and 1× Complete protease inhibitors cocktail (Roche) and then washed 6 times in RIPA buffer and once in Tris–EDTA Buffer (Tris 10 mM, EDTA 1 mM). Elution buffer (Tris 50 mM, EDTA 10 mM, SDS 1%) was added and samples were incubated at 65 °C, 1400 rpm, in a Thermomixer for 30 min to remove beads. Chromatin in the supernatant was then reverse cross-linked by incubating 10 h at 65 °C. DNA was purified with a QIAquick PCR purification kit (Qiagen). Quantitative PCR analysis was performed using the FAST SYBR Green Master Mix (Applied Biosystems). The following primers were used for PCR amplification: were as follow: 5′-GCCAGTAACGCAAGGCAACA-3′ and 5′-CGTAGCAGGGAAACGATAAG-3′ for St8sia4 and 5′-TCTGGCTCAGCTCAGTCTCC-3′ and 5′-GCTTTTCCTGCCCGAGA-3′ for St8sia2. Quantification was performed with a standard curve method, using input DNA for normalization.
Table 1.
Antibodies used in Chromatin immunoprecipitation experiments.
| Target | Company | Reference |
|---|---|---|
| Histone H3 | Millipore | 17-10046 |
| Acetyl-Histone H3 (Lys9) | Millipore | 17-658 |
| Acetyl-Histone H3 (Lys14) | Millipore | 17-10051 |
| Acetyl-Histone H3 (Lys36) | Millipore | 07-540 |
| Trimethyl-Histone H3 (Lys27) | Millipore | 07-449 |
| Histone H4 | Millipore | 17-10047 |
| Monomethyl-Histone H4 (Lys20) | Millipore | 17-651 |
| Acetyl-Histone H4 (Lys16) | Millipore | 17-10101 |
| CBP | Santa Cruz | sc-369 X |
| GCN5 | Santa Cruz | sc-20698 X |
| MOF | Bethyl | A300-992A |
| TIP60 | Santa Cruz | sc-5725 X |
| RNA polymerase II CTD-phospho Ser5 (Pol2S5P) | Abcam | ab5131 |
2.6. Total RNA extraction, RT and qPCR analysis
Quantitative PCR analysis was performed using inventoried TaqMan Gene Expression Assays and the TaqMan FAST Universal PCR Master Mix (Applied Biosystems). TaqMan assay IDs for the mRNA analyzed were as follow: Mm00458911_m1 for Mof, Mm01292231_m1 for St8sia4, Mm00456815_m1 for Ncam1, Mm00456289_m1 for St8sia2, Mm00450174_m1 for Gne, Mm02342429_g1 for Ppia. For each reaction, 1 μl of a 1:10 dilution of the RT product was used in a final volume of 10 μl and each sample was assayed in triplicate. PCR amplification was run on a StepOne Plus RealTime PCR System (Applied Biosystems) and the StepOne Gene Expression Software was used for gene expression analysis. Quantification was done using the comparative Ct method and Ppia was used for normalization.
2.7. PSA ELISA assay
Brains were promptly removed and the mediobasal hypothalamic regions dissected. Tissue lysis was performed in RIPA lysis buffer using the TissueLyser system (Qiagen) and 5 mm stainless steel beads (Qiagen). Homogenates were then centrifuged 5 min at 5000 g and supernatants were collected for PSA assay using an enzyme-linked immunosorbent assay kit (PSA NCAM ELISA kit; Eurobio). Total protein concentration was determined using the Protein Assay Kit ll (Bio-Rad).
2.8. Statistical analysis
All data are displayed as mean ± SEM. Equality of variances and normality of distribution were checked prior to analysis. Data sets with two independent groups with similar variances were analyzed for statistical significance using an unpaired two-tailed Student's t test. Otherwise, a Mann Whitney test was applied. Data sets with more than two groups were analyzed using one-way analysis of variance (ANOVA) followed by a Dunnett's post-hoc test. For statistical analyses in Figure 6C and E, a two-way ANOVA was performed. All p values below 0.05 were considered significant. Significant differences were indicated as follow: *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 6.
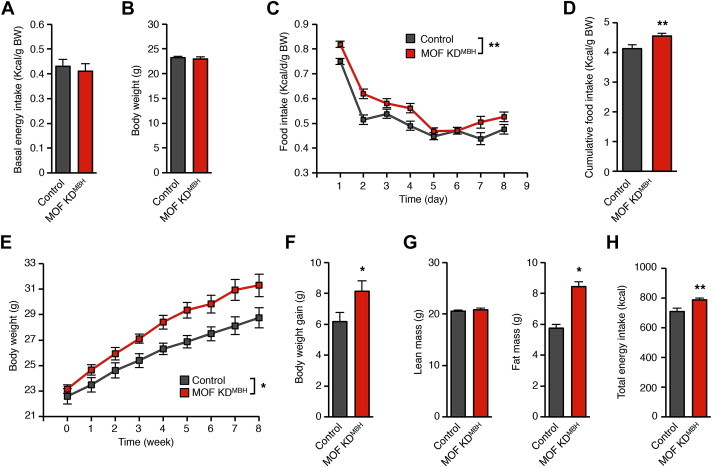
Lentiviral-mediated Mof knock-down in the hypothalamus accelerates the onset of DIO. (A) Energy intake of control and MOF KDMBH mice fed a standard diet, 3 weeks after the injection of the shRNA-containing lentiviral particles. Control: n = 12; MOF KDMBH: n = 14. (B) Body weight of control and MOF KDMBH mice fed a standard diet, 3 weeks after the injection of the shRNA-containing lentiviral particles. Control: n = 12; MOF KDMBH: n = 14. (C)–(H) Effect of Mof knock-down on food intake and body weight during short-term and long-term HFD. Control and MOF KDMBH mice were fed a HFD for 8 weeks. (C) Analysis of the effect of Mof knock-down on the homeostatic feeding response to HFD. Daily energy intake of control and MOF KDMBH mice was recorded for 8 days. (D) Cumulative energy intake over 8 days of HFD. (E) Body weight of control and MOF KDMBH mice fed a HFD for 8 weeks. (F) Body weight gain of control and MOF KDMBH mice after 8 weeks of HFD feeding. (G) Lean mass and fat mass of control and MOF KDMBH mice after 8 weeks of HFD. (H) Total energy intake of control and MOF over 8 weeks of HFD feeding. Control: n = 7; MOF KDMBH: n = 8. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
3. Results
3.1. PSA removal in the hypothalamus accelerates the onset of DIO
To assess the role of hypothalamic polysialylation in the long-term maintenance of body weight of mice on HFD, we generated PSA-depleted mice. We used endoneuraminidase N (endoN), a bacterial enzyme that specifically removes PSA residues from cell adhesion molecules [29]. EndoN remains active at least eight days after a single injection in the brain parenchyma [8]. We chronically depleted PSA in this area by bilateral intraparenchymal injections of endoN once a week during one month in mice fed with HFD (Figure 1A). The effect of the drug was checked by post-mortem analysis. We observed that the repeated injections of endoN reduced by 80–90% the PSA level in the MBH (Figure 1B). The repeated injections of endoN did not modify body weight in mice fed a standard diet for one month (Figure 1C–E, and Supplementary Figure 2). However, the persistent loss of hypothalamic PSA caused by endoN dramatically increased the weight gain of mice on HFD, in comparison to vehicle-treated mice (Figure 1C). As a result, body weight gain and fat mass measured after one month in mice on HFD were higher in endoN-treated mice relative to vehicle-treated mice (Figure 1D and E). Thus, PSA removal in the hypothalamus accelerated the onset of DIO in mice.
Figure 1.
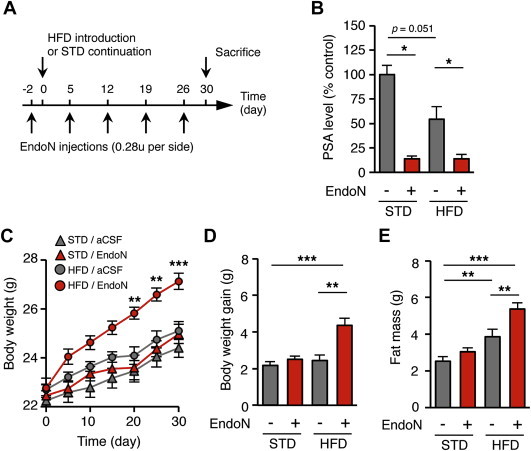
PSA removal in the hypothalamus accelerates the onset of DIO. (A) Picture showing the injection protocol of endoN in the mediobasal hypothalamus of mice fed either standard or a high-fat diet for one month. Five weekly bilateral injections of endoN (0.28 units/side) were performed. Control mice received artificial cerebrospinal fluid. (B) Effect of endoN injections on PSA level in the mediobasal hypothalamus, at the end of the treatment (n = 4 STD/aCSF; n = 5 STD/endoN; n = 5 HFD/aCSF; n = 6 HFD/endoN; *: p < 0.05; Mann Whitney test). (C) Body weight of endoN-treated and control mice fed a standard diet or a HFD for one month (n = 10 STD/aCSF; n = 9 STD/endoN; n = 13 HFD/aCSF; n = 14 HFD/endoN; **: p < 0.01. ***: p < 0.001; two-way ANOVA and Bonferroni post-hoc test). (D) Body weight gain of endoN-treated and control mice fed a standard diet or a HFD for one month (**: p < 0.01. ***: p < 0.001; Mann Whitney test). (E) Fat mass of endoN-treated and control mice fed a standard diet or a HFD for one month (**: p < 0.01. ***: p < 0.001; Mann Whitney test). All results are mean ± SEM.
3.2. Reduction of polysialylation in the hypothalamus increases the body weight gain on HFD
PST-1 is the major polysialyltransferase in adult hypothalamus [30,31]. To test whether hypothalamic PST-1 is involved in the long-term maintenance of body weight, we examined the metabolic consequences of St8sia4 knock-down in the hypothalamus of mice fed a HFD. We used lentiviral vectors-mediated RNA interference for specifically knocking down St8sia4 expression in the hypothalamus of adult mice. Control mice received lentiviral vectors carrying a shRNA sequence directed against a non-mammalian target. Three weeks after surgery, St8sia4 mRNA level was reduced by 21% in St8sia4 knock-down (KD) animals (PST-1 KDMBH) mice compared to control mice (Figure 2A). The expression of St8sia2, Ncam, and Gne was not altered (Figure 2A), indicating that the RNAi-mediated knockdown of St8sia4 was specific and did not modify expression of other genes involved in the signaling pathway of PSA. Body weight gain and energy intake of control and PST-1 KDMBH mice fed a standard diet for 2 months were similar (Supplementary Figure 3). To investigate whether a reduction of polysialylation in the hypothalamus increased the vulnerability to DIO, PST-1 KDMBH mice were fed a HFD for two months. In these mice, knock-down efficiency was determined by post-mortem analysis of PSA level in MBH biopsies. Only mice with more than 5% reduction of hypothalamic PSA were further analyzed, considering otherwise that we failed to target the MBH. After screening, we detected a significant reduction of the PSA level by 25.7% in the MBH of PST-1 KDMBH mice, in comparison to control mice (Figure 2B). A retrospective analysis showed that PST-1 KDMBH mice gained progressively more weight during the 2-month HFD than control mice (Figure 2C). Body weight gain and adiposity of PST-1 KDMBH mice on HFD were therefore significantly increased (Figure 2D and E). Thus, a limited polysialylation in the hypothalamus accelerated the onset of DIO.
Figure 2.
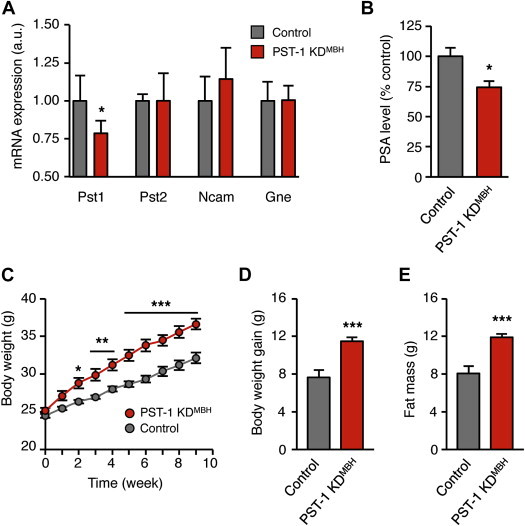
Reduction of polysialylation in the hypothalamus increases the body weight gain on HFD. St8sia4 knockdown mice, designated as PST-1 KDMBH mice, were stereotactically injected in the mediobasal hypothalamus with ∼7.5 × 105 shRNA-expressing lentiviral vectors against a Pst1 sequence. Control mice received lentiviral vectors targeting a non-mammalian sequence. (A) Effect of the lentiviral vectors-mediated RNA interference on relative mRNA expression of genes involved in the PSA signaling, assessed by RT-qPCR on mediobasal hypothalamus extracts from standard diet-fed mice (n = 4 control; n = 5 PST-1 KDMBH; *: p < 0.05; unpaired t test. (B) PSA level in the mediobasal hypothalamus of PST-1 KDMBH mice fed a HFD for 2 months, compared to control mice (n = 9 control; n = 9 PST-1 KDMBH; *: p < 0.05; unpaired t test). (C) Kinetic of the body weight of PST-1 KDMBH and control mice during 2-month HFD (*: p < 0.05, **: p < 0.01. ***: p < 0.001; two-way ANOVA and Bonferroni post-hoc test). (D) Body weight gain of PST-1 KDMBH and control mice after 2-month HFD (***: p < 0.001; unpaired t test). (E) Fat mass of PST-1 KDMBH and control mice after 2 month-HFD (***: p < 0.001; unpaired t test). All results are mean ± SEM.
3.3. Short-term HFD activates the transcription of the St8sia4 gene in the MBH
We next investigated the regulation of St8sia4 expression in the hypothalamus of HFD-fed mice. We determined if HFD exposure triggered chromatin remodeling and transcriptional activation of the St8sia4 gene in the MBH. Chromatin modifications were analyzed on the promoter of St8sia4 by ChIP with chromatin extracts obtained from the MBH of mice fed a HFD for 24 h–72 h (Figure 3 and Supplementary Figure 4). After 24 h on HFD, an increase in H3 and H4 acetylation and a decrease in H4 methylation were observed (Figure 3A; 2.25, 1.86 and 1.83 fold for H3K14Ac, H4K16Ac and H4K20Me respectively). The levels of H4K16Ac, H4K20Me and H3K14Ac returned to basal values after 72 h on HFD, indicating that these post-translational modifications were transient (Supplementary Figure 4B). HFD feeding for 48 h triggered a dissociation of histones H3 and H4 from the promoter of this gene (Figure 3B; 1.66 fold and 1.74 fold decrease respectively). The decrease in histone interaction with St8sia4 promoter preceded an increase in Pol2-S5P level -the activated form of RNA polymerase II- indicating an activation of St8sia4 transcription (Figure 3B). St8sia4 mRNA levels were also significantly higher after 3 days on HFD (Figure 3C). Additional analysis revealed no post-translational histone modifications, no histone dissociation and no change in RNA polymerase II phosphorylation on the promoter of the St8sia2 gene, encoding the PST-2 polysialyltransferase (Supplementary Figure 4C and 4D). The chromatin remodeling events detected in the MBH after HFD introduction were thus specific to the St8sia4 polysialyltransferase gene.
Figure 3.
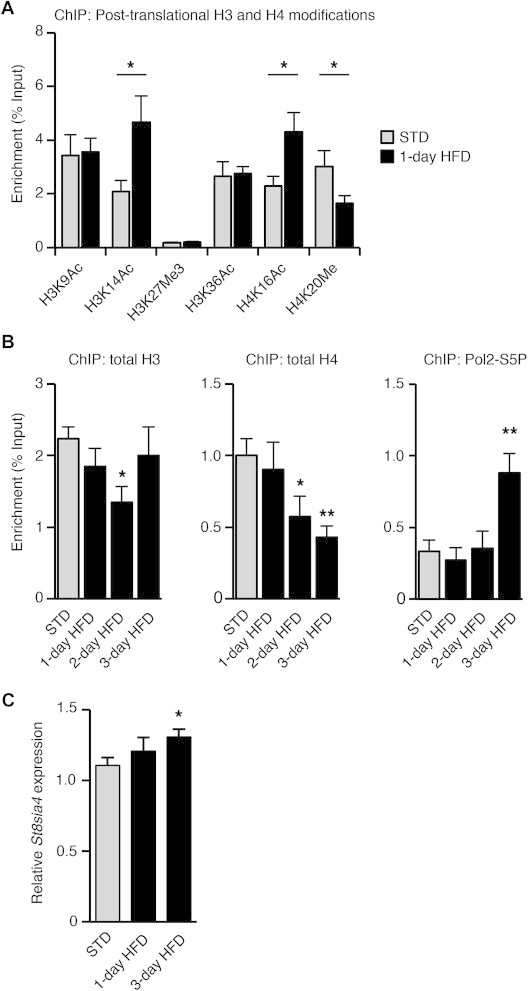
HFD exposure activates St8sia4 gene transcription. (A) Histones H3 and H4 post-translational modifications on the promoter of St8sia4 were analyzed by ChIP. Chromatin extracts were obtained from MBH of mice fed either a standard diet (STD) or a HFD for 24 h (1-day HFD). (B) Association of total histones H3 and H4 and of Pol2-S5P with the promoter of St8sia4 was analyzed by ChIP on MBH extracts obtained after 1, 2 or 3 days of HFD exposure and compared to levels in MBH of mice fed a standard diet (STD). (C) St8sia4 mRNA expression analyzed by RT-qPCR on MBH of mice fed a either a standard diet (STD) or a HFD for 1 or 3 day(s). STD: n = 9–12; HFD: n = 5–10. Data are expressed as mean ± SEM. *p < 0.05.
3.4. Short-term HFD elicits the recruitment of the histone acetyltransferase MOF on the St8sia4 promoter in the MBH
In order to identify the histone acetyltransferases (HAT) that trigger the increase in histone acetylation and the subsequent activation of St8sia4 transcription in the MBH of HFD-fed mice, the recruitment of CBP, GCN5, MOF and TIP60 was analyzed by ChIP. These HATs are representative members of the 3 different families of histone lysine acetyltransferases and are known to acetylate either H3K14 or H4K16 or both [32–34]. Their interaction with St8sia4 was analyzed in MBH chromatin extracts from mice fed a HFD for 6 h or 10 h (Figure 4). After 6 h on HFD, a 2.20 fold increase in MOF was detected on St8sia4 promoter. This increase was persistent after 10 h of HFD exposure. At this time point, GCN5 was also recruited. The early recruitment of MOF suggested that this HAT plays a critical role in mediating the effects of HFD on the activation of St8sia4 transcription in the MBH.
Figure 4.
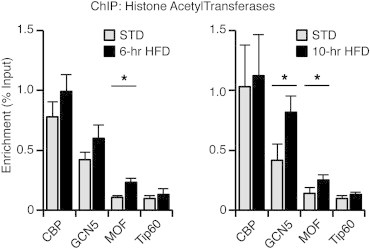
Histone acetyltransferases are recruited at the promoter of St8sia4 upon HFD exposure. The recruitment of the histone acetyltransferases CBP, GCN5, MOF and TIP60 on the promoter of St8sia4 was analyzed by ChIP. MBH chromatin extracts were prepared from mice either fed a standard diet (STD) or exposed to a HFD for 6 h (6-hr HFD) and 10 h (10-hr HFD). STD: n = 9–10; HFD 6 h: n = 9–10; HFD 10 h: n = 8. Data are expressed as mean ± SEM. *p < 0.05.
3.5. MOF is required for HFD-induced transcription of the St8sia4 gene and the associated PSA synthesis in the MBH
To determine the role of MOF in HFD-induced hypothalamic polysialylation, we knocked down MOF in the hypothalamus and tested whether St8sia4 transcription and PSA levels could still be increased upon HFD exposure. Lentiviral particles containing a MOF-targeting shRNA sequence were stereotactically injected in the MBH of adult mice. Control mice were injected with lentiviral particles containing a non-mammalian target shRNA. After 3 weeks of recovery, a 26.73% decrease in Mof mRNA level was detected in the hypothalamus of silenced mice (MOF KDMBH mice) (Figure 5A). Since H4K16Ac has been identified as the main substrate of MOF in many cell types and organisms [32,35–37], we tested whether Mof knock-down altered the HFD-induced increase in H4K16Ac level on St8sia4 in the MBH (Figure 5B). After 24 h of HFD feeding, the level of H4K16Ac on St8sia4 was reduced by 25.39% in MOF KDMBH mice in comparison to control mice. The level of H4K16Ac in HFD-fed MOF-KDMBH mice was similar to the level in standard-diet fed mice (Figure 3A), indicating that Mof knock-down abolished the HFD-induced acetylation of H4K16. After 3 days on HFD, the level of Pol2-S5P detected on St8sia4 was strongly reduced in MOF-KDMBH mice in comparison to the control group (38.33% reduction, Figure 5C). Again, Pol2-S5P level in MOF-KDMBH mice was similar to the level in standard-diet fed mice (Figure 3B). We finally assessed polysialylation capacities in MOF-KDMBH mice by measuring both St8sia4 mRNA level and PSA level in the MBH after exposure to HFD for 3 days (Figure 5C and D). St8sia4 mRNA level was reduced by 38.73% and PSA level was reduced by 14.10% in MOF-KDMBH mice. Altogether, these results indicated that MOF is crucial for activating the polysialylation pathway in the MBH in response to HFD.
Figure 5.
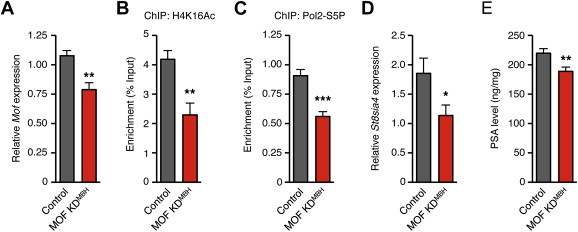
Mof knock-down impairs St8sia4-dependent polysialylation in the mediobasal hypothalamus. Mof knock-down was obtained by stereotaxic injection of lentiviral particles (∼1.0 × 106 particles) containing Mof targeting shRNA (MOF KDMBH) or a shRNA targeting a non-mammalian RNA sequence (control) in the MBH. All analyses were performed 3 weeks after the delivery of lentiviral particles to the MBH. (A) RT-qPCR analysis of hypothalamic Mof mRNA levels in MOF KDMBH and control mice. Control: n = 6; MOF KDMBH: n = 7. (B) ChIP analysis of H4K16Ac interaction with the St8sia4 promoter in the MBH of control and MOF KDMBH mice after 24 h of HFD. Control: n = 7; MOF KDMBH: n = 7. (C) ChIP analysis of Pol2-S5P interaction with St8sia4 promoter in control and MOF KDMBH mice fed a HFD for 3 days. Control: n = 7; MOF KDMBH: n = 7. (D) RT-qPCR analysis of hypothalamic St8sia4 mRNA levels in MOF KDMBH and control mice. Control: n = 6; MOF KDMBH: n = 7. (E) PSA levels in control and MOF KDMBH mice fed a HFD for 8 days. Control: n = 9; MOF KDMBH: n = 9. Data are expressed as mean ± SEM. *p < 0.05.
3.6. MOF-depleted mice are prone to obesity
We finally examined the consequences of MOF knock-down on the PSA-dependent short-term feeding response to HFD and the long-term regulation of body weight on HFD. The short-term feeding response to HFD was assessed over 8 days of HFD exposure. When fed a standard diet, control and MOF KDMBH mice displayed similar body weight and daily energy intake (Figure 6A and B). In both control and MOF KDMBH mice, HFD caused a transient increase in energy intake (Figure 6C). However the normalization of energy intake was slightly but significantly altered in MOF KDMBH mice. Consequently, the cumulative energy intake of MOF KDMBH mice during the first week of HFD was higher in this group (Figure 6D). HFD feeding revealed an alteration of the homeostatic control of food intake in MOF KDMBH mice. To assess the role of MOF in the long-term regulation of energy homeostasis, control and MOF KDMBH mice were fed a HFD for 8 weeks. Food intake and body weight were recorded daily during this period. Only mice with more than 5% reduction of hypothalamic Mof mRNA level were further analyzed, considering otherwise that we failed to target the MBH. As a reference, body weight of control and MOF KDMBH mice fed a standard diet was measured over 6 weeks after surgery. No significant differences were observed in body weight gain between control mice and MOF KDMBH mice (+2.49 ± 0.28 g vs 2.96 ± 0.52 g). However, upon HFD, a dramatic increase in body weight was observed in MOF KDMBH mice (Figure 6E). The body weight gain of MOF KDMBH mice over the 8 weeks of HFD was 31.85% higher than the one of the control group (Figure 6F). This increase in body weight gain was due to an increase in fat mass in MOF KDMBH mice (Figure 6G). The higher cumulative energy intake of MOF KDMBH mice in comparison to control mice over the 8-week period likely explained this phenotype (Figure 6H, MOF KDMBH mice: 786.83 ± 14.06 kcal and control mice: 708.41 ± 21.79 kcal).
4. Discussion
Obesity is on the rise in both developed and developing countries, and it is now considered as a worldwide epidemic [38]. It has been proposed that obesity could be a brain disease [39], and recently, several molecular brain targets of therapeutic interest have been identified [40]. Investigation of central regulatory pathways involved in the control of appetite and body weight could probably help with the fight against obesity. We previously identified polysialylation as an essential process in the nutritionally regulated synaptic plasticity of the melanocortin system [8]. We now show that polysialylation is essential to the long-term regulation of body weight. We found that a defect in polysialylation may be a primary cause of obesity. Furthermore, the experimental data presented here unveil an unexpected role for the histone acetyltransferase MOF in the brain control of energy balance.
Developmental malformation of the melanocortin system, i.e. disruption of neural projection pathways within the hypothalamus, could contribute to the obese phenotype and related metabolic disorders [41]. Moreover, the wiring of arcuate POMC neurons of rats kept on standard diet predicts vulnerability to weight gain on HFD, suggesting that accurate connectivity in this neuronal system is crucial to maintain energy homeostasis [10]. Besides, an abnormal synaptic organization of feeding circuits that can be acutely reversed by hormonal or pharmacological treatments has been found in obese mice and rats [42,43]. Thus, it has been suggested that the altered synaptic plasticity in feeding circuits, in addition to steady neuroanatomical defects, might promote DIO [44]. Nevertheless, whether synaptic inadequacy is cause or consequence of the metabolic imbalance is still not clear [9]. Here, we report that the lack of hypothalamic PSA strongly accelerated the onset of DIO in mice. Given the role of PSA in synaptic plasticity, our results suggest that the inflexibility of feeding circuits is a risk factor for obesity. This is in line with the well-documented adaptive function of the plasticity in feeding circuits in response to metabolic fluctuations [4,6].
The removal of hypothalamic PSA was sufficient to cause severe overweight in mice fed a HFD for 1–2 months, suggesting that the inability to properly rewire feeding circuits depending on the nutritional conditions, makes mice prone to obesity. Since the human hypothalamus is able to undergo hormone-dependent structural plasticity [45], our findings could be relevant to the etiology of obesity in humans as well. Indeed, large-scale studies on the genetic determinants of body mass index have revealed the strong influence of some genes involved in neuronal plasticity [46,47]. Interestingly, the polymorphism in Negr1, the gene coding the neuronal growth regulator 1, a cell adhesion molecule involved in the brain remodeling, seems to be causal in high body weight [47]. Moreover, genetic defects reducing the levels of plasticity-related molecules such as the brain-derived neurotrophic factor (BDNF) or its receptor TrkB cause severe early-onset obesity [48,49]. Therefore, it is conceivable that an altered synaptic plasticity could predispose humans to obesity.
To gain insight into the molecular sequence underlying the synaptic plasticity-dependent control of energy balance, we explored the chromatin remodeling events underlying the activation of the St8sia4 gene expression upon high fat diet (HFD). We found that the HFD induced a decrease in the level of monomethylated H4K20 and an increase in acetylation of histones H3K14 and H4K16 on the St8sia4 gene. In mammals, histones localized in silent heterochromatic regions are usually characterized by high levels of methylation at certain specific sites and low levels of acetylation [50]. H4K20Me1 marks are mostly found in regions of facultative heterochromatin and H4K20 demethylation has been associated with transcription activation in different models [51–53]. Both H3K14 and H4K16 acetylation impact chromatin structure and allow switching from a repressive state to a transcriptionally active state [54–57]. In our study, the decrease in H4K20Me1 and the increase in H3 and H4 acetylation upon HFD were followed by their dissociation. The level of RNA polymerase II phosphorylated on the serine 5 of the carboxy-terminal domain increased subsequently. Moreover, the phosphorylation of the serine 5 of the RNA polymerase II is acquired during the transcription initiation phase [58]. Altogether, our results clearly demonstrate that HFD triggers an activation of St8sia4 transcription.
Tracing back the signaling cascade leading to the activation of St8sia4 transcription, we identified two histone acetyltransferases, GCN5 and MOF, recruited on St8sia4 within the few hours following the introduction of HFD. Transcription activation relies on cross-regulation of histone post-translational modifications through the stepwise or concomitant recruitment of histone modifying enzymes [59]. Of note, H3K14 and H4K16 are major acetylation targets for GCN5 and MOF, respectively [32,35,37,55,60]. Additionally, MOF depletion in drosophila or human cells resulted in a genome-wide loss of H4K16Ac [32,37,61,62]. In our study, we did not investigate interactions of histone demethylases with St8sia4 promoter. However, PHF8 is the only histone H4K20Me1 demethylase identified up to now and its activity is associated with a positive regulation of gene expression [52]. PHF8 could thus contribute, in cooperation with GCN5 and MOF to the activation of St8sia4 transcription in the hypothalamus upon high fat feeding.
In our study, since MOF recruitment was already detected after 6 h of HFD feeding, we focused on the function of this HAT in the activation of St8sia4 expression upon HFD exposure. The depletion of MOF in the MBH completely abolished the HFD-induced increase in H4K16 acetylation on St8sia4 and the subsequent recruitment of the RNA polymerase II on this gene. The early recruitment of MOF on St8sia4 upon HFD exposure, the absence of activation of St8sia4 transcription and the decrease in PSA level after MOF knock-down strongly suggested a crucial role for MOF in mediating the effect of HFD exposure on the establishment of PSA-dependent synaptic plasticity in the hypothalamus. MOF KDMBH mice displayed an impairment of the short-term feeding response to HFD, which is actually a PSA-dependent process [8]. In addition, we show that MOF KDMBH mice displayed a higher propensity to develop DIO. Reducing polysialylation by St8sia4 silencing or PSA removal from the hypothalamus accelerated the onset of a DIO phenotype as well. MOF was originally identified as required for dosage compensation in Drosophila [63,64]. Afterwards, studies performed on mammalian cells and tumor biopsies indicated that MOF is crucial to several cell functions such as cell proliferation and differentiation, DNA damage repair, autophagy and tumorigenesis [65–67]. In vivo, a Purkinje cells-specific deletion of MOF in the cerebellum produced neurological abnormalities including impaired motor coordination, ataxia and a backward-walking phenotype [68]. The present work demonstrates that mediobasal hypothalamic MOF is involved in the regulation of energy homeostasis through an activation of polysialylation. Whereas some studies suggest that MOF is globally involved in the activation of transcription, others indicate that this HAT rather activates defined subset of genes and has a function in transcription regulation in specific cell types only [32,35,37,69,70]. Though we cannot rule out that a larger MOF-dependent transcriptomic program contributes to the accelerated onset of diet-induced obesity in MOF-depleted mice, the subsequent deficit in hypothalamus polysialylation capacities of these mice might largely contribute to their increased vulnerability to DIO.
In few recent studies, MOF has been detected in enhancer regions on mammalian genes [71,72]. In addition, interactions of MOF with transcription factors have been identified. For instance, MOF is recruited to FOXP3-target genes by directly interacting with FOXP3 and the transcription factor p53 is an acetylation target of MOF [73,74]. This suggests that MOF could be recruited to the St8sia4 gene by interacting directly with a transcription factor activating St8sia4 transcription upon high fat feeding. Otherwise, its recruitment could involve Mediator complex which is known to convey regulatory information to the basal RNA polymerase II transcription machinery by interacting both with gene-specific transcription factors and with histone modifying enzymes [75,76]. The mechanism by which MOF is targeted to St8sia4 upon HFD feeding remains to be determined.
In conclusion, the present work clearly indicated the causal role of altered PSA signaling in accelerating the development of obesity. Given the role of PSA in the modulation of cell-to-cell interactions these findings strengthen the concept that individual capacity to synaptic plasticity is a key factor in the development of overweight with obesogenic foods [10,44].
Author contributions
A.B., X.B., and C.R. designed experiments; C.R., X.B., E.N., A.La., A.G., S.C., A.Le., F.D., J.G. and A.B. performed experiments; C.R., X.B., T.K., L.P. and A.B. performed data analysis; C.R., X.B., and A.B. wrote the manuscript.
Acknowledgments
We thank the animal facility of the CSGA, Rita Gerardy-Schahn for fruitful discussion, Christelle Boileau for the editing of the manuscript, Anne Lefranc and Michel Tavan for their technical assistance. This work was funded by CNRS, INRA, the Région Bourgogne (FEDER 2010 O0431SGO003S03440), and Agence Nationale de la Recherche (ANR-13-JSV1-0003-01).
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data related to this article:
Supplementary Figure 1.
Traces of injectors in stained brain sections. A. Traces of the cannula guide (CG) and the injectors (I) used for repeated injections of EndoN in the MBH. Injectors were inserted at −5.6 mm below the skull surface. Note that the permanent cannula guide was shorter than the injectors to minimize injury. The injectors were labeled with DiI (in yellow) to visualize their traces. Slides were stained with DAPI (in blue) to reveal nuclei. B. Traces of the nanofil (NF) used for single injection of lentiviral particles in the MBH. Nanofil was inserted at −5.6 mm below the skull surface. Slides were stained with DAPI (in blue) to reveal nuclei.
Supplementary Figure 2.
Total energy intake and body weight gain of endoN-treated and control mice during 1 month. Mice received 5 weekly bilateral stereotactic injections of endoN or vehicle (artificial cerebrospinal fluid) in the MBH under gaseous anesthesia. Animals were kept on standard diet. Food intake and body weight were monitored daily during one month after the first injection.
Supplementary Figure 3.

Total energy intake and body weight gain of PST-1 KDMBH and control mice during 2 months. Mice received shRNA-expressing lentiviral vectors against Pst-1 or control vectors, by bilateral stereotactic injections in the MBH. After a 3-week recovery period, mice were further maintained on standard diet for an additional 2 month-period. Food intake and body weight were monitored daily during this period.
Supplementary Figure 4.
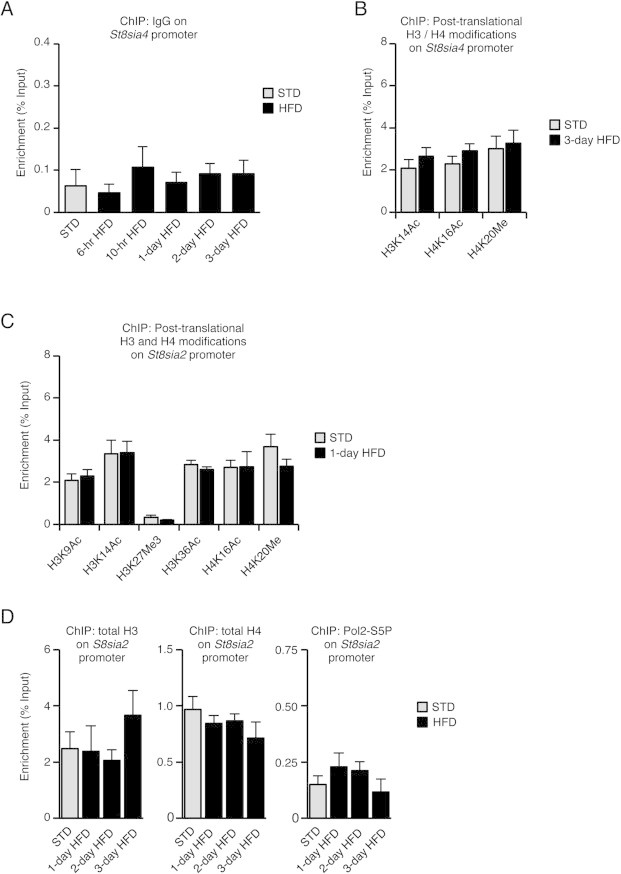
Specificity and reversibility of St8sia4 chromatin remodeling upon HFD exposure in MBH. A. Control ChIP with a non-specific isotype IgG antibody. B. Histones H3 and H4 post-translational modifications on the promoter of St8sia4 were analyzed by ChIP. Chromatin extracts were obtained from MBH of mice fed either a standard diet (STD) or a HFD for 3 days (3-day HFD). C. Histones H3 and H4 post-translational modifications on the promoter of St8sia2 were analyzed by ChIP. Chromatin extracts were obtained from MBH of mice fed either a standard diet (STD) or a HFD for 24 h (1-day HFD). D. Association of total histones H3 and H4 and of Pol2-S5P with the promoter of St8sia2 was analyzed by ChIP on MBH extracts obtained after 1, 2 or 3 days of HFD exposure and compared to levels in MBH from mice fed a standard diet (STD). n = 6–8. Data are expressed as mean ± SEM. *p < 0.05.
References
- 1.Morton G.J., Cummings D.E., Baskin D.G., Barsh G.S., Schwartz M.W. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 2.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 3.Warne J.P., Xu A.W. Metabolic transceivers: in tune with the central melanocortin system. Trends in Endocrinology and Metabolism: TEM. 2013;24(2):68–75. doi: 10.1016/j.tem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu T., Kong D., Shah B.P., Ye C., Koda S., Saunders A. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron. 2012;73(3):511–522. doi: 10.1016/j.neuron.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vong L., Ye C., Yang Z., Choi B., Chua S., Jr., Lowell B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71(1):142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y., Atasoy D., Su H.H., Sternson S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146(6):992–1003. doi: 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath T.L. The hardship of obesity: a soft-wired hypothalamus. Nature Neuroscience. 2005;8(5):561–565. doi: 10.1038/nn1453. [DOI] [PubMed] [Google Scholar]
- 8.Benani A., Hryhorczuk C., Gouaze A., Fioramonti X., Brenachot X., Guissard C. Food intake adaptation to dietary fat involves PSA-dependent rewiring of the arcuate melanocortin system in mice. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32(35):11970–11979. doi: 10.1523/JNEUROSCI.0624-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeltser L.M., Seeley R.J., Tschop M.H. Synaptic plasticity in neuronal circuits regulating energy balance. Nature Neuroscience. 2012;15(10):1336–1342. doi: 10.1038/nn.3219. [DOI] [PubMed] [Google Scholar]
- 10.Horvath T.L., Sarman B., Garcia-Caceres C., Enriori P.J., Sotonyi P., Shanabrough M. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutishauser U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nature Reviews. Neuroscience. 2008;9(1):26–35. doi: 10.1038/nrn2285. [DOI] [PubMed] [Google Scholar]
- 12.Becker C.G., Artola A., Gerardy-Schahn R., Becker T., Welzl H., Schachner M. The polysialic acid modification of the neural cell adhesion molecule is involved in spatial learning and hippocampal long-term potentiation. Journal of Neuroscience Research. 1996;45(2):143–152. doi: 10.1002/(SICI)1097-4547(19960715)45:2<143::AID-JNR6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 13.Venero C., Herrero A.I., Touyarot K., Cambon K., Lopez-Fernandez M.A., Berezin V. Hippocampal up-regulation of NCAM expression and polysialylation plays a key role on spatial memory. The European Journal of Neuroscience. 2006;23(6):1585–1595. doi: 10.1111/j.1460-9568.2006.04663.x. [DOI] [PubMed] [Google Scholar]
- 14.El Maarouf A., Kolesnikov Y., Pasternak G., Rutishauser U. Neural cell adhesion molecule and its polysialic acid moiety exhibit opposing and linked effects on neuropathic hyperalgesia. Experimental Neurology. 2012;233(2):866–870. doi: 10.1016/j.expneurol.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Maarouf A., Kolesnikov Y., Pasternak G., Rutishauser U. Polysialic acid-induced plasticity reduces neuropathic insult to the central nervous system. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(32):11516–11520. doi: 10.1073/pnas.0504718102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theodosis D.T., Bonhomme R., Vitiello S., Rougon G., Poulain D.A. Cell surface expression of polysialic acid on NCAM is a prerequisite for activity-dependent morphological neuronal and glial plasticity. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1999;19(23):10228–10236. doi: 10.1523/JNEUROSCI.19-23-10228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoyk Z., Parducz A., Theodosis D.T. The highly sialylated isoform of the neural cell adhesion molecule is required for estradiol-induced morphological synaptic plasticity in the adult arcuate nucleus. The European Journal of Neuroscience. 2001;13(4):649–656. doi: 10.1046/j.1460-9568.2001.01427.x. [DOI] [PubMed] [Google Scholar]
- 18.Guan Z., Giustetto M., Lomvardas S., Kim J.H., Miniaci M.C., Schwartz J.H. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111(4):483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- 19.Leslie J.H., Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Progress in Neurobiology. 2011;94(3):223–237. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.West A.E., Greenberg M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harbor Perspectives in Biology. 2011;3(6) doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson E.D., Monteggia L.M. Epigenetics in the mature mammalian brain: effects on behavior and synaptic transmission. Neurobiology of Learning and Memory. 2011;96(1):53–60. doi: 10.1016/j.nlm.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zocchi L., Sassone-Corsi P. Joining the dots: from chromatin remodeling to neuronal plasticity. Current Opinion in Neurobiology. 2010;20(4):432–440. doi: 10.1016/j.conb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogel-Ciernia A., Matheos D.P., Barrett R.M., Kramar E.A., Azzawi S., Chen Y. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nature Neuroscience. 2013;16(5):552–561. doi: 10.1038/nn.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Golden S.A., Christoffel D.J., Heshmati M., Hodes G.E., Magida J., Davis K. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nature Medicine. 2013;19(3):337–344. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bousiges O., Neidl R., Majchrzak M., Muller M.A., Barbelivien A., Pereira de Vasconcelos A. Detection of histone acetylation levels in the dorsal hippocampus reveals early tagging on specific residues of H2B and H4 histones in response to learning. PLoS ONE. 2013;8(3):e57816. doi: 10.1371/journal.pone.0057816. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Graff J., Rei D., Guan J.S., Wang W.Y., Seo J., Hennig K.M. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer A., Sananbenesi F., Wang X., Dobbin M., Tsai L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447(7141):178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 28.LaPlant Q., Nestler E.J. CRACKing the histone code: cocaine's effects on chromatin structure and function. Hormones and Behavior. 2011;59(3):321–330. doi: 10.1016/j.yhbeh.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vimr E.R., McCoy R.D., Vollger H.F., Wilkison N.C., Troy F.A. Use of prokaryotic-derived probes to identify poly(sialic acid) in neonatal neuronal membranes. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(7):1971–1975. doi: 10.1073/pnas.81.7.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ong E., Nakayama J., Angata K., Reyes L., Katsuyama T., Arai Y. Developmental regulation of polysialic acid synthesis in mouse directed by two polysialyltransferases, PST and STX. Glycobiology. 1998;8(4):415–424. doi: 10.1093/glycob/8.4.415. [DOI] [PubMed] [Google Scholar]
- 31.Hildebrandt H., Becker C., Murau M., Gerardy-Schahn R., Rahmann H. Heterogeneous expression of the polysialyltransferases ST8Sia II and ST8Sia IV during postnatal rat brain development. Journal of Neurochemistry. 1998;71(6):2339–2348. doi: 10.1046/j.1471-4159.1998.71062339.x. [DOI] [PubMed] [Google Scholar]
- 32.Smith E.R., Cayrou C., Huang R., Lane W.S., Cote J., Lucchesi J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Molecular and Cellular Biology. 2005;25(21):9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuo Y.M., Andrews A.J. Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PLoS One. 2013;8(2):e54896. doi: 10.1371/journal.pone.0054896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto N., Izumi H., Noguchi T., Nakajima Y., Ohmiya Y., Shiota M. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. The Journal of Biological Chemistry. 2008;283(26):18218–18226. doi: 10.1074/jbc.M802332200. [DOI] [PubMed] [Google Scholar]
- 35.Conrad T., Cavalli F.M., Holz H., Hallacli E., Kind J., Ilik I. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Developmental Cell. 2012;22(3):610–624. doi: 10.1016/j.devcel.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Gupta A., Hunt C.R., Pandita R.K., Pae J., Komal K., Singh M. T-cell-specific deletion of Mof blocks their differentiation and results in genomic instability in mice. Mutagenesis. 2013;28(3):263–270. doi: 10.1093/mutage/ges080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taipale M., Rea S., Richter K., Vilar A., Lichter P., Imhof A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Molecular and Cellular Biology. 2005;25(15):6798–6810. doi: 10.1128/MCB.25.15.6798-6810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman J.M. Obesity in the new millennium. Nature. 2000;404(6778):632–634. doi: 10.1038/35007504. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz M.W., Porte D., Jr. Diabetes, obesity, and the brain. Science. 2005;307(5708):375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 40.Yeo G.S., Heisler L.K. Unraveling the brain regulation of appetite: lessons from genetics. Nature Neuroscience. 2012;15(10):1343–1349. doi: 10.1038/nn.3211. [DOI] [PubMed] [Google Scholar]
- 41.Bouret S.G., Draper S.J., Simerly R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304(5667):108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- 42.Pinto S., Roseberry A.G., Liu H., Diano S., Shanabrough M., Cai X. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304(5667):110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 43.Cristino L., Busetto G., Imperatore R., Ferrandino I., Palomba L., Silvestri C. Obesity-driven synaptic remodeling affects endocannabinoid control of orexinergic neurons. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(24):E2229–E2238. doi: 10.1073/pnas.1219485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dietrich M.O., Horvath T.L. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends in Neurosciences. 2013;36(2):65–73. doi: 10.1016/j.tins.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Baroncini M., Jissendi P., Catteau-Jonard S., Dewailly D., Pruvo J.P., Francke J.P. Sex steroid hormones-related structural plasticity in the human hypothalamus. Neuroimage. 2010;50(2):428–433. doi: 10.1016/j.neuroimage.2009.11.074. [DOI] [PubMed] [Google Scholar]
- 46.Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nature Genetics. 2009;41(1):18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 47.Willer C.J., Speliotes E.K., Loos R.J., Li S., Lindgren C.M., Heid I.M. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics. 2009;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han J.C., Liu Q.R., Jones M., Levinn R.L., Menzie C.M., Jefferson-George K.S. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. The New England Journal of Medicine. 2008;359(9):918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeo G.S., Connie Hung C.C., Rochford J., Keogh J., Gray J., Sivaramakrishnan S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nature Neuroscience. 2004;7(11):1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 50.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Sims J.K., Houston S.I., Magazinnik T., Rice J.C. A trans-tail histone code defined by monomethylated H4 Lys-20 and H3 Lys-9 demarcates distinct regions of silent chromatin. The Journal of Biological Chemistry. 2006;281(18):12760–12766. doi: 10.1074/jbc.M513462200. [DOI] [PubMed] [Google Scholar]
- 52.Qi H.H., Sarkissian M., Hu G.Q., Wang Z., Bhattacharjee A., Gordon D.B. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466(7305):503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466(7305):508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karmodiya K., Krebs A.R., Oulad-Abdelghani M., Kimura H., Tora L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012;13:424. doi: 10.1186/1471-2164-13-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnsson A., Durand-Dubief M., Xue-Franzen Y., Ronnerblad M., Ekwall K., Wright A. HAT-HDAC interplay modulates global histone H3K14 acetylation in gene-coding regions during stress. EMBO Reports. 2009;10(9):1009–1014. doi: 10.1038/embor.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shogren-Knaak M., Ishii H., Sun J.M., Pazin M.J., Davie J.R., Peterson C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 57.Zippo A., Serafini R., Rocchigiani M., Pennacchini S., Krepelova A., Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138(6):1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 58.Hsin J.P., Manley J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes & Development. 2012;26(19):2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Latham J.A., Dent S.Y. Cross-regulation of histone modifications. Nature Structural & Molecular Biology. 2007;14(11):1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 60.Lafon A., Petty E., Pillus L. Functional antagonism between Sas3 and Gcn5 acetyltransferases and ISWI chromatin remodelers. PLoS Genetics. 2012;8(10):e1002994. doi: 10.1371/journal.pgen.1002994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta A., Sharma G.G., Young C.S., Agarwal M., Smith E.R., Paull T.T. Involvement of human MOF in ATM function. Molecular and Cellular Biology. 2005;25(12):5292–5305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dou Y., Milne T.A., Tackett A.J., Smith E.R., Fukuda A., Wysocka J. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121(6):873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 63.Hilfiker A., Hilfiker-Kleiner D., Pannuti A., Lucchesi J.C. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. The EMBO Journal. 1997;16(8):2054–2060. doi: 10.1093/emboj/16.8.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akhtar A., Becker P.B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Molecular Cell. 2000;5(2):367–375. doi: 10.1016/s1097-2765(00)80431-1. [DOI] [PubMed] [Google Scholar]
- 65.Fullgrabe J., Lynch-Day M.A., Heldring N., Li W., Struijk R.B., Ma Q. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500(7463):468–471. doi: 10.1038/nature12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rea S., Xouri G., Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007;26(37):5385–5394. doi: 10.1038/sj.onc.1210607. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y., Han X., Guan J., Li X. Regulation and function of histone acetyltransferase MOF. Frontiers of Medicine. 2014;8(1):79–83. doi: 10.1007/s11684-014-0314-6. [DOI] [PubMed] [Google Scholar]
- 68.Kumar R., Hunt C.R., Gupta A., Nannepaga S., Pandita R.K., Shay J.W. Purkinje cell-specific males absent on the first (mMof) gene deletion results in an ataxia-telangiectasia-like neurological phenotype and backward walking in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3636–3641. doi: 10.1073/pnas.1016524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feller C., Prestel M., Hartmann H., Straub T., Soding J., Becker P.B. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Research. 2012;40(4):1509–1522. doi: 10.1093/nar/gkr869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horikoshi N., Kumar P., Sharma G.G., Chen M., Hunt C.R., Westover K. Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integrity. 2013;4(1):3. doi: 10.1186/2041-9414-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taylor G.C., Eskeland R., Hekimoglu-Balkan B., Pradeepa M.M., Bickmore W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Research. 2013;23(12):2053–2065. doi: 10.1101/gr.155028.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chelmicki T., Dundar F., Turley M., Khanam T., Aktas T., Ramirez F. MOF-associated complexes ensure stem cell identity and Xist repression. eLife. 2014:e02024. doi: 10.7554/eLife.02024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katoh H., Qin Z.S., Liu R., Wang L., Li W., Li X. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Molecular Cell. 2011;44(5):770–784. doi: 10.1016/j.molcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sykes S.M., Mellert H.S., Holbert M.A., Li K., Marmorstein R., Lane W.S. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular Cell. 2006;24(6):841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carlsten J.O., Zhu X., Gustafsson C.M. The multitalented Mediator complex. Trends in Biochemical Sciences. 2013;38(11):531–537. doi: 10.1016/j.tibs.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 76.Poss Z.C., Ebmeier C.C., Taatjes D.J. The Mediator complex and transcription regulation. Critical Reviews in Biochemistry and Molecular Biology. 2013;48(6):575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]