

Abstract
IMPORTANCE
The US Food and Drug Administration (FDA) evaluates high-risk medical devices such as cardiac implantable electronic devices (CIEDs), including pacemakers, implantable cardioverter-defibrillators, and cardiac resynchronization therapy devices, via the premarket approval (PMA) process, during which manufacturers submit clinical data demonstrating safety and effectiveness. Subsequent changes to approved high-risk devices are implemented via “supplements,” which may not require additional clinical testing.
OBJECTIVE
To characterize the prevalence and characteristics of changes to CIEDs made through the PMA supplement process.
DESIGN
Using the FDA’s PMA database, we reviewed all CIEDs approved as original PMAs or supplements from 1979 through 2012. For each supplement, we collected the date approved, type of supplement (panel-track, 180-day, real-time, special, and 30-day notice), and the nature of the changes. We calculated the number of supplements approved per PMA and analyzed trends relating to different supplement regulatory categories overtime. For supplements approved via the 180-day regulatory pathway, which often involve significant design changes, from 2010-2012, we identified how often additional clinical data were collected.
RESULTS
From 1979-2012, the FDA approved 77 original and 5829 supplement PMA applications for CIEDs, with a median of 50 supplements per original PMA (interquartile range [IQR], 23-87). Excluding manufacturing changes that do not alter device design, the number of supplements approved each year was stable around a mean (SD) of 2.6 (0.9) supplements per PMA per year. Premarket approvals remained active via successive supplements over a median period of 15 years (IQR, 8-20), and 79% of the 77 original PMAs approved during our study period were the subject of at least 1 supplement in 2012. Thirty-seven percent of approved supplements involved a change to the device’s design. Among 180-day supplements approved from 2010-2012, 23% (15/64) included new clinical data to support safety and effectiveness.
CONCLUSIONS AND RELEVANCE
Many CIED models currently used by clinicians were approved via the PMA supplement process, not as original PMAs. Most new device models are deemed safe and effective without requiring new clinical data, reinforcing the importance of rigorous postapproval surveillance of these devices.
In the United States, the Food and Drug Administration (FDA) reviews high-risk medical devices–those that support human life, prevent illness, or present an unreasonable risk1–via the premarket approval (PMA) pathway, through which manufacturers collect preclinical and clinical data as necessary to provide “reasonable assurance” of the device’s safety and effectiveness.2 Medical device regulation earned scrutiny in recent years after several device recalls, including the Medtronic Sprint Fidelis and St Jude Medical Riata implantable cardioverter-defibrillator (ICD) leads, which were approved through the PMA process.3,4 However, these leads were not original PMA applications and were not tested clinically in human trials prior to approval. Rather, these ICD leads were design changes to prior-marketed devices and were “supplements” to PMA applications originally submitted almost a decade earlier.5
Premarket approval supplements are commonly used to approve changes to existing high-risk devices. A Government Accountability Office report found that from 2003 to 2007, the FDA authorized 170 PMA applications and 664 supplements for high-risk devices.6 Supplements allow patients to benefit from incremental innovation in device technology by providing efficient and inexpensive FDA review pathways for smaller device changes. Supplements may include major or minor design changes as well as routine changes in labeling, materials, or packaging.7 By statute, the FDA must seek only the “least burdensome” supporting data necessary for review.8
Given the frequent use of PMA supplements for high-risk devices, we performed an in-depth study of supplements related to cardiac implantable electronic devices (CIEDs), including pacemakers, ICDs, and cardiac resynchronization therapy (CRT) devices. Cardiac implantable electronic devices provide a useful case study because they have been the subject of substantial evolution over the past 30 years.9 We reviewed original and supplement PMAs for CIEDs approved 1979-2012 to identify the number of PMA supplements emerging from each original PMA, characterize the nature of the changes in each supplement, and understand the data supporting these changes.
Methods
We searched for original PMA applications filed under CIED-related product codes (DSZ, DTB, DTD, DXY, KRG, LOT, LWO, LWP, LWS, LWW, MRM, NIK, NKE, NVN, NVY, NVZ, OJX, OSR) in the FDA’s public PMA database, which categorizes approved PMA applications by approval date, device type, and application type (original vs supplement).5,10 The PMA database links each PMA application to records of its supplements.
PMA Supplement Types
Five different review tracks exist for PMA supplements based on the type of change being implemented (Table). Each type requires different supporting data.7 “Panel-track” supplements represent the most significant changes (eg, new indications), are always accompanied by clinical testing, and are reviewed by apanel of subject matter experts at the FDA. When PMA was introduced in 1976, all PMA applications required panel reviews; however, the law was amended in 1990 so the FDA could internally review PMA applications (including supplements) without a panel.11 In such cases, the PMA review team consists of FDA staff members, which may include a clinician specialist as needed. Changes to a device’s design that do not require panel review are approved via a “180-day” process. Preclinical testing is commonly sufficient for 180-day supplements, although clinical tests may also be required.7 Starting in 1997, the FDA began allowing minor changes to a device’s design to gain approval through an even more efficient “real-time” review process.12 To qualify for real-time review, the design change must not require additional clinical study.7 Alterations to devices’ labeling to improve safety (eg, adding a contraindication) are administered via “special” supplements. In 1997, Congress created a fifth pathway for changes to manufacturing processes that do not alter device design–the “30-day notice” supplement. Manufacturers may enact these changes 30 days after the notice if there is no FDA action. If the FDA deems a 30-day notice application insufficient, it may request a lengthier 135-day review and require additional information to adequately ensure device safety and effectiveness.
Table.
FDA Regulatory Categories for Premarket Approval Supplements
| Type of Supplement | Types of Changes to Device | Data Required | User Fee (Fiscal Year 2014), $ | Reviewer | Year Category Formally Introduced |
|---|---|---|---|---|---|
| Panel-track | Significant design change; new indication | Clinical; limited preclinical data in some cases | 193 890 | Panel of subject matter experts and FDA staff | 1990 |
| 180-day | Significant design change; labeling change | Preclinical; confirmatory clinical data in some cases | 38 778 | FDA staff | 1986 |
| Real-time | Minor design change | Preclinical only | 18 096 | FDA staff | 1997 |
| Speciala | Labeling change that enhances device safety | No specific data requirements | NA | FDA staff | 1986 |
| 30-day noticeb | Manufacturing change | No specific data requirements | 4136 | FDA staff | 1997 |
Abbreviations: FDA, Food and Drug Administration; NA, not applicable.
A manufacturer may temporarily enact the change before the FDA issues approval.
A manufacturer may enact the change 30 days after the FDA files the notice if there is no FDA action. Alternatively, the FDA may request a 135-day review if 30-day notice is deemed insufficient.
The device manufacturer decides the supplement type covering its requested change, although the FDA reserves the right to change categorizations if necessary. User fees, collected since 2002, vary based on the complexity of the change, from $193 890 for a panel-track supplement, to $38 778 for 180-day, to $18 096 for real-time.13,14
Manufacturers may submit a single change that affects multiple devices approved under different PMAs (eg, a connector change that affects ICD, CRT, and pacemaker models). The change is bundled under a single review at the FDA.15 The PMA database contains a unique entry for each affected device, regardless of whether the change was bundled with and reviewed at the same time as other applications.
Data Extraction
For each PMA application for a CIED, we extracted the approval date, manufacturer, device type, and number of approved supplements (through December 2012). If it was available, we also downloaded the Summary of Safety and Effectiveness Data (SSED), which describes the device and supporting data. Using this information, 2 authors (B.N.R., D.B.K.) classified each original PMA as a pacemaker, ICD, or CRT.
For each PMA supplement, we extracted the date of approval, type (panel-track, 180-day, real-time, special, or 30-day notice), and reason for the supplement. The supplement reason described in the database allowed us to identify those supplements that included a change to a device’s design, compared with supplements for labeling or manufacturing changes. Since the mid-1990s, the FDA’s database has included a short paragraph describing the change for most approved supplements. Starting in 2010, the FDA published longer “review memos” to accompany approval of 180-day supplements. Panel-track supplements are generally accompanied by SSED reports, similar to original PMA applications. We used these descriptive paragraphs, review memos, and SSEDs accompanying any supplement to determine the nature of the approved changes.
Data Analysis
We calculated the median number of supplements per PMA. For each PMA, we calculated the time during which that PMA was active, defined as the amount of time (in years) between initial approval and the PMA’s most recent supplement. For example, an original PMA approved in January 1985 and most recently supplemented in December 2000 was active for 16 years.
We then identified the number of PMA supplements approved in each of the 5 regulatory categories each year from 1979-2012. Because original PMAs for CIEDs were added over time, we divided the number of supplements approved in a given year by the number of active PMAs in that year.
Finally, we identified examples of changes made to CIEDs by 180-day and real-time supplements. According to FDA’s industry guidance, these 2 categories encompass changes to a device’s design that are not reviewed by an independent panel of experts.7 Among CIED models advertised in July 2013 on the websites of the 5 manufacturers with the most approved PMA applications (Biotronik, Boston Scientific, Medtronic, Sorin Group, and St Jude Medical), we searched the PMA database to determine how and when each device model was approved. We also determined manufacturer-level trends in the number of 180-day vs real-time supplements approved since 1997 (the year the real-time pathway was introduced) for the 6 device manufacturers that received the most supplement approvals for CIEDs.
Results
From 1979-2012, the FDA approved 77 PMA applications for CIEDs, including 46 pacemaker devices (32 pulse generators, 11 leads, and 3 combined systems), 19 ICDs (7 pulse generators, 3 leads, and 9 combined systems), and 12 CRT devices (1 CRT-defibrillator and 1 CRT-pacemaker pulse generator, 4 CRT-defibrillator and 3 CRT-pacemaker combined systems, and 3 left ventricular leads).
These PMAs were the subject of 5829 PMA supplement applications. Nearly half (2754, 47%) were approved as 30-day notice supplements for manufacturing changes. The FDA required a 135-day review for 346 (13%) of these approved 30-daynotice applications. The next-largest categories were 180-day supplements (1538,26%) and real-time supplements (1312, 23%). Small fractions were panel-track supplements (15, 0.3%) or special supplements (108, 2%). Data were missing for 102 supplements (2%).
The number of supplements approved for each PMA ranged from 0 to 366, with a median of 50 (interquartile range [IQR], 23-87). Excluding 30-day notice supplements, each PMA led to a median of 30 supplements (range, 0-177; IQR, 10-50).
Variations in Supplement Approval Over Time
In the last decade, the number of supplements approved annually increased nearly 10-fold, from 77 to 704. Excluding 30-day notice supplements, which have increased markedly since their introduction in 1997, there was still a 3-fold increase over the past decade, from 60 supplements to 194.
Figure 1 displays the number of supplements approved each year, adjusted for the number of active PMAs in that year. Excluding 30-day notice supplements, the number of supplements approved per active PMA has remained stable around a mean (SD) of 2.6 (0.9) supplements per PMA per year (range, 1.1-4.6). Before 1986, most PMA supplements were not explicitly classified. From 1986 to 1996, 93% were 180-day supplements. Starting in 1997, minor design changes previously classified as 180-day supplements were diverted into the real-time pathway.
Figure 1. Trends in Premarket Approval Supplements for Cardiac Implantable Electronic Devices, 1980-2012.
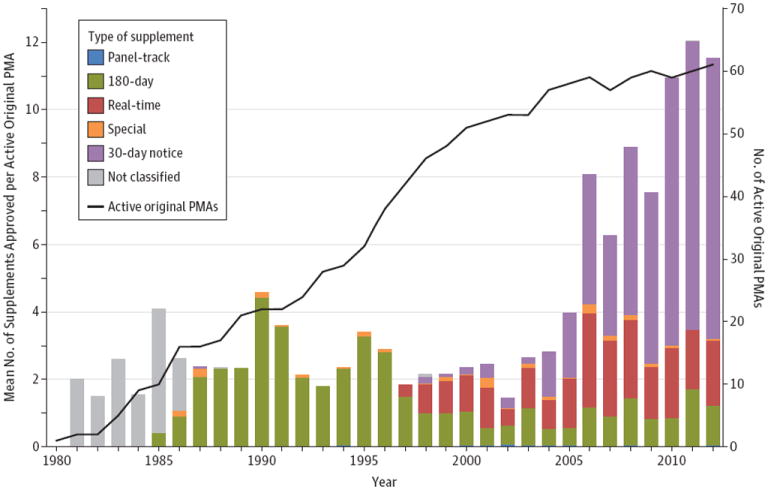
Mean number of premarket approval (PMA) supplements, by type, approved per active PMA each year from 1980-2012. The first supplement for a cardiac implantable electronic device was approved in 1981. The right-side axis (black line) displays the number of active PMAs, which increased steadily to 61 in 2012.
PMA Life Span
Figure 2 shows the active time period of each of the 77 PMAs (median, 15 years; range, 0-31; IQR, 8-20). We found that 61 (79%) received at least 1 supplement in 2012. The most recent original PMA for any transvenous ICD system was approved in 2000, indicating that all ICD models released since then have been supplements to existing PMAs.
Figure 2. Life Span of 77 Premarket Approvals for Cardiac Implantable Electronic Devices.
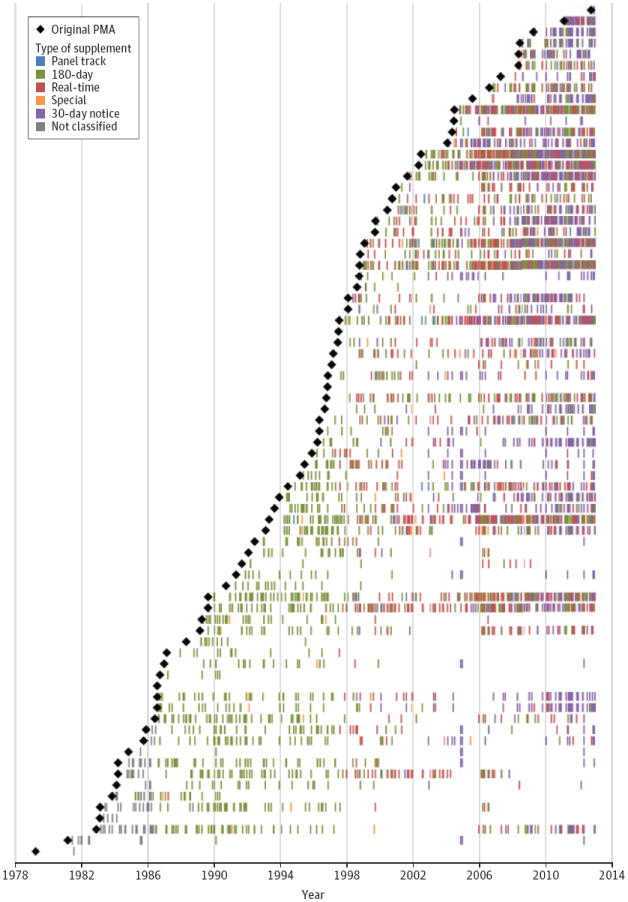
Each black diamond represents 1 of the 77 original premarket approvals (PMAs) for a cardiac implantable electronic device, organized by date of approval from oldest (bottom) to newest (top). The first original PMA was approved in 1979. Each colored mark depicts the approval of one of the types of supplements for each PMA. Three original PMAs underwent 0 supplements.
For example, the St Jude Riata ICD lead was a real-time supplement to a PMA from May 1996 that yielded 78 supplementary changes (including 45 non-30-day notice supplements) through October 2012. St Jude originally marketed the PMA in 1996 as “TVL Lead System,” and subsequent models have been marketed under 4 additional names, including the Riata and Riata ST lead models, which were introduced as realtime supplements in 2002 and 2005, respectively, and recalled in 2011. Similarly, the Medtronic Sprint Fidelis lead, recalled in 2007, originated as a PMA approved in 1993 for the Transvene Lead System. Through 2012, that PMA has been successively supplemented 91 times (38, excluding 30-day notices), including the 180-day supplement approved for the Sprint Fidelis model in 2004.
We found that all ICD leads and pulse generators currently advertised on the websites of major CIED manufacturers were either 180-day or real-time supplements (eTable 1 in the Supplement). These devices were the culmination of successive iterations dating back to PMAs from 3.7 to 19.0 years prior.
Characteristics of 180-Day and Real-Time Supplements
Among 180-day and real-time supplements, 76% (2155) represented a change to a device’s design or materials; the remaining 24% were related to changes to labeling, manufacturing, and postapproval study protocols. Since the 1997 introduction of real-time review, the FDA has approved 542 (32%) 180-day supplements and 1170 (68%) real-time supplements for changes to a device’s design. The 6 manufacturers with the most 180-day and real-time supplements collectively accounted for 95% of such supplements. Among these 6 manufacturers, the use of real-time supplements for design changes ranged from 58% (Biotronik) to 85% (Boston Scientific). Medtronic used the real-time process to approve 67% of device design changes, while St Jude used the real-time process 77% of the time.
Review memos were available for 157 of 223180-day supplements (70%) from 2010-2012. Changes to device design described in these memos included changes in lead insulation material, new software features, and new lead connections. Because proposed changes could affect more than 1 device, many of the review memos were duplicates; there were 64 unique review memos for the 157 cases, indicating that a single FDA review included changes to an average of 2.5 devices. Among the 180-day supplement review memos, 15 (23%) mentioned the collection of new clinical data supporting the altered device. Among the 15 review memos that included clinical data, 5 described the study design as prospective, multicenter, nonrandomized clinical trials, and 1 memo described a retrospective analysis to support a labeling change. The remaining 9 did not describe the study design. Seven memos with clinical data described the number of patients in the study (median, 175; range, 111-539).
Discussion
To our knowledge, this analysis is the first to characterize the PMA supplement process, as past studies of high-risk device approvals have focused exclusively on original PMA applications.16-18 Since the first CIED was approved via PMA in 1979, the FDA has authorized 5829 supplements for 77 PMA applications, a median of 50 supplements per PMA. More than one-third (37%; 2163) of supplements represented at least minor alterations to the device’s design or materials. In the vast majority of these cases, the FDA deemed that new clinical data were not necessary for approval.
The PMA supplement process allows manufacturers to update devices via incremental innovations rather than encouraging them to wait and release a package of more substantial changes that might necessitate a new PMA application. For patients, this means that useful technological advances can be rapidly integrated into clinical care. The FDA reviews supplements in the context of the original application, previous supplements, and postmarket device experiences. If a new device was approved in Europe before the United States, the FDA can review European postmarket data. In the review process, the FDA consults staff experts (including clinicians) to make a determination as to what types of data are necessary for approval. In some cases, preclinical testing may be superior to clinical testing in assessing changes.19 For example, mechanical testing of ICD leads can simulate years of clinical conditions relatively quickly, and animal studies may allow for repeated induction of arrhythmias that would be impossible in a human model.19 Thus, our results should not be interpreted to indicate that the FDA is failing to review PMA supplement applications to determine safety and effectiveness.
However, clinicians and patients should also be aware of our finding that clinical data are rarely collected as part of PMA supplement applications prior to marketing. The recalled Medtronic Sprint Fidelis and St Jude Riata ICD leads were both PMA supplements–Fidelis a 180-day supplement and Riata a real-time supplement. Neither lead was studied in human trials prior to FDA approval. The FDA’s approval of many supplements without new human trials, as in the case of these recent ICD changes, highlights the importance of collecting rigorous postapproval performance data.
Awareness by physicians of how incremental device innovation is facilitated by the PMA supplement process may have implications for the rapid adoption of newly approved CIED technologies. One study found that nearly two-thirds of ICDs implanted in the United States are a manufacturer’s most current model.20 Physicians should weigh the potential benefits offered by new device models against possible harms in the context of established alternatives, particularly for life-saving or life-sustaining interventions. Indeed, greater attention to this issue by the clinical community may motivate manufacturers to conduct much-needed comparative effectiveness studies, because currently little prospective testing of this sort exists to guide physicians’ decision making.
In contrast to the SSED reports provided with each original PMA, the evidence supporting PMA supplements has traditionally not been as accessible to clinicians or the public. Since 2010, the FDA has improved transparency by providing abbreviated review memos for approved 180-day supplements. For example, in the last few years, the FDA has approved novel 4-pole connection systems for ICD leads and generators as 180-day supplements without requiring new clinical studies.3 Medtronic’s 4-pole ICD connection system was approved in January 2012, and the review memo published by the FDA outlines the preclinical testing performed, including a 6-month animal study.21
We also found differences in how device manufacturers used 180-day and real-time PMA supplements to modify their devices. It is uncertain whether these differences reflect development and marketing strategies, with some companies releasing updated products that each offer smaller innovations over the previous generation more quickly than others. Alternatively, manufacturers may simply have varying thresholds for deciding which device changes should be submitted for realtime review instead of the normal 180-day process. Regulators and industry should ensure that standards for selecting PMA supplement pathways are consistently applied.
Our analysis of PMA supplements has several limitations. We relied on the FDA’s PMA database, which may contain errors in classifying supplements and provides only limited information about the nature of changes to medical devices and the data used to support those changes. Some data predate the FDA’s digital efforts, while other data may be considered proprietary. In the past 3 years, the FDA has started providing insight into the clinical data submitted to justify supplement approval, although greater transparency would be useful about the rationale behind the approved study designs and the outcomes of any require d postmarket analyses. In addition, the PMA database contains an entry for each PMA supplement application, even if multiple applications were bundled together under a single review. Consequently, our results represent the number of applications, which is greater than the number of reviews performed by the FDA. However, bundled applications often include changes to more than 1 device, and so our analysis approximates the total number of altered device models even if similar changes are reviewed at the same time.
Our study excludes any CIEDs approved via FDA pathways other than PMA. For example, implantable pacemakers were in use before 1976, and until the mid-1990s the FDA continued to approve some new pacemakers via the 510(k) pathway as substantially equivalent to preamendment devices. We also limited our analysis to the premarket device review process and did not consider postapproval studies that may be required for PMA supplement-approved devices, as these are uncommon and only evaluate safety and effectiveness after a device may have gained widespread clinical use. Although PMA applications are used to regulate many types of high-risk devices, including some ophthalmologic, orthopedic, and urologic implants, our analysis was limited to cardiovascular devices, as has been done in other studies of the PMA pathway.16,17 Cardiovascular devices make up almost half of all PMA and PMA supplement applications.
Although recent media and policy-making attention around medical devices has focused on the 510(k) process,22-24 we found that many high-risk cardiovascular devices, including those in widespread use today, are approved as PMA supplements. Many supplements involve minor design changes or changes to labeling, packaging, or manufacturing, and in these cases the supplement process provides a speedy and inexpensive avenue for regulatory review. The long active life of CIED PMA approvals suggests that minor design changes may accumulate over time and in some cases may add up to substantial changes from the device approved in the original PMA application. To guard against this outcome, the FDA could mandate an expert panel review of each PMA every 5 to 7 years in which it is active to evaluate the extent to which clinical data from older models still apply to newer ones. Another possibility would be more widespread implementation of rigorous postmarket studies to evaluate device performance once approved for clinical use. The rise of unique device identifiers and device registries may permit more comparative effectiveness research between older and newer versions of devices that emerge via the PMA supplement pathway.25
Conclusions
The PMA process supporting FDA approval has long been considered the gold standard for rigorously establishing the safety and effectiveness of high-risk medical devices. Our findings demonstrate that the PMA supplement process provides a commonly used review pathway for FDA authorization of changes to existing high-risk devices. Many of the pacemaker, ICD, and CRT models currently used by clinicians have come via the PMA supplement process. Most device models approved as PMA supplements are deemed safe and effective without requiring new clinical data, reinforcing the importance of postapproval surveillance of these devices. In making decisions about use of these high-risk devices, clinicians and patients should consider the strengths and limitations of the PMA supplement approval processes.
Supplementary Material
Acknowledgments
Funding/Support: Mr Rome was supported by a Harvard Medical School fellowship. Dr Kramer is the Lois Green Scholar at the Hebrew SeniorLife Institute for Aging Research and is supported by a career development award from the Harvard Catalyst Clinical and Translational Research Center and a Paul B. Beeson career development award in aging (1K23AG045963). Dr Kesselheim is supported by a Robert Wood Johnson Foundation Investigator Award in Health Policy Research, a Greenwall Faculty Scholarship in Bioethics, and a career development award from the Agency for Healthcare Research and Quality (K08HS18465-01).
Role of the Sponsor: The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Mr Romeand Dr Kesselheim had full access to all of the data in the study and take responsibility forthe integrity of the data and the accuracy of the data analysis.
Study concept and design: Rome, Kramer, Kesselheim.
Acquisition of data: Rome.
Analysis and interpretation of data: Rome, Kramer, Kesselheim.
Drafting of the manuscript: Rome.
Critical revision of the manuscript for important intellectual content: Rome, Kramer, Kesselheim.
Statistical analysis: Rome.
Obtained funding: Rome, Kesselheim.
Administrative, technical, and material support: Kramer, Kesselheim.
Study supervision: Kramer, Kesselheim.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr Kramer reported serving as a consultant to the FDA’s Circulatory Systems Advisory Panel. Drs Kramer and Kesselheim reported previously publishing research funded by the FDA on comparative medical device regulation. No other disclosures were reported.
References
- 1.Premarket approval (PMA) US Food and Drug Administration; [December, 26, 2013]. http://www.fda.gov/medicaldevices/deviceregulationandguidance/howtomarketyourdevice/premarketsubmissions/premarketapprovalpma/ [Google Scholar]
- 2.Medical Device Amendments of 1976 [Pub L No. 94-295] US Government Printing Office; [December, 26, 2013]. http://www.gpo.gov/fdsys/pkg/STATUTE-90/pdf/STATUTE-90-Pg539.pdf. [Google Scholar]
- 3.Hauser RG, Almquist AK. Learning from our mistakes? testing new ICD technology. N Engl J Med. 2008;359(24):2517–2519. doi: 10.1056/NEJMp0805359. [DOI] [PubMed] [Google Scholar]
- 4.Kay GN, Ellenbogen KA. An ICD lead advisory: a plea for more diligence and more data. Pacing Clin Electrophysiol. 2012;35(6):648–649. doi: 10.1111/j.1540-8159.2012.03398.x. [DOI] [PubMed] [Google Scholar]
- 5.Premarket approval (PMA) database. US Food and Drug Administration; [January 4, 2013]. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pma.cfm. [Google Scholar]
- 6.Crosse MG. US Government Accountability Office; [December 26, 2013]. Medical devices: shortcomings in FDA’s premarket review, postmarket surveillance, and inspections of device manufacturing establishments. http://www.gao.gov/products/GAO-09-370T. [Google Scholar]
- 7.Guidance for industry and FDA staff: modifications to devices subject to premarket approval (PMA): the PMA supplement decision-making process [2008] US Dept of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, Center for Biologics Evaluation and Research. [December 26, 2013]; http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM089360.pdf.
- 8.The least burdensome provisions of the FDA Modernization Act of 1997: concept and principles: final guidance for FDA and industry. US Dept of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, Office of Device Evaluation, Center for Biologics Evaluation and Research. [December 26, 2013]; http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm085994.htm.
- 9.DiMarco JP. Implantable cardioverter-defibrillators. N Engl J Med. 2003;349(19):1836–1847. doi: 10.1056/NEJMra035432. [DOI] [PubMed] [Google Scholar]
- 10.Product classification. US Food and Drug Administration. [January 20, 2013]; http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm.
- 11.Safe Medical Devices Act of 1990 [Pub L No.101-629] US Government Printing Office; [December 26, 2013]. http://www.gpo.gov/fdsys/pkg/STATUTE-104/pdf/STATUTE-104-Pg4511.pdf. [Google Scholar]
- 12.“Real-time” review program for premarket approval application (PMA) supplements. US Food and Drug Administration, Office of Device Evaluation. 1997 [Google Scholar]
- 13.Medical Device User Fee and Modernization Act of 2002 [Pub L No. 107-250] US Government Printing Office; [December 26, 2013]. http://www.gpo.gov/fdsys/pkg/PLAW-107publ250/content-detail.html. [Google Scholar]
- 14.PMA review fees. US Food and Drug Administration; [October 26, 2013]. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/ucm048161.htm. [Google Scholar]
- 15.Guidance for industry and FDA staff: bundling multiple devices or multiple indications in a single submission. US Dept of Health and Human Services, Center for Devices and Radiological Health; 2003. [Google Scholar]
- 16.Dhruva SS, Bero LA, Redberg RF. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. JAMA. 2009;302(24):2679–2685. doi: 10.1001/jama.2009.1899. [DOI] [PubMed] [Google Scholar]
- 17.Kramer DB, Mallis E, Zuckerman BD, Zimmerman BA, Maisel WH. Premarket clinical evaluation of novel cardiovascular devices: quality analysis of premarket clinical studies submitted to the Food and Drug Administration 2000-2007. Am J Ther. 2010;17(1):2–7. doi: 10.1097/MJT.0b013e3181ca8105. [DOI] [PubMed] [Google Scholar]
- 18.Chen CE, Dhruva SS, Redberg RF. Inclusion of comparative effectiveness data in high-risk cardiovascular device studies at the time of premarket approval. JAMA. 2012;308(17):1740–1742. doi: 10.1001/jama.2012.14491. [DOI] [PubMed] [Google Scholar]
- 19.Shein MJ, Schultz DG. Testing new ICD technology. N Engl J Med. 2008;359(24):2610. doi: 10.1056/NEJMc086467. [DOI] [PubMed] [Google Scholar]
- 20.Lampert R, Wang Y, Curtis JP. Variation among hospitals in selection of higher-cost, “higher-tech,”implantable cardioverter-def ibrillators: data from the National Cardiovascular Data Registry (NCDR) Implantable Cardioverter/Defibrillator (ICD) Registry. Am Heart J. 2013;165(6):1015–1023. doi: 10.1016/j.ahj.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Premarket approval (PMA) database: 2012;S176:010031. US Food and Drug Administration; [December 27, 2013]. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=26238. [Google Scholar]
- 22.Medical devices and the public’s health: the 510(k) clearance process at 35 years. Institute of Medicine; [December 26, 2013]. http://www.iom.edu/Reports/2011/Medical-Devices-and-the-Publics-Health-The-FDA-510k-Clearance-Process-at-35-Years.aspx. [Google Scholar]
- 23.Zuckerman DM, Brown R, Nissen SE. Medical device recalls and the FDA approval process. Arch Intern Med. 2011;171(11):1006–1011. doi: 10.1001/archinternmed.2011.30. [DOI] [PubMed] [Google Scholar]
- 24.Ardaugh BM, Graves SE, Redberg RF. The 510(k) ancestry of a metal-on-metal hip implant. N Engl J Med. 2013;368(2):97–100. doi: 10.1056/NEJMp1211581. [DOI] [PubMed] [Google Scholar]
- 25.Food and Drug Administration, HHS. Uniquedevice identification system: final rule. Fed Regist. 2013;78(185):58785–58828. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.