

Abstract
High homocysteine (Hcys) levels are suspected to contribute to the pathogenesis of cardiovascular disease and of other chronic conditions. Failure of B vitamins to reduce the incidence of cardiovascular events while lowering the Hcys levels, has prompted the search for alternative treatments. We tested the ability of anethole dithiolethione (ADT) to lower the Hcys levels in rats and we explored possible underlying mechanisms. Parenteral administration of 10 mg/kg ADT to normal rats for 3 days lowered the Hcys levels between 51.4% and 31.5% in kidneys, liver, testis and plasma. Concomitantly, glutathione (GSH) increased between 112% and 28% in kidneys, brain, liver and plasma whereas protein thiolation index decreased 30%. In hyperhomocysteinemic rats, the plasma Hcys levels dropped 70% following a single IP injection of 10 mg/kg ADT, while they decreased 55% following oral administration of 2 mg/kg/day ADT for one week. Significant additive effects occurred when sub-therapeutic doses of ADT and folic acid were used in combination. To test the possible mechanism(s) of these actions, we perfused isolated rat livers and kidneys with albumin-bound Hcys, the prevalent form of plasma Hcys, and physiological thiols and disulfides at different ratios. In both organ preparations, the elimination rate of albumin-bound Hcys was progressively faster as the amount of reduced thiols was increased in the perfusate. These findings indicate that ADT shifts the redox ratio of GSH and other thiols with their oxidized forms toward the reduced forms, thus favoring the dissociation of albumin-bound Hcys and its transfer to renal and hepatic cells for further processing.
Keywords: hyperhomocysteinemia, anethole dithiolethione, protein-bound homocysteine, thiols
1. Introduction
Elevated plasma levels of homocysteine (Hcys) are linked to atherosclerotic cardiovascular disease [1] and to other common pathologic conditions including the metabolic syndrome and diabetes [2,3], renal failure [4,5] and neurodegenerative disorders [6]. Even moderate elevations of the plasma Hcys levels, within the concentration range of the general population, are suspected to contribute to the pathogenesis of these disease states [7] and considerable effort is devoted to finding effective pharmacologic treatments for hyperhomocysteinemia.
Since folic acid deficiency and excessive dietary intake of methionine (Met) or proteins are well-known causes of hyperhomocysteinemia [6,8,9], efforts to reduce the Hcys levels have primarily focused until recently on the pharmacological supplementation of folic acid and other B vitamins. However, while effective at lowering the Hcys levels, these vitamins do not seem capable of decreasing the incidence of cardiovascular events in humans [10,11]. This explains the aim of more recent investigation toward discovery of new therapeutic agents and/or modalities.
Thiol molecules have generated interest as a possible new class of Hcys-lowering agents based on evidence that up to 80% of circulating Hcys is complexed with albumin via disulfide bond, which prevents its free exchange between extra- to intracellular space [12,13]. Reduced thiols can break the Hcys-albumin bond and release free Hcys thus facilitating its internalization and intracellular metabolism and elimination [14]. In a recent clinical trial, however, one-month oral intake of N-acetylcysteine failed to consistently decrease Hcys, probably because of poor bioavailability of the drug [15].
As an alternative pharmacologic approach, we recently reported that both the sulfur-containing compounds 5-[4-hydroxyphenyl]-3H-1,2-dithiole-3-thione (ADT-OH) and its derivative 2-acetyloxybenzoic acid 4-(3-thioxo-3H-1,2-dithiol-5-yl)phenyl ester (S-aspirin or ACS14) lower the concentration of Hcys and raise those of GSH and Cys in the rat [16]. The ADT-OH and ACS14 molecules share the presence of a phenyl-dithiolethione group which is also characteristic of anethole dithiolethione (ADT), a natural constituent of certain cruciferous vegetables and a marketed drug approved in several countries as a choleretic and sialogogue [17,18]. Thus we suspect that ADT, similarly to ADT-OH and ACS14, may have the ability to lower plasma concentration of tHcys.
With the present work, we formally tested the hypothesis that ADT is indeed capable of lowering Hcys in both plasma and solid organs. In addition we explored possible mechanisms of action of ADT through modulation of GSH and other thiols metabolism.
2. Material and Methods
2.1 Materials
ADT was purchased from Hubei Zhonghecheng Chemical Co., Ltd, (Hu Bei, China), Monobromobimane (mBrB) was obtained from Calbiochem, Milan, Italy. HPLC grade solvents were purchased from Mallinckrodt-Baker (Milan, Italy). All other reagents were obtained from Sigma-Aldrich, Milan, Italy.
2.2 Experimental model
Sprague-Dawley rats (300 g) were purchased from Charles River (Calco, Milan, Italy) and were kept in a controlled environment (22–24°C, relative humidity 40–50%, under a 12-h light/dark cycle). Food was ad libitum for 2–3 weeks before experimentation and activity was unrestricted before and during the experiments. In the studies that called for high basal levels of blood homocysteine, this was induced by enriching the standard chow with 1.5% (w/w) Met [19]; on this diet, the blood levels of total homocysteine (tHcys) peaked at 50–80 μM after 1–2 days and then stabilized around 40 μM (not shown). In the studies that required repeated blood collection, the jugular vein was cannulated two days before experimentation with PE-50 tubing connected to a double valve (model 607, 15 mm × 16 mm, Danuso Instruments, Milano, Italy) under pentobarbital anesthesia, as previously described [20]. All animal manipulations were made in accordance with the European Community guidelines for the use of laboratory animals. The experiments were authorized by the local ethical committee of the University of Siena.
2.3 Experimental protocols
2.3.1 Effect of ADT on the blood and tissue levels of low molecular mass thiols (LMM-SH)
Rats were injected in the peritoneum (ip) with 10 mg/kg ADT or vehicle [dimethyl sulfoxide (DMSO)] once daily for three days. Twenty-four hours after the last injection, the rats were anesthetized with pentobarbital (50 mg/kg) and blood was collected from the abdominal aorta into tubes containing 50 mg/ml K3EDTA (40:1) and immediately centrifuged at 14 000g for 20 seconds to separate plasma and erythrocytes. Organs were then rapidly removed in the following order: liver, kidneys, lungs, heart, testis and brain, washed with physiologic saline, weighed and immediately frozen in liquid nitrogen and stored at −80° until analysis.
2.3.2 Acute effects of single ADT injection
Rats were injected ip with a single dose of ADT (10 mg/kg) followed by serial collection of blood (200 μl aliquots) from the jugular vein catheter (see above) 0, 1, 2, 4, 8, 24, 48, and 72 hours post-injection and by immediate blood processing for separation of plasma as described above.
2.3.3 Effect of ADT on H2S
Rats were given a single ip dose of 1, 2, 5, or 10 mg/kg ADT followed by serial collection of blood (100 μl aliquots) from the jugular vein catheter (see above) 0, 1, 2, 4, 8, 16, 24, 32, 48, and 72 hours post-injection and by immediate blood processing for separation of plasma as described above.
2.3.4 Effect of ADT on Protein Thiolation Index (PTI) and reduced/disulfide thiol ratio
Rats were given intraperitoneally 1, 2, 5, or 10 mg/day ADT for three days, followed by blood collection from the abdominal aorta and processing for separation of plasma as described above.
2.3.5 Effect of ADT on hyperhomocysteinemia
Rats were fed a high Met diet for 2–3 days before a single ip injection of ADT (2, 5, or 10 mg/kg). Two hundred μl of blood was collected from the jugular vein catheter 24, 48, 72 hours after injection of ADT and processed for plasma separation as above.
2.3.6 Ability of ADT to prevent a Met-induced rise in tHcys
Rats were administered a single ip injection of 2, 5, or 10 mg/kg ADT followed after 24 hours by a single ip injection of 100 mg/kg Met. Two hundred μl of blood was collected from the jugular vein catheter at 0, 24, 48, 72 hours and processed for plasma separation as above.
2.3.7 Effect of orally administered ADT on tHcys
Rats were fed with a Met-enriched diet (see above) starting 3 days before daily oral administration by gavage of 1, 2, 5, or 10 mg/kg ADT for one week, followed by blood collection from the abdominal aorta as above.
2.3.8 Effect of combined ADT and folic acid on tHcys
Rats were fed with a Met-enriched diet starting two days before daily oral administration of ADT (0.2 mg/kg), folic acid (10 μg/kg), or their combination (0.1 mg/kg ADT with 5 μg/kg folic acid, or 0.2 mg/kg ADT with 10 μg/kg folic acid) for three weeks, followed by blood collection from the abdominal aorta as described above.
2.4 Analysis of thiols and disulfides
Plasma total thiols were measured by reduction of disulfides by reaction with dithiothreitol (DTT) and labeling of the –SH group by reaction with mBrB [16]. Briefly, 25 μl samples were incubated with 2 mM (final concentration) dithiothreitol (DTT) for 5 before addition of three volumes of a solution consisting of 6% (w/v) trichloroacetic acid (TCA) and 1 mM K3EDTA and centrifuged at 10 000g for 2 min, to precipitate proteins. Twenty-five μl of clear supernatant were adjusted to pH 8.0 with 5 μl of 2 M Tris, spiked with 0.5 μl of 40 mM mBrB in methanol, incubated for 10 min in the dark, and then acidified with 1 μl of 1M HCl and centrifuged for injection into the HPLC apparatus.
Erythrocyte thiols were measured by washing three times the cell pellets with ice cold physiologic saline containing 20 mM Na+/K+ phosphate, pH 7.4, and 5 mM glucose followed by hemolysis with 10 volumes of 10 mM Na+/K+ phosphate buffer, pH 7.4. Intracellular thiols were measured by labeling the –SH group by reaction with mBrB [21]. Briefly, one hundred μl of the cell lysate were incubated for 10 minutes with 2.5 μl of 40 mM mBrB in the dark, then acidified with 5.5 μl of 60% (w/v) TCA in preparation for HPLC analysis.
Tissue thiols were analyzed after homogenization of the samples and labeling of the –SH group by reaction with mBrB [22,23]. Briefly, samples were homogenized in five volumes of an ice cold solution consisting of 8% (w/v) TCA and 1 mM K3EDTA using a glass/teflon tissue grinder. After centrifugation, 100 μl of protein-free acid supernatant were brought to pH 8.0 with 15 μl of 2 M Tris, spiked with 3 μl of 40 mM mBrB, incubated for 10 min in the dark and then acidified with 4 μl of 1M HCl and centrifuged for HPLC analysis [3,4].
An Agilent series 1100 HPLC (Agilent Technologies Italia, Milan, Italy) equipped with diode array and a fluorimetric detector was used for all determinations.
2.5 Analysis of PTI and low molecular mass thiols (LMM-SH)/low molecular mass disulfides (LMM-SS)
PTI was calculated based on analysis of the –SH groups by colorimetric reaction with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) [24] and of the total thiolated protein by our method which is based on spectrophotometric determination of ninhydrin [25]. Total LMM-SH and total LMM-SS were measured in acid extracted plasma with DTNB before and after thiol reaction with NEM and disulfide reduction with NaBH4 [26].
2.6 Analysis of enzyme activity
Enzyme activity was assayed in solid organ homogenates and in hemolyzed erythrocytes (see above). All enzymatic activities were measured in ultracentrifugation supernatants (105 000 g for 1 h at 4°C). Glutathione S-transferase activity was determined by colorimetric assay of the GSH-DNB conjugate at 340 nm [27]. Glutathione reductase activity was determined by colorimetric assay of NADPH oxidation [28]. γ-Glutamyl-cysteinyl synthase activity was determined by measuring the production of γ-Glutamylcysteine (γ-Glu-Cys) over time by HPLC [16]. Cystathionine β-synthase (CB synthase) activity was measured based on HPLC quantification of cystathionine product [29].
2.7 Protein assay
Protein concentration in rat organs was analysed with the Bradford method [30] using bovine serum albumin as reference standard. Red blood cells (RBCs) hemoglobin concentration was analysed by spectrophotometry using a wavelength range of 500–700 nm and a peak height of 541 nm (ε=13.8 mM−1 cm−1) [31].
2.8 H2S measurement
H2S was analyzed in plasma by a modification of the methylene blue method [32] coupled with HPLC detection, as previously described [33]. Briefly, 20 μl of fresh plasma was added to 40 μl of 20% (w/v) TCA solution containing 1 mM K3EDTA and 0.2 mM diethylene triamine pentaacetic acid. After 30 s centrifugation at 18,000g 40 μl of the supernatant were added with 5 μl of N,N-dimethyl-p-phenylenediamine sulfate (DPD, 20 mM in 7.2 N HCl) and then 5 μl of FeCl3 (30 mM in HCl 1.2 N); after 20′ incubation in the dark, samples were separated by HPLC using a Zorbax Eclipse XDBC18 column (4.6×150 mm, 5 μm, Agilent Technologies, Milan, Italy). The methylene blue formed from the reaction of H2S with DPD in the presence of FeCl3 was monitored at 667 nm wavelength by means of a diode array detector.
2.9 Experiments in isolated organs
Experiments in the isolated perfused liver (IPL) and kidney (IPK) were carried out in the recirculation mode following established procedures [34,35]. For the IPL studies, after clarification of the effluent, recirculated perfusion was started using 150 ml of perfusion buffer enriched with 20 mg/ml bovine serum albumin (BSA, fraction V containing 25 μM homocysteine bound by disulfide bond), cysteine (Cys, 5, 10, or 20 μM), GSH (0.5, 1, or 2 μM), 25 μM cystine (Cyss), and 25 μM glutathione disulfide (GSSG), with or without 20 μM ADT (all final concentrations). At the indicated times a 0.5 ml aliquot of recirculating buffer was collected for analyses of total Hcys. Bile samples were collected at 10 min intervals. For the IPK studies, after clarification of the effluent, inulin (60 mg/100 mL) was added to the perfusion medium and, then after a 5-min equilibration period, recirculated perfusion was started with 150 mL of perfusate consisting of Krebs–Henseleit bicarbonate buffer (pH 7.4) with 1 mM BSA containing 25 μM homocysteine bound by disulfide bond, 5 mM D-glucose, 2 mM glycine, 0.5 mM L-glutamic acid, Cys (5, 10, or 20 μM, GSH (0.5, 1, or 2 μM), 25 μM Cyss and 25 μM GSSG, with or without 20 μM ADT (all final concentrations). At the indicated times a 0.5 ml aliquot of recirculating buffer was collected for analyses of total Hcys. Urine samples were collected at 10 min intervals.
2.10 Inulin analysis
Inulin was hydrolyzed to its fructose monomers by mixing samples with perchloric acid, fructose was then converted to glucose-6-phospate, which was detected by measuring production of NADPH as previously described [36].
2.11 Preparation of homocysteinylated bovine serum albumin (BSA)
Completely reduced BSA (rBSA) was prepared by incubating 80 mg/mL protein solution (BSA dissolved in 50 mM phosphate buffer, pH 7.40) with 5 mM GSH at room temperature for 4h, followed by dialysis against the same buffer with dialysate exchange every 4h for a total of three times. Finally, rBSA was extensively dialyzed against water. Homocysteinylated BSA was obtained by reacting rBSA with 2 mM homocystine for 24 h at room temperature. The excess homocystine was then eliminated by extensive dialysis against water. Before its use, BSA was extensively dialyzed against IPL or IPK buffers and adjusted to the final BSA concentration.
2.12 Statistics
Data are expressed as mean ± SD. Differences between groups were evaluated using one-way analysis of variance (ANOVA) with Neuman-Keuls post-hoc test. p<0.05 was considered statistically significant.
3. Results
3.1 Effect of treatment with ADT on homocysteine and H2S in rat tissues
Parenteral administration of ADT for three days markedly decreased Hcys in several organs and tHcys in plasma as compared to controls (Fig. 1). The largest drops were in the kidney (51.4%; p<0.01), liver (41.5%; p<0.01), testis (31.8%; p<0.05) and in plasma (31.5%; p<0.05). Even a single ip injection of ADT was able to induce a sustained stable decrease of circulating tHcys as compared to vehicle. The tHcys levels dropped 29.9% and 45.8%, respectively, 2 and 8 hours after ip injection and they maintained comparably low levels for 72 hours (Fig 2). Furthermore, since the effects of ADT on Hcys and Cys (see below) are likely to involve catalysis within the transsulfuration pathway (TSP) and the latter is responsible for production of H2S, rats were also tested for circulating H2S. Fig. 3 shows that plasma H2S was increased early after ADT treatment as compared to vehicle, peaking at about 0.5 μM, 4 hours after administration of the highest ADT dose (p<0.01) and maintaining similar levels until the end of the 72 hours of observation. Even with lower doses, a slight temporary increase of H2S was observed early on followed by a drop to the initial values after 12–24 hours. Despite this evidence, the effects of ADT on Hcys did not seem to be mediated by an action on CB synthase, the rate limiting enzyme of Hcys catabolism within the TSP, since the enzyme activity was not affected by ADT (Table 1).
Figure 1.

Levels of Hcys in several rat tissues after daily ip injection of 10 mg/kg ADT (grey bar) or vehicle (black bar) for 3 days. The unit measures are pmoles Hcys/g tissue for the solid organs, nmoles Hcys/g Hb for erythrocytes, and μmoles total Hcys/l for plasma. Data represent mean ± SD of 4 animals per treatment; * indicates p<0.05 ADT vs vehicle; ** indicates p<0.01 ADT vs vehicle.
Figure 2.

Levels of plasma total Hcys in rats treated ip with a single administration of 10 mg/kg ADT (white circles) and in control rats treated with DMSO (black triangle). Data represent mean ± SD of 4 animals per treatment; **indicates p<0.01 vs. vehicle.
Figure 3.

Levels of plasma hydrogen sulfide in rats treated ip with a single administration of 1 (black square), 2 (white diamond), 5 (white triangle down), 10 (black circles) mg/kg ADT. Control rats were treated with vehicle (black triangles, DMSO). Data represent mean of 4 animals per treatment; SD and statistical significance were omitted for some data points for graphical reasons. **indicates p<0.01 vs vehicle.
Table 1.
Enzymatic activities in tissues and erythrocytes of rats after 72 h from ip administration of ADT (10 mg/kg/day for three days). Control animals were administered with vehicle (DMSO) only. Data are expressed as mean ±SD (n=4 for each treatment).
| Treatment | RBCs | Liver | Kidney | Lung | Heart | Testis | Brain |
|---|---|---|---|---|---|---|---|
|
CBS1
(mU/mg protein)
|
|||||||
| ADT | 0 | 0.094±0.007 | 0.037±0.005 | 0 | 0 | 0 | 0.017±0.004 |
| Vehicle | 0 | 0.087±0.005 | 0.032±0.006 | 0 | 0 | 0 | 0.021±0.005 |
|
|
|||||||
|
γ–GCS (mU/mg protein)
|
|||||||
| ADT | 0.785±0.037 | 38.0±8.2 | 45.4±2.6 | 5.94±0.37 | 5.84±0.86 | 13.1±2.8 | 18.7±1.6 |
| Vehicle | 0.723±0.148 | 33.7±6.5 | 42.6±4.3 | 6.48±0.53 | 4.90±0.57 | 11.7±4.6 | 16.6±3.0 |
|
| |||||||
|
GR (mU/mg protein)
|
|||||||
| ADT | 1.21±0.37 | 54.5±6.85 | 213±35 | 64.9±10.4 | 27.3±5.4 | 19.7±1.0 | 13.2±2.2 |
| Vehicle | 1.08±0.23 | 43.8±9.58 | 237±47 | 70.6±8.3 | 24.0±3.3 | 22.8±3.8 | 11.6±3.8 |
|
| |||||||
|
GST (mU/mg protein)
|
|||||||
| ADT | 3.12±0.59 | 659±96 | 141±18 | 68.1±5.94 | 72.7±5.2 | 541±73 | 23.7±4.5 |
| Vehicle | 2.84±0.45 | 587±64 | 137±9 | 64.2±6.66 | 69.2±4.4 | 604±87 | 26.1±3.8 |
Abbreviations: CBS: cystathionine-β-synthase; γ–GCS; γ-glutamyl-cysteine synthase; GR: glutathione reductase; GST: glutathione transferase
3.2 Effect of ADT on GSH and on the total thiol redox balance in plasma
Besides the above described effects of ADT on Hcys and H2S, ip administration of this compound for three days also increased significantly GSH and its precursors Cys and γ-glutamyl-cysteine (γ-Glu-Cys) content in several rat tissues. The increment of GSH varied depending on the organ being analyzed (Fig 4A), with the largest rise observed in kidney (112%; p<0.01 vs. vehicle), brain (70%; p<0.01 vs. vehicle) and liver (30%; p<0.05 vs. vehicle). Lungs and heart experienced smaller yet significant increments. Plasma too was characterized by a significant rise in total GSH (28.0%; p<0.01 vs vehicle, inset). Similar but stronger effects were also noted for Cys (Fig 4B) and γ-Glu-Cys (Fig 4C) in several tissues and in plasma. Besides this remarkable effect on thiols, Fig. 5 shows that ADT also affected the systemic redox balance, that was evaluated by measuring the thiol to disulfide ratio in plasma. In fact, the intracellular volume of the body is only two thirds of the total, the remaining being represented by extracellular fluids. Indeed, in vitro data indicate that variations in extracellular redox state have significant impact on critical cell functions and variations in the extracellular thiol/disulfide pools are expected to contribute to the susceptibility of tissues to oxidative stress [37]. Indeed, the plasma protein thiolation index (PTI; a marker of oxidative stress [25]) was approximately 30% lower in ADT-treated than in control animals (Fig. 5A; p<0.01 vs vehicle for 2,5,10 mg /kg doses). Conversely, the ratio between LMM-SH and LMM disulfide (LMM-SS), that express the low molecular mass thiol/disulfide pools in plasma, was about 50% higher in ADT-treated animals (Fig 5, B; p<0.01 for 2 and 10 mg/kg and p<0.05 for 5 mg/kg). However, these remarkable effects on GSH concentration and redox balance were not associated with change in the activity of several major enzymes involved in GSH metabolism including, GSH reductase, γ-glutamylcysteine synthase and glutathione S-transferase (Table 1).
Figure 4.

Levels of glutathione (panel A), cysteine (panel B), γ-glutamylcysteine (panel C) in several rat tissues after daily ip injection of 10 mg/kg ADT (grey bar) or vehicle (black bar) for 3 days. The unit measures are pmoles /g tissue for the solid organs, pmoles /g Hb for erythrocytes, and μmoles total thiols/l for plasma. Data represent mean ± SD of 4 animals per treatment; *indicates p<0.05 ADT vs. vehicle; **p<0.01 ADT vs. vehicle.
Figure 5.

Values of Protein Thiolation Index (panel A) and of LMM-SH to LMM-SS ratio in plasma. Rats were ip treated with 1, 2, 5, 10 mg/kg/day of ADT for three days. Control rats were treated with vehicle (DMSO). Data represent mean ± SD of 4 animals per treatment; *indicates p<0.05 ADT vs vehicle; **p<0.01 ADT vs. vehicle.
3.3 Effect of ADT on Hcys levels in a rat model of hyperhomocysteinemia
The ability of ADT to ameliorate hyperhomocysteinemia was evaluated in rats with diet-induced high blood levels of Hcys (41±6 μM) (see Methods). As shown in Fig. 6, hyperhomocysteinemic rats responded to a single ip injection of 2, 5, or 10 mg/kg ADT with a dramatic dose-dependent decrease of circulating tHcys. Two and 5 mg/kg ADT, respectively, lowered the tHcys concentration transiently at 24 and from 24 through 48 hours after injection, while 10 mg/kg ADT lowered tHcys by approximately 70% (p<0.01 vs vehicle) starting at 24 hours and throughout the 72 hours of observation. A preventive action of ADT on diet-induced hyperhomocysteinemia was then evaluated. Fig 7 shows that rats pretreated ip with 2, 5 or 10 mg/kg ADT did not experience a rise in tHcys following ip injection of 100 mg/kg Met (a classical model of hyperhomocysteinemia). Since ADT is available as an oral formulation, rats with diet-induced hyperhomocysteinemia were treated orally with 1, 2, 5 and 10 mg/kg/day ADT, which is commensurate to the recommended human therapeutic dose. Fig. 8 shows that one-week treatment with ADT caused a dose-dependent drop of circulating tHcys with approximately 55% drop elicited by 2 mg/kg or higher ADT doses (p<0.01 vs vehicle). Finally, hyperhomocysteinemic rats were tested for the combined effect of ADT and folic acid. Fig. 9 shows that separate administration of low doses ADT or folic acid (0.2 mg/kg and 10 μg/kg, respectively) failed to lower tHcys, but a combination of these two compounds caused approximately 55% drop of circulating tHcys (p<0.01 vs vehicle).
Figure 6.

Plasma levels of tHcys in rats fed with a Met enriched diet. After achieving stable tHcys levels, the rats received a single ip injection of 0 (dark circle), 2 (white circle), 5 (dark triangle), or 10 mg/kg (dark square) ADT. Levels of tHCys are shown at baseline and 24, 48, 72h after ADT injection. Data represent mean ± SD of 4 animals per treatment; * indicates p<0.05 ADT vs. control rats; ** indicates p<0.01 ADT vs. control rats; ## indicates p<0.01 ADT vs. the lowest dose ADT; §§indicates p<0.01 ADT vs. intermediate dose of ADT.
Figure 7.

Plasma levels of tHcys in rats pre-treated with a single ip injection of 2 mg/kg (white diamond), 5 mg/kg (black square) or 10 mg/kg (white triangle) ADT, or vehicle (dark circle), followed after 24 hours by the ip injection of 100 mg/kg Met. Data represent mean ± SD of 4 animals per treatment; **indicates p<0.01 ADT vs. control rats.
Figure 8.

Plasma levels of tHcys in rats fed with a Met-enriched diet and treated daily with 0, 1, 2, 5, or 10 mg/kg oral ADT for one week. Data represent mean ± SD of 4 animals per treatment; *indicates p<0.05 ADT vs. vehicle; **p<0.01 ADT vs. vehicle.
Figure 9.

Levels of plasma total Hcys in rats fed with a Met-enriched diet orally treated with ADT, folic acid (FA) or a combination of both daily for three weeks. Control rats were treated with vehicle (DMSO). Data represent mean ± SD of 4 animals per treatment; **indicates p<0.01 ADT vs. vehicle.
3.4 Effect of the thiol redox balance on the hepatic and renal metabolism of circulating Hcys
Liver and kidney are critical to the metabolism of both Hcys and GSH and these two organs demonstrated the largest response to treatment with ADT. Therefore, we used models of isolated perfused rat liver (IPL) and kidney (IPK) to investigate possible mechanisms by which ADT lowers circulating Hcys. Specifically, we tested whether the relative concentrations of thiols and disulfides in the extracellular fluids affect the hepatic and renal efficiency of elimination of a given amount of albumin-bound Hcys. Isolated livers or kidneys were studied with perfusate containing approximately half, equal or double LMM-SH concentrations as compared to physiological human concentrations. Specifically, the perfusate contained 25 μM homocysteinylated BSA (with 20 or 65 mg/mL of unthiolated BSA), variable amounts of Cys (5, 10, or 20 μM) and GSH (0.5, 1, or 2 μM), 25 μM Cyss and 25 μM GSSG (see further details in the 2.9 Section). Both perfused liver and kidney displayed elimination rates of Hcys that were greater at higher thiol/disulfide ratios (Fig 10, panels A and B). However, the levels of bile and urine tHcys did not change in response to variation of the thiol to disulfide ratio (Fig 10, panels A and B insets) and addition of ADT to the perfusate had no detectable effect on the bile, urine or perfusate levels of tHcys.
Figure 10.

Efficiency of elimination of tHcys in the isolated perfused rat liver (IPL; panel A) and isolated perfused rat kidney (IPK, panel B). The IPL perfusate included 20 mg/mL BSA containing 25 μM homocysteine bound by disulfide bond. The IPK perfusate consisted of 65 mg/mL BSA containing 25 μM homocysteine bound by disulfide bond. Both the IPL and IPK perfusate contained variable concentrations and relative ratios of Cys and GSH: low concentration, Cys 5 μM and GSH 0.5 μM (white circle), intermediate concentration, Cys 10 μM and GSH 1 μM (white triangle), high concentration, Cys 20 μM and GSH 2 μM (black circles). 25 μM of cystine and of GSSG occur in all perfusates. The downward pointing dark arrows represent perfusate with low concentration Cys/GSH plus 20 μM ADT. Insets: tHcys levels in bile and urine collected at 10-min intervals. Data represent the mean of 4 experiments per treatment. SD and statistical significance were omitted for some data points for graphical reasons. **indicates p<0.01 high vs. low and intermediate concentration Cys/GSH; ##p<0.01 intermediate vs. low concentration Cys/GSH.
4. Discussion
In the present study we show that ADT has dose-dependent Hcys-lowering action that is: (1) sustained during several days of administration, (2) applicable to a model of diet-induced hyperhomocysteinemia, (3) elicited by both parenteral and oral administration, and (4) synergistic with that of folic acid. In addition, we present evidence that (5) the Hcys-lowering effect of ADT associates with higher tissue levels of GSH and increased plasma thiol to disulfide ratio. Furthermore, since most circulating Hcys (up to 80%) is complexed with albumin by disulfide bond (Hcyssp) [12], these data have also led us to consider the possibility that the Hcys-lowering effect of ADT may be the consequence of a primary action to raise the tissue levels of GSH and/or its precursors. This effect on GSH, in turn, would promote a rightward shift of reaction (1) with formation of free Hcys.
| (1) |
Since free Hcys is more membrane-permeable than the albumin-Hcys complex, it can be more easily internalized and degraded by cells.
This hypothesis is supported by our studies in the rat IPL and IPK for which we used perfusate which mimicked plasma as to thiol and disulfide content and ratios (Fig 10). The data indicate that perfusate with higher thiol-disulfide ratio, i.e. a more reduced thiol redox balance, leads to faster degradation rates of Hcys by both liver and kidney. We hypothesize that a similar mechanism is at play in vivo: administration of ADT causes a rise in circulating LMM-SH in particular GSH and Cys, followed by change in redox balance, dissociation of albumin-Hcys complex, hepatic and renal internalization of free Hcys and, finally, intracellular processing of Hcys to Cys by the TSP (or to methionine by the Methyl Activated Cycle [MAC]) [14]. This hypothesis is also supported by our observation that ADT raises the concentrations of Cys and GSH in the kidneys and liver more than in other organs. In fact, increased hepatic and renal internalization of Hcys makes this amino acid more available for degradation to Cys. The latter, in turn, is the rate limiting amino acid for formation of γ-Glu-Cys (catalyzed by the γ-Glu-Cys synthase) and then for the synthesis of GSH. Since the kM of γ-Glu-Cys synthase for Cys (kM ~0.4 mM) [38] is almost one order of magnitude higher than the cell concentration of the substrate (80–300 μM), one can expect that the catalytic rate of γ-Glu-Cys formation will increase nearly linearly as the Cys concentration increases in kidney and liver, which are the major organs of GSH production.
A possible fate of Cys, γ-Glu-Cys and GSH in both kidney and liver is their transfer extracellularly into plasma thus increasing the plasma thiol-to-disulfide ratio, thereby promoting dissociation of Hcys from albumin and other proteins. The diagram in Figure 11 summarizes the proposed mechanism by which ADT could lower Hcys by activating a self-sustained cycle of Hcys release from complexed to more degradable forms and of utilization of Hcys for the synthesis of new GSH. Maintenance of low intracellular Hcys is probably critical for cell function and viability [39] and cells accomplish this by degrading Hcys via the TSP [14,40] and by exporting it via specialized transporters rather than involving it in the MAC [39]. Thus, intracellular Hcys levels are the product of a dynamic balance between formation, utilization and export of the amino acid, whereas the extracellular/plasma levels depend exclusively on import/export of cellular Hcys [41]. This said, it is remarkable that the extracellular fluid is the main repository of Hcys with an approximate amino acid concentration of 10 μM and a total volume of 10 L, compared with 0–3 μM in other tissues and organs. In plasma, the major fraction of Hcys is the mixed disulfide with albumin (up to 80%), a form that crosses cell membranes with difficulty [12]. Since urinary excretion of Hcys is negligible, metabolism is the only means for eliminating this amino acid. Liver and kidney are the organs with highest TSP enzymes activity and patients suffering from renal or liver failure have increased plasma levels of total Hcys. Hyperhomocysteinemia in these patients is not due to the insufficient biliary or urinary excretion but to inefficient metabolism by either kidney or liver tissue.
Figure 11.
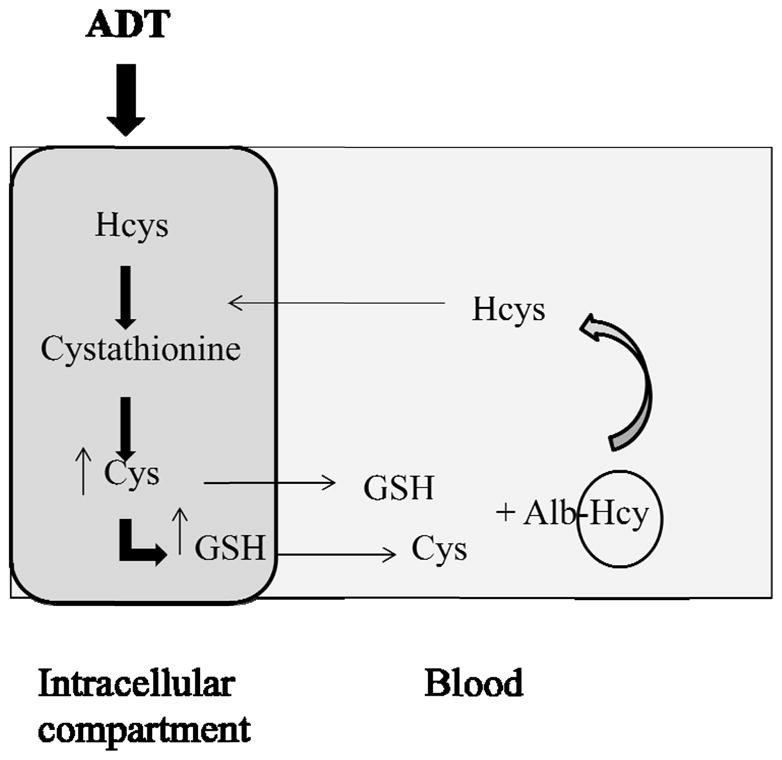
Proposed mechanism of the Hcys lowering effect of ADT. ADT may increase the intracellular levels GSH by enhancing the TSP pathway, by means of which Hcys is converted to Cys. In fact, Cys availability is rate-limiting for GSH synthesis. Consequently, an increased output from intracellular compartment to blood of both Cys and GSH can occur, leading to a rise of the thiol to disulfide ratio in plasma. Thiols can react with the protein-bound form of Hcys (Alb-Hcys in the scheme), favoring the delivery of free Hcys. Released Hcys, that is able to cross membranes, can be internalized and undergo further metabolic processing.
Despite prior evidence of pharmacological effects of ADT on GSH [for review, 17], the molecular mechanisms of this action remain undefined. For example, we could not confirm in this or in earlier work [16] the report of another laboratory about ADT-induced activation of several enzymes that are involved in GSH metabolism [42]. Specifically we show here that the GSH synthase and reductase and the γ-Glu-Cys synthase are not affected by ADT. Despite this, it is possible that ADT may affect GSH through other antioxidant enzymes. For example growing evidence supports that ADT is able to stimulate the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signal pathway, including activation of the antioxidant response element (ARE) which is present in the promoter region of a vast array of genes that codify antioxidant enzymes and it operates essentially as a master regulator of antioxidant cell functions [43]. Since ADT induces early transient generation of free radicals [16] and excess free radicals can directly activate Nrf2 [44], it is possible that ADT may modulate through Nrf2 other antioxidant enzymes besides those tested in the present study, such as haem oxygenase 1, peroxiredoxin, NAD(P)H:quinone oxidoreductase-1, superoxide dismutase 1. Furthermore, in cultured cells from the CNS, ADT inhibits very efficiently the monoamine oxidase B [45], which is suspected to contribute to oxidative stress and neurodegeneration. It remains undefined whether these findings may be applicable to other tissues and what the molecular mechanism(s) of this action might be, although it is known that activity of both monoamine oxidase A and B is affected by interaction with cysteine residues involved in the catalytic mechanism of the enzyme [46]. These possibilities need to be tested with in vitro and in vivo studies including, for example, investigation about the impact of ADT on intracellular GSH/GSSG ratio and the relation with Nrf2 processing and/or modulation of targeted specific enzymatic activities by protein thiolation/dethiolation.
Poorly understood are also the molecular mechanism(s) by which hyperhomocysteinemia results in tissue damage. An interesting possibility proposed by Loscalzo [7] is that elevation of plasma Hcys, even when mild, may adversely affect the endothelium possibly via oxidative damage. Based on this construct, transition metal enzymes would catalyze the oxidation of Hcys to the disulfide form which, in turn, would generate a variety of reactive oxygen species. In this context, administration of ADT, through a rise of GSH and a drop of Hcys, may synergistically prevent tissue damage from hyperhomocysteinemia.
Our findings about therapeutic effects of ADT in the rat may have clinical implications. First, ADT is a natural constituent of certain cruciferous vegetables [17,42] and therefore it is accessible by humans through the diet. Second, ADT is marketed in several countries as a choleretic and sialogogue drug, with a good record of safety and tolerability [17]. Thus, ADT may be applicable for the treatment of human conditions associated to hyperhomocysteinemia. This possibility is particularly well-timed since recent trials that tested prospectively other more publicized Hcys-reducing approaches in humans, including those with folic acid, vitamin B6 and B12 have all shown lack of effectiveness or paradoxical detrimental actions. For example, a recent multicenter clinical trial in patients with moderately advanced diabetic nephropathy found that combined treatment with pharmacological doses of folic acid, vitamin B6 and B12 lowers circulating Hcys by approximately 14%, but it also causes more rapid progression of renal disease and worse composite cardiovascular outcome as compared to placebo treatment [47]. It should be emphasized that besides their effect on Hcys, none of these B complex vitamins is known to have major effects on other thiols including GSH. ADT may therefore be superior to the B-complex vitamins specifically because of its positive effects on the thiols redox balance. Also, since ADT increases GSH primarily in the kidney of our rats (Fig. 4), it is can be inferred that this compound may be capable of protecting this organ from oxidative damage which, for example, is known to occur in diabetic nephropathy. The clinical significance of our animal studies, however, needs to be further demonstrated by human investigation that both confirms the Hcys-lowering effect of ADT and demonstrates long-term cardiovascular benefits.
In conclusion, our data indicate that ADT is able to reduce Hcys in normal and hyperhomocysteinemic animals and to concomitantly increase GSH in solid tissues and plasma. As a therapeutic agent, ADT is supposed to have both the ability to lower the Hcys levels and to protect from Hcys-induced oxidative damage. These pharmacological features position ADT as a promising Hcys-lowering drug with good potential for application in humans. This study can set the stage for future prospective clinical investigation aimed at verifying the therapeutic potential of ADT in humans.
Acknowledgments
This work was supported in part by grants to PF from NIH-NCCAM (#AT004490) and from the VA (Merit Review #1I01CX000264).
References
- 1.Wilcken DEL, Wilcken B. The pathogenesis of coronary artery disease: a possible role for methionine metabolism. J Clin Invest. 1976;57:1079–82. doi: 10.1172/JCI108350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becker A, Smulders YM, van Guldener C, Stehouwer CD. Epidemiology of homocysteine as a risk factor in diabetes. Metab Syndr Relat Disord. 2003;1:105–20. doi: 10.1089/154041903322294434. [DOI] [PubMed] [Google Scholar]
- 3.Vayá A, Carmona P, Badia N, Pérez R, Hernandez-Mijares A, Corella D. Homocysteine levels and the metabolic syndrome in a Mediterranean population: a case-control study. Clin Hemorheol Microcirc. 2011;47:59–66. doi: 10.3233/CH-2010-1366. [DOI] [PubMed] [Google Scholar]
- 4.Suliman ME, Qureshi AR, Barany P, Stenvinkel P, Filho JC, Anderstam B, Heimbürger O, Lindholm B, Bergström J. Hyperhomocysteinemia, nutritional status, and cardiovascular disease in hemodialysis patients. Kidney Int. 2000;57:1727–35. doi: 10.1046/j.1523-1755.2000.00018.x. [DOI] [PubMed] [Google Scholar]
- 5.Arnadottir M, Hultberg E. Homocysteine in renal disease. In: Carmel R, Jacobsen DW, editors. Homocysteine in health and disease. Cambridge, UK: Cambridge University Press; 2001. pp. 321–30. [Google Scholar]
- 6.Herrmann W, Herrmann M, Obeid R. Hyperhomocysteinemia: a critical review of old and new aspects. Curr Drug Metab. 2007;8:17–31. doi: 10.2174/138920007779315008. [DOI] [PubMed] [Google Scholar]
- 7.Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia. J Clin Invest. 1996;98:5–7. doi: 10.1172/JCI118776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guttormsen AB, Schneede J, Fiskerstrand T, Ueland PM, Refsum HM. Plasma concentrations of homocysteine and other aminothiol compounds are related to food intake in healthy human subjects. J Nutr. 1994;124:1934–41. doi: 10.1093/jn/124.10.1934. [DOI] [PubMed] [Google Scholar]
- 9.Stolzenberg-Solomon RZ, Miller ER, Maguire MG, Selhub J, Appel LJ. Association of dietary protein intake and coffee consumption with serum homocysteine concentrations in an older population. Am J Clin Nutr. 1999;69:467–75. doi: 10.1093/ajcn/69.3.467. [DOI] [PubMed] [Google Scholar]
- 10.Maron BA, Loscalzo J. The treatment of hyperhomocysteinemia. Annu Rev Med. 2009;60:39–54. doi: 10.1146/annurev.med.60.041807.123308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham JM, Cho L. The homocysteine hypothesis: still relevant to the prevention and treatment of cardiovascular disease? Cleve Clin J Med. 2010;77:911–18. doi: 10.3949/ccjm.77a.10036. [DOI] [PubMed] [Google Scholar]
- 12.Ueland PM. Homocysteine species as components of plasma redox thiol status. Clin Chem. 1995;41:340–2. [PubMed] [Google Scholar]
- 13.Urquhart BL, Freeman DJ, Spence JD, House AA. Mesna as a nonvitamin intervention to lower plasma total homocysteine concentration: implications for assessment of the homocysteine theory of atherosclerosis. J Clin Pharmacol. 2007;47:991–7. doi: 10.1177/0091270007303767. [DOI] [PubMed] [Google Scholar]
- 14.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–37. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 15.Friedman AN, Bostom AG, Laliberty P, Selhub J, Shemin D. The effect of N-acetylcysteine on plasma total homocysteine levels in hemodialysis: a randomized controlled study. Am J Kidney Dis. 2003;41:442–6. doi: 10.1053/ajkd.2003.50054. [DOI] [PubMed] [Google Scholar]
- 16.Giustarini D, Del Soldato P, Sparatore A, Rossi R. Modulation of thiol homeostasis induced by H2S-releasing aspirin. Free Radic Biol Med. 2010;48:1263–72. doi: 10.1016/j.freeradbiomed.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Christen MO. Anethole dithiolethione: biochemical considerations. Methods Enzymol. 1995;252:316–23. doi: 10.1016/0076-6879(95)52034-1. [DOI] [PubMed] [Google Scholar]
- 18.Ansher SS, Dolan P, Bueding E. Chemoprotective effects of two dithiolthiones and of butylhydroxyanisole against carbon tetrachloride and acetaminophen toxicity. Hepatology. 1983;3:932–5. doi: 10.1002/hep.1840030608. [DOI] [PubMed] [Google Scholar]
- 19.Chang L, Xu J, Yu F, Zhao J, Tang X, Tang C. Taurine protected myocardial mitochondria injury induced by hyperhomocysteinemia in rats. Amino Acids. 2004;27:37–48. doi: 10.1007/s00726-004-0096-2. [DOI] [PubMed] [Google Scholar]
- 20.Rossi R, Milzani A, Dalle-Donne I, Giannerini F, Giustarini D, Lusini L, Di Simplicio P. Different metabolizing ability of thiol reactants in human and rat blood: biochemical and pharmacological implications. J Biol Chem. 2001;276:7004–10. doi: 10.1074/jbc.M005156200. [DOI] [PubMed] [Google Scholar]
- 21.Giustarini D, Dalle-Donne I, Milzani A, Rossi R. Oxidative stress induces a reversible flux of cysteine from tissues to blood in vivo in the rat. FEBS J. 2009;276:4946–58. doi: 10.1111/j.1742-4658.2009.07197.x. [DOI] [PubMed] [Google Scholar]
- 22.Giustarini D, Dalle-Donne I, Milzani A, Rossi R. Low molecular mass thiols, disulfides and protein mixed disulfides in rat tissues: influence of sample manipulation, oxidative stress and ageing. Mech Ageing Dev. 2011;132:141–8. doi: 10.1016/j.mad.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Giustarini D, Dalle-Donne I, Milzani A, Fanti P, Rossi R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat Protoc. 2013;8:1660–9. doi: 10.1038/nprot.2013.095. [DOI] [PubMed] [Google Scholar]
- 24.Ellman G, Lysko H. A precise method for the determination of whole blood and plasma sulfhydryl groups. Anal Biochem. 1979;93:98–102. [PubMed] [Google Scholar]
- 25.Giustarini D, Dalle-Donne I, Lorenzini S, Selvi E, Colombo G, Milzani A, Fanti P, Rossi R. Protein thiolation index (PTI) as a biomarker of oxidative stress. Free Radic Biol Med. 2012;53:907–15. doi: 10.1016/j.freeradbiomed.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Svardal AM, Mansoor MA, Ueland PM. Determination of reduced, oxidized, and protein bound glutathione in human plasma with precolumn derivatization with monobromobimane and liquid chromatography. Anal Biochem. 1990;184:338–46. doi: 10.1016/0003-2697(90)90691-2. [DOI] [PubMed] [Google Scholar]
- 27.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–9. [PubMed] [Google Scholar]
- 28.Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–90. doi: 10.1016/s0076-6879(85)13062-4. [DOI] [PubMed] [Google Scholar]
- 29.Kraus JP. Cystathionine beta-synthase (human) Methods Enzymol. 1987;143:388–94. doi: 10.1016/0076-6879(87)43068-1. [DOI] [PubMed] [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 31.Di Iorio EE. Preparation of derivatives of ferrous and ferric hemoglobin. Methods Enzymol. 1981;76:57–72. doi: 10.1016/0076-6879(81)76114-7. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M, Moore PK. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–8. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- 33.Sparatore A, Perrino E, Tazzari V, Giustarini D, Rossi R, Rossoni G, Erdman K, Schröder H, Del Soldato P. Pharmacological profile of a novel H2S-releasing aspirin. Free Radic Biol Med. 2009;46:586–92. doi: 10.1016/j.freeradbiomed.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Mancinelli A, Evans AM, Nation RL, Longo A. Uptake of L-carnitine and its short-chain ester propionyl-L-carnitine in the isolated perfused rat liver. J Pharmacol Exp Ther. 2005;315:118–24. doi: 10.1124/jpet.105.087890. [DOI] [PubMed] [Google Scholar]
- 35.Giustarini D, Dalle-Donne I, Paccagnini E, Milzani A, Rossi R. Carboplatin induced alteration of the thiol homeostasis in the isolated perfused rat kidney. Arch Biochem Biophys. 2009;488:83–9. doi: 10.1016/j.abb.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 36.Fischer PA, Bogoliuk CB, Ramirez AJ, Sánchez RA, Masnatta LD. A new procedure for evaluation of renal function without urine collection in rat. Kidney Int. 2000;58:1336–41. doi: 10.1046/j.1523-1755.2000.00290.x. [DOI] [PubMed] [Google Scholar]
- 37.Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr. 2004;24:481–509. doi: 10.1146/annurev.nutr.24.012003.132208. [DOI] [PubMed] [Google Scholar]
- 38.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–35. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 39.Refsum H, Guttormsen AB, Fiskerstrand T, Ueland PM. Hyperhomocysteinemia in terms of steady-state kinetics. Eur J Pediatr. 1998;157 (Suppl 2):S45–9. doi: 10.1007/pl00014303. [DOI] [PubMed] [Google Scholar]
- 40.Kruger WD. The transsulfuration pathway. In: Carmel R, Jacobsen DW, editors. Homocysteine in health and disease. Cambridge, UK: Cambridge University Press; 2001. pp. 153–61. [Google Scholar]
- 41.Van Guldener C, Donker AJ, Jakobs C, Teerlink T, de Meer K, Stehouwer CD. No net renal extraction of homocysteine in fasting humans. Kidney Int. 1998;54:166–9. doi: 10.1046/j.1523-1755.1998.00983.x. [DOI] [PubMed] [Google Scholar]
- 42.Ansher SS, Dolan P, Bueding E. Biochemical effects of dithiolthiones. Food Chem Toxicol. 1986;24:405–5. doi: 10.1016/0278-6915(86)90205-x. [DOI] [PubMed] [Google Scholar]
- 43.Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171–84. doi: 10.1016/j.canlet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 44.Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–16. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 45.Drukarch B, Flier J, Jongenelen CA, Andringa G, Schoffelmeer AN. The antioxidant anethole dithiolethione inhibits monoamine oxidase-B but not monoamine oxidase A activity in extracts of cultured astrocytes. J Neural Transm. 2006;113:593–8. doi: 10.1007/s00702-005-0350-0. [DOI] [PubMed] [Google Scholar]
- 46.Hubalek F, Pohl J, Edmondson DE. Structural comparison of human monoamine oxidases A and B: mass spectrometry monitoring of cysteine reactivities. J Biol Chem. 2003;278:28612–8. doi: 10.1074/jbc.M303712200. [DOI] [PubMed] [Google Scholar]
- 47.House AA, Eliasziw M, Cattran DC, Churchill DN, Oliver MJ, Fine A, Dresser GK, Spence JD. Effect of B-vitamin therapy on progression of diabetic nephropathy: a randomized controlled trial. JAMA. 2010;303:1603–9. doi: 10.1001/jama.2010.490. [DOI] [PubMed] [Google Scholar]