

Abstract
Upregulation of cytokines and chemokines is a frequent finding in multiple myeloma (MM). CCL3 (also known as MIP-1α) is a pro-inflammatory chemokine whose levels in the MM microenvironment correlate with osteolytic lesions and tumor burden. CCL3 and its receptors, CCR1 and CCR5, contribute to the development of bone disease in MM by supporting tumor growth and regulating osteoclast (OC) differentiation. Here, we identify inhibition of osteoblast (OB) function as an additional pathogenic mechanism in CCL3-induced bone disease. MM-derived and exogenous CCL3 represses mineralization and osteocalcin production by primary human bone marrow stromal cells and HS27A cells. Our results suggest that CCL3 effects on OBs are mediated by ERK activation and subsequent downregulation of the osteogenic transcription factor osterix. CCR1 inhibition reduced ERK phosphorylation and restored both osterix and osteocalcin expression in the presence of CCL3. Finally, treating SCID-hu mice with a small molecule CCR1 inhibitor suggests an upregulation of osteocalcin expression along with OC downregulation. Our results show that CCL3, in addition to its known catabolic activity, reduces bone formation by inhibiting OB function and therefore contributes to OB/OC uncoupling in MM.
Keywords: Myeloma, CCL3, Osteoblast, Osteocalcin, ERK, Osterix
INTRODUCTION
More than 40% of multiple myeloma (MM) patients experience skeletal complications that jeopardize their quality of life and greatly increase the risk of death (1). Treatments preventing bone disease may therefore result in prolonged survival. However, effective therapies require the identification of the underlying pathogenic events. In MM, studies on bone biopsies identified a dual mechanism of bone destruction, consisting of osteoclast (OC) upregulation and inhibition of osteoblast (OB) activity (2).
OC activation in MM has been extensively studied and CCL3 is a critical player in this context. CCL3, or MIP-1α in the old nomenclature, is a pro-inflammatory cytokine belonging to the CC chemokine subfamily. CCL3 modulates OC differentiation by binding to G-protein coupled receptors, CCR1 and CCR5, and activating ERK and AKT signaling pathways. In the tumor niche, malignant plasma cells and OCs are the main sources for CCL3 that promotes MM cell migration and survival, along with stimulation of osteoclastogenesis (3, 4). Importantly, a clinical grade small molecule CCR1 antagonist, MLN3897, inhibits CCL3-induced osteoclastogenesis and OC support of MM cells (5).
OB impairment is a frequent finding in MM, but the underlying mechanism has yet to be fully clarified. OBs express CCL3 receptors and similar to other inflammatory cytokines, such as TNF-α and TWEAK, CCL3 may also inhibit OB in MM (6-8). However, no studies have addressed direct effects of CCL3 on osteoblastogenesis.
Here, we describe for the first time that CCL3 in addition to its known bone resorptive effects, may reduce bone formation by inhibiting OB function. Our data suggests that CCL3 impairs matrix mineralization and suppresses osteocalcin (OCN) production. In particular, CCL3 upregulates ERK activity and downregulates the osteogenic transcription factor osterix. Importantly, CCR1 inhibition via MLN3897 blocks ERK phosphorylation and restores, at least partially, OCN expression in vitro and in vivo. This data therefore suggests that the pro-inflammatory chemokine CCL3 may contribute to OB/OC uncoupling characteristic of MM bone disease, prompting the development of targeted strategies against CCL3.
MATERIALS AND METHODS
Reagents
CCL3 (R&D Systems, Inc., Minneapolis, MN) was used at concentrations of 50 ng/ml (unless otherwise specified), according to the manufacturer’s instructions. A neutralizing antibody against CCL3 (R&D Systems, Inc, Minneapolis, MN) was used at concentrations of 5 μg/ml. PD098059 (Sigma Aldrich, Saint Louis, MO), a specific inhibitor of the activation of mitogen activated ERK kinase (MEK) 1/2, was reconstituted in ethanol and stored at −20 C and used at a concentration of 30 μM. MLN3897, an orally active, small molecule specific antagonist of human CCR1, was kindly provided by Millennium Pharmaceuticals (Cambridge, MA) (5). In vivo stable drug levels were obtained by means of twice daily oral gavage with 2 mg/kg MLN3897 diluted in 0.5% methylcellulose and 0.2% tween 80. The peak plasma concentrations reached by this schedule of administration corresponded to 50 nM, subsequently used in the in vitro studies.
MM and BMSC cells
MM cell lines
Dexamethasone sensitive (MM.1S) and resistant (MM.1R) human MM cell line was kindly provided by Dr Steven Rosen (Northwestern University, Chicago, IL) (9). INA-6 human IL-6–dependent MM cell line was provided by Dr. Renate Burger (10) (University of Kiel, Kiel, Germany) and cultured in the presence of 2.5 ng/mL IL-6 (R&D Systems, Inc. Minneapolis, MN). RPMI cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD). All MM cell lines were cultured in RPMI 1640 media (Sigma Chemical, Saint Louis, MO) containing 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Grand Island, NY).
BMSC
We generated human BMSCs from MM patients by placing bone marrow mononuclear cells in 75-mm2 culture flasks in RPMI 1640 media (Sigma Chemical, Saint Louis, MO) containing 20% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Grand Island, NY). Once confluent, the cells were trypsinized and passaged as needed. Only cells between the second and the fourth passage were used. We also used the BMSC cell line HS27A, obtained from the ATCC (Rockville, MD) and cultured in 10% RPMI 1640 media.
OB differentiation
We cultured BMSC or HS27A cells (5000 cells/well) in the presence of osteoblastogenic media as previously described (11).
Patient samples
We studied bone marrow (BM) plasma from 22 MM patients at diagnosis for CCL3 and OCN expression by ELISA. All patients provided written informed consent per the Declaration of Helsinki and approval was obtained by the institutional review board of the Massachusetts General Hospital Cancer Center (Boston, MA).
ELISA
CCL3 and osteopontin (OPN) expression was analyzed by ELISA assays purchased from R&D Systems (Minneapolis, MN) and OCN levels were assessed using the N-MID osteocalcin ELISA kit (Immunodiagnostic Systems Inc, Fountain Hills, AZ). Supernatants of MM cell lines and mature OBs, seeded in 10% FBS RPMI 1640 or 20% FBS α-MEM, respectively, for 48 or 72 hours were assayed. All measurements were performed in duplicates.
Flow cytometry
For evaluation of surface receptor expression, cells were suspended in Dulbecco’s phosphate-buffered saline and incubated for 30 minutes at 4°C with the primary antibody anti-CCR1 and anti-CCR5 (phycoerythrin-conjugated, R&D Systems, Inc. Minneapolis, MN). Data was acquired using the CellQuest software on a BD FACS Calibur (BD Biosciences, California, USA) and analyzed with FlowJo software. Results are expressed as fold change of mean fluorescence intensity compared to isotype control.
Real-time quantitative PCR
Gene expression was analyzed by quantitative-PCR on RNA extracted from OBs as previously described. (11) Primers for ALPL (alkaline phosphatase), COL1A1 (collagen type 1 alpha 1), IBSP (bone sialoprotein), Bglap1 (osteocalcin), ATF4, RUNX2, SP7 (osterix) were obtained from SuperArray (Frederick, MD). Transcript levels were normalized to β-actin.
Immunofluorescence and immunohistochemistry
OCN protein expression was assessed by immunofluorescence (IF) and immunohistochemistry (IHC) after incubating the samples overnight with 1:80 primary antibody clone OC4-30 (Abcam, Cambridge, MA). IF was performed on OB differentiated on culture slides (BD Biosciences San Jose, California) for 2 weeks in the presence or absence of CCL3, media was changed twice weekly. Cells were then fixed, stained with primary antibody and secondary anti-rabbit Alexa Fluor 488 conjugated antibody. Counterstain was performed with DAPI. IHC was performed on paraffin-embedded sections from human fetal bones. Antigen retrieval consisted of incubation with proteinase K (ROCHE, Indianapolis, IN). After incubation with the primary antibody, slides were probed with biotinylated secondary antibody (1:300) and signal detected with the Streptavidin-HRP system provided by the TSA kit (Perkin Elmer-Life Sciences, MA, USA), according to the manufacturer's guidelines. Immunostained areas were estimated by determining a threshold on the basis of two positive control images and the threshold was used to analyze all the IHC images (12). The values were then corrected per trabecular bone area.
Western blotting
HS-27A differentiated for 2 weeks with osteogenic media were stimulated with CCL3 in the absence or presence of MLN3897 or PD098059 and lysed in lysis buffer as previously described (5). Samples were then subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis, transferred to PVDF membrane, and immunoblotted with antibodies against pERK, ERK, pAKT, pP38, pβcatenin and βcatenin (Cell Signaling Technology, Beverly, MA). Antigen-antibody complexes were detected by enhanced chemiluminescence (Amersham, Arlington Heights, IL). The membrane was stripped and reprobed with anti-actin antibody to ensure equal protein loading.
In vivo mouse model
All animal studies were conducted according to protocols approved by the Institutional Animal Care and Use Committee of Dana Farber Cancer Institute (Boston, MA). The SCID-hu mouse model was generated as previously described (13). We treated 6 mice with oral MLN3897 (2 mg/kg twice daily for 6 days/week) and 5 mice with vehicle for four weeks. Tumor cell growth was monitored with weekly ELISA for soluble human IL6 Receptor (sHuIL6R) (R&D Systems, Minneapolis, MN) in blood sera. At week 3 of treatment three mice were sacrificed (2 in the treatment and 1 in the control group) because of increase in fetal bone graft size. Since our primary endpoint was bone analysis, the remaining eight mice were sacrificed at end of the treatment (week 4) and bone grafts harvested. After 72 hours fixation in 4% paraphormaldehyde, the bones were decalcified in 20% EDTA for 3 weeks and paraffin-embebbed. Cross-sections of each bone at different levels were performed and all bones analyzed for bone area, tumor invasion, OC and OB number. Trabecular bone area and tumor area as proportion of total tissue area were quantified on H&E stained slides using a Nikon Eclipse 80i microscope and Image J analysis software (14). OCs were identified after TRAP staining and hematoxylin counterstaining and expressed as number per mm2 of trabecular bone. OBs were counted on H&E stained sections as bone-lining cells, active OBs and osteocytes per mm2 of trabecular bone.
Statistical analysis
Each experiment was performed in quadruplicate and results have been expressed as mean ± SD of at least three different experiments. Statistical comparisons of continuous variables were performed by Student’s two-tailed t test. Association between OCN and CCL3 levels in BM plasma from MM patients was assessed by Pearson correlation analysis.
RESULTS
MM-DERIVED PRIMARY OSTEOBLASTS AND HS27A STROMAL CELL LINE EXPRESS CCL3 AND ITS RECEPTORS, CCR1 and CCR5
We first evaluated ligand and receptor expression levels in MM patient-derived OBs at different stages of maturation. In vitro osteogenesis consists of three principal periods of differentiation: i) mesenchymal stem cells proliferation during the first week of OB differentiation, that we defined as immature OBs, ii) extracellular matrix maturation during the second week of differentiation, that we defined as mature OBs, and iii) finally the third week of mineralization (15). As shown in figure 1A, the average levels of CCL3 detected in 72h-culture supernatant of mature MM-derived OBs ranged between 1 and 231 pg/ml (average 103.63 pg/ml). CCL3 production was 8 to 10 times lower compared to MM cell lines (801.2 pg/ml ± 407.7) that represent the main source of CCL3 in the tumor niche (5, 16). To evaluate the potential effect of CCL3 on OB development and function, we assessed the receptor expression levels both by RNA and protein levels. Mature OB express higher levels of CCR1 and CCR5 mRNA compared to immature OB (2.5± 1.75 vs 1.52± 0.8 fold increase, Figure S1). Similarly, assessment by flow cytometry suggested that osteogenic differentiation upregulated both receptors with a 2.1 ± 0.81 and 1.7 ± 0.38 fold increase in MFI, respectively, compared to isotype control at day 14 (Figure 1B) (7). We also characterized the CCL3 pathway in a human stromal cell line, HS-27A that recapitulates the features of primary human BMSC. HS-27A cells show preOB features (17, 18) and, in the presence of osteogenic media, differentiate to ALP positive cells that mineralize in sheets rather than nodules (Figure S2). CCL3 secretion was upregulated with OB differentiation and, similar to patient-derived OBs, the average CCL3 production by mature HS27A-OB was 112 ± 48 pg/ml (Figure 1C). We also observed that CCR1 and CCR5 expression in HS27A cells is increased by osteogenic differentiation (1.82 ± 1.28 and 1.64± 0.96 fold increase in MFI compared to isotype control, Figure 1D).
Figure 1. Differentiated osteoblasts secrete CCL3 and express its receptors.

(A) ELISA detection of CCL3 in 72h-culture supernatant from several myeloma (MM) cell lines and patient-derived osteoblasts (OB). (B) (upper panel) Fold increase of CCR1 and CCR5 surface expression during OB differentiation assessed by flow cytometry. (Lower panel) Representative flow cytometric analysis of receptor expression on OB at day 7 and day 14 of differentiation compared to isotype control. (C) CCL3 expression levels from culture supernatant of HS-27A cells during OB differentiation and (D) CCR1 and CCR5 surface expression detected by flow cytometry.
CCL3 INHIBITS OB MINERALIZATION ACTIVITY
The known upregulation of CCL3 in the MM niche (16) and the expression of the receptors by OBs prompted us to evaluate its effects on OB development and activity. Using patient derived cells we observed that CCL3 (50 ng/ml) did not affect alkaline phosphatase protein (ALP) activity, a well established marker for OB differentiation (Figure 2A). On the contrary, matrix mineralization was significantly impaired in OB cultures continuously exposed to CCL3, as shown by a 50% decrease in calcium deposition compared to untreated OBs (Figure 2B, p=0.01). This data suggests that CCL3 preferentially impairs OB function rather than OB differentiation of MM-derived BMSC. Similarly, treating mature receptor-positive HS27A-OB with increasing concentrations of CCL3 for one week inhibited mineralization to basal control levels (Figure 2C), suggesting that late exposure to CCL3 inhibits mineralization and OCN expression as continuous treatment. As shown in figure 1A, MM cell lines produce high amounts of CCL3. To confirm the relevance of this chemokine in MM-induced OB impairment, we exposed mature HS27A-OB to MM cell culture supernatant in the presence or absence of neutralizing CCL3 antibodies. The impaired mineralization observed in the presence of MM supernatant was reversed by treatment with the neutralizing antibody against CCL3 (Figure 2D), suggesting that CCL3 may mediate MM inhibition of OB activity.
Figure 2. CCL3 inhibits OB mineralization.
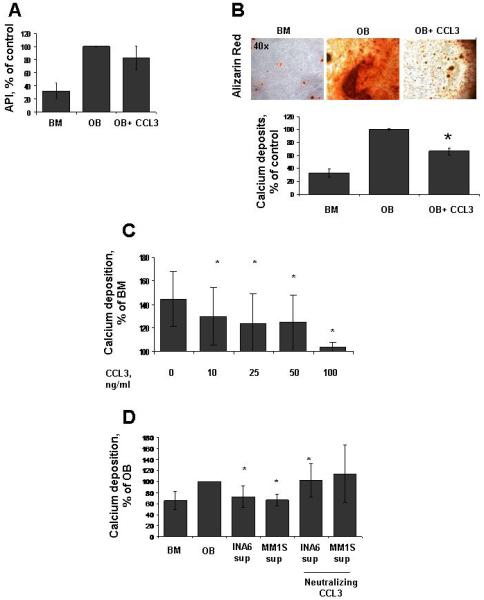
(A) The ratio of alkaline phosphatase activity relative to the amount of viable cells (API) in 2-week differentiated OBs with or without CCL3 50 ng/ml. (B) (upper panel) Representative image of alizarin red staining to detect mineralization from OB exposed to CCL3 for 3 weeks. (Lower panel) Quantification of calcium deposition in OB differentiated with or without CCL3 50 ng/ml for 3 weeks. (C) Alizarin Red assessment of HS27A-derived mature OB treated for one week with CCL3 from 10 to 100 ng/ml. (D) Quantification of mineralization via alizarin red in HS27A-derived mature OB exposed to INA6 and MM.1S cell supernatant for one week with or without neutralizing antibody against CCL3 (*, p < 0.05).
CCL3 DOWNREGULATES OSTEOCALCIN EXPRESSION VIA ERK AND OSTERIX MODULATION
OB function is a complex process consisting mainly of matrix formation and mineralization, and requiring the concerted expression of bone-specific proteins. OCN in particular is the most abundant noncollagen protein in the bone matrix and stimulates bone mineral maturation by stimulating the growth of apatite crystals (19, 20). CCL3 treatment downregulated both RNA and protein levels of OCN by 40% and 23%, respectively (Figure 3A, p<0.05). In contrast, collagen type I alpha I, ALP, and bone sialoprotein gene expression as well as osteopontin protein levels were not affected by exposure to CCL3 (Figure S3). CCL3 BM plasma levels are increased in MM patients with extensive bone disease (21). Notably, we observed in patients with lytic lesions (n=14) a borderline significant, negative correlation between BM plasma CCL3 and OCN expression (Figure S4, r=−0.53; p=0.05).
Figure 3. CCL3 downregulates osteocalcin expression via ERK and osterix modulation.

(A) Osteocalcin (OCN) expression in mature MM-derived OBs, with or without CCL3 (50 ng/ml), detected via (left panel) quantitative PCR analysis, (middle panel) ELISA on culture supernatant, and (right panel) immunofluorescent detection of OB cultures (OCN is stained with Alexa Fluo 488, counterstain with DAPI). (B) Western blot detection of pERK, pAKT, pP38, ERK1/2 on HS27A-derived OB exposed to CCL3 50 ng/ml for the specified time points. (C) Western blot detection of pERK and ERK1/2 in HS27A-derived OB after dose-dependent exposure to CCL3. A densitometric analysis averaging more than 3 experiments is shown. (D) Osterix gene expression assessed by quantitative PCR in OB exposed to increasing time and doses of CCL3. (E) RUNX2 and ATF4 gene expression evaluation on mature OBs in the presence of CCL3 (4h, 50 ng/ml) (* p<0.05, ** p<0.01)
CCL3/CCR interactions activate several downstream effectors, including the MAPK and AKT signaling pathways (4). Western blot analysis of these effectors was used to determine their contribution to CCL3 inhibitory effects on OB function. We observed early upregulation of phospho-ERK in mature HS27A-OBs, associated with P38 and AKT signaling activation at later time-points (Figure 3B). No changes in βcatenin and phospho-βcatenin were observed (Figure S5A). Upon time and dose-dependent exposure to CCL3, we noted increase in phospho-ERK expression as early as 30’ and starting from 10 ng/ml of CCL3. Overall a 2-fold upregulation after 4 hour treatment with 50 ng/ml of CCL3 was observed (p<0.01, Figure 3C). Importantly, undifferentiated HS27A did not show any ERK pathway activation by CCL3 treatment, providing further evidence that CCL3 targets mainly mature OB (Figure S5B). Downstream of ERK signaling pathway a cascade of transcription factors critical to OB function have been identified. Gene expression analysis of osteoblastic transcription factors in CCL3-treated HS27A-OBs revealed a time and dose-dependent osterix downregulation, with a 2-fold decrease after 4 hours treatment with 50 ng/ml of CCL3 (p=0.01, figure 3D). On the contrary, exposure to CCL3 did not significantly alter the expression levels of RUNX2 or ATF4 (Figure 3E).
CCR1 INHIBITION AT LEAST PARTIALLY REVERSES CCL3 EFFECTS IN VITRO AND IN VIVO
To provide additional evidence for the inhibitory effects of the CCL3/CCR1 pathway on OB function, we blocked CCR1 using the small molecule human CCR1 antagonist, MLN3897, previously studied for its anti-OC effects (5). Pretreatment of HS27A-OB with the CCR1 inhibitor at 50 nM suppressed CCL3-induced ERK activation (Figure 4A). In addition, osterix transcription inhibited by CCL3 was at least partially restored by pretreatment with the CCR1 inhibitor (Figure 4B). Similarly, pretreatment of HS27A-OB with the MEK1 inhibitor, PD098059 (Sigma Aldrich, St. Louis, MO) rescued osterix gene downregulation in the presence of CCL3 (from 0.29 ± 0.27 to 1.72 ± 1.5 in PD098059 treated cells) (Figure 4C). Finally, OCN expression was increased by MLN3897 in the presence of CCL3 (from 0.39 ± 0.19 to 3.4 ±2.6 in the presence of MLN3897) (Figure 4D). This data suggests that by modulation of the ERK signaling pathway CCL3 inhibits OB function via osterix inhibition and downregulation of OCN expression. CCR1 is at least partially responsible for these effects, although the role of other receptors such as CCR5 should not be excluded.
Figure 4. CCR1 mediates at least partially CCL3 inhibitory effects on OBs.

(A) Western blot detection of pERK and ERK signaling in HS27A-derived OB treated with CCL3 in the presence of a small molecule CCR1 inhibitor (50 nM). (B) Osterix gene expression by quantitative PCR in OB treated for 4 hours with CCL3 with or without the CCR1 inhibitor (50 nM) (left panel) or MEK1 ihibitor, PD098059 (30 μM, 1h pretreatment) (right panel). (C) Osteocalcin gene expression by quantitative PCR in OB treated for 24 hours with CCL3 with or without the CCR1 inhibitor (50 nM) (* p<0.05).
We then investigated the effects of CCR1 inhibition on tumor burden and bone structure in an in-vivo setting using a humanized model of MM bone disease. 11 mice subcutaneously implanted with fetal human bone chips and injected with INA6 MM cells, were treated with MLN3897 or vehicle. Analysis of tumor burden during treatment by serial quantification of sHuIL6R revealed delayed tumor growth in the treated group (Figure 5A, left panel). No weight loss or other toxicities were observed. Importantly, the tumor infiltration of the bone specimens decreased from 40.6% in the control to 26% in the treated mice confirming a reduction in MM burden (Figure 5A, right panel). Treatment with the CCR1 inhibitor induced a 2-fold increase in trabecular bone area per tissue area (15.9 ±5.59 vs 27±14.9) (Figure 5B) and a 1.5 fold decrease in OC number per trabecular bone area compared to the control group (46.2 ±26/mm2 vs 29±12/mm2) (Figure 5C), confirming our previous in vitro findings on the important role of CCR1 in osteoclastogenesis (5). We then evaluated the effects of CCR1 inhibition on the OB compartment of the tumor milieu by assessing OB number and activity via detection of OCN in the trabecular bone. No differences in the number of OB per trabecular bone area were observed (575± 94.4/mm2 in the control group vs 506± 62.1/mm2 in the treated group) (Figure 5D, left panel). However, the ratio of the OCN positive areas per trabecular bone area was increased in the MLN3897 treated animals (0.26 ±0.28 vs 0.53±0.34) (Figure 5D, middle and right panel). These results suggest that CCR1 inhibition may impact OB activity indirectly via OC inhibition. Furthermore, based on our in vitro data CCR1 inhibition also directly affects OBs via impairment of CCL3 signaling. Future studies will confirm these effects with larger sample size.
Figure 5. In vivo CCR1 inhibition in a MM mouse model.

(A) (Left panel) Weekly assessment of tumor burden via soluble human IL6 receptor (shuIL6R) levels during treatment. Values are expressed as fold increase. (Right panel) Tumor invasion was also assessed and expressed as % of tissue area (B) (Left panel) Representative image of H&E staining on MM-injected bone chips from control and treated mice. (Right panel) Bone area was counted and represented as % of total tissue area. (C) (Left panel) Representative image of immunocytochemistry for TRAP activity on MM-injected bone chips from control and treated mice. (Right panel) The number of TRAP+ cells per bone area was counted. (D) (Left panel) OB number per bone area was assessed on both control and treated bones. (Middle panel) Representative image of immunohistochemical detection of OCN on the MM-injected bone chips. (Right panel) The percentage of OCN positive area per bone area was analyzed.
DISCUSSION
Understanding the cellular interactions within the tumor-bone milieu is critical to the development of novel agents simultaneously targeting myeloma cells and reversing bone disease. The proinflammatory chemokine CCL3 plays a critical role in tumor development and disruption of bone homeostasis via OC stimulation. Here, we demonstrate for the first time that CCL3 may contribute to the OB/OC uncoupling not only by promoting OC activity but also by inhibiting OB activity. We investigated the CCR/CCL3 pathway in OBs and observed that exogenous and tumor-derived CCL3 inhibits OB mineralization and OCN expression. Our data shows that modulation of ERK signaling and osterix expression mediate CCL3 effects, and CCR1 inhibition may reverse at least partially these effects.
Both receptors for CCL3 have been identified on BMSC and OB surface, although expression levels vary according to cell type and species (7, 22, 23). Here, we observed upregulation of both CCR1 and CCR5 during osteogenesis from primary cells and the human stromal cell line, HS27A. The receptor expression observed on mature cells may account for the preferential inhibition of OB function rather than differentiation; explaining also the different effects on ALP, an early OB gene, and OCN, produced typically by mature cells. Notably, specific CCR1 inhibition only partially reverses CCL3 effects, while neutralizing antibody against CCL3 completely restores OB activity, suggesting that CCR5 may also play a key role in mediating CCL3 effects on OBs. Targeted receptor inhibition will be used to address this question in future studies.
Inhibition of OB is a frequent finding in bone biopsies of MM patients and correlates with tumor burden (2). MM cells block OB differentiation by means of contact-mediated RUNX2 inhibition (24), tumor cell secretion of β-catenin antagonists such as DKK1 (25) and tumor induced production of OB inhibitors by BMSC, such as activin A (11). As observed with other inflammatory cytokines such as TWEAK and TNFα, here we observed that tumor-derived CCL3 downregulates OB mineralization activity, at least partially, via OCN inhibition (8, 26).
Myeloma patients with extensive bone disease have high serum levels of CCL3 (21). In addition, high expression levels of the cytokine have been identified on tumor plasma cells (27), correlating with extent of osteolysis. Interestingly, we observed a negative correlation between CCL3 and OCN levels in patients with high osteolytic burden. Although increased tumor burden and OC activity decreased OCN levels via OB inhibition, this data may also reflect a direct negative regulation of OCN by CCL3. Therefore, CCL3 contribution to development of osteolytic lesions relies on a dual effect on OC and OB. In addition, our in vitro and in vivo data suggests that CCL3 pathway inhibition restores OB function and therefore targeted therapies against CCL3 may be of benefit by reversing the OB/OC uncoupling typical of MM.
The MAPK signaling pathway represents an important modulator of OB differentiation: the concerted activation and inhibition of ERK, p38 and JNK regulate the expression of several osteogenic transcription factors (28). In particular, the ERK pathway exerts opposite effects on OBs according to the differentiation stage. Continuous inhibition of ERK signaling promotes OB development, whereas activation during late stages of differentiation inhibits osteogenesis. (28-31). Our data suggests that exposing differentiated OBs to CCL3 activates ERK signaling, which in turn downregulates osterix expression, as previously observed with TNF-α (26). Interestingly, one of the mechanisms of OB inhibition by proinflammmatory cytokines is sclerostin upregulation (8), whose role in mediating CCL3 effects on OBs will be assessed in future studies. CCL3 has been mainly considered as a pro-osteoclastogenic cytokine promoting catabolic activity in the pathogenesis of bone disease. Our results identify a novel effect of CCL3, as an inhibitor of OB function and a mediator of OB/OC uncoupling in MM. Therefore, we provide further evidence for the important pathogenic role played by CCL3 and its receptors in MM and for the need to assess targeted inhibitors of this pathway in upcoming clinical trials.
Supplementary Material
ACKNOWLEDGEMENTS
Grant support: IMF junior award (SV, SP, LS), ASCO CDA, MMRF, LLS CDA, NIH P50 CA 100707-06 (NR).
MLN3897 was kindly provided by Millennium Pharmaceuticals, Cambridge MA.
Footnotes
DISCLOSURE OF CONFLICT OF INTEREST
The author NR is consultant for Celgene and Novartis, member of the advisory committee for Celgene and Amgen, and she received research funding from Astra Zeneca and Acetylon. P.V. is an employee of Millennium Pharmaceuticals.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
REFERENCES
- 1.Saad F, Lipton A, Cook R, Chen YM, Smith M, Coleman R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer. 2007 Oct 15;110(8):1860–1867. doi: 10.1002/cncr.22991. [DOI] [PubMed] [Google Scholar]
- 2.Bataille R, Chappard D, Marcelli C, Dessauw P, Sany J, Baldet P, et al. Mechanisms of bone destruction in multiple myeloma: the importance of an unbalanced process in determining the severity of lytic bone disease. J Clin Oncol. 1989 Dec;7(12):1909–1914. doi: 10.1200/JCO.1989.7.12.1909. [DOI] [PubMed] [Google Scholar]
- 3.Han JH, Choi SJ, Kurihara N, Koide M, Oba Y, Roodman GD. Macrophage inflammatory protein-1alpha is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor kappaB ligand. Blood. 2001 Jun 1;97(11):3349–3353. doi: 10.1182/blood.v97.11.3349. [DOI] [PubMed] [Google Scholar]
- 4.Lentzsch S, Gries M, Janz M, Bargou R, Dorken B, Mapara MY. Macrophage inflammatory protein 1-alpha (MIP-1 alpha ) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood. 2003 May 1;101(9):3568–3573. doi: 10.1182/blood-2002-08-2383. [DOI] [PubMed] [Google Scholar]
- 5.Vallet S, Raje N, Ishitsuka K, Hideshima T, Podar K, Chhetri S, et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007 Nov 15;110(10):3744–3752. doi: 10.1182/blood-2007-05-093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbas S, Zhang YH, Clohisy JC, Abu-Amer Y. Tumor necrosis factor-alpha inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine. 2003 Apr;22(1-2):33–41. doi: 10.1016/s1043-4666(03)00106-6. [DOI] [PubMed] [Google Scholar]
- 7.Yano S, Mentaverri R, Kanuparthi D, Bandyopadhyay S, Rivera A, Brown EM, et al. Functional expression of beta-chemokine receptors in osteoblasts: role of regulated upon activation, normal T cell expressed and secreted (RANTES) in osteoblasts and regulation of its secretion by osteoblasts and osteoclasts. Endocrinology. 2005 May;146(5):2324–2335. doi: 10.1210/en.2005-0065. [DOI] [PubMed] [Google Scholar]
- 8.Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, et al. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res. 2009 Aug;24(8):1434–1449. doi: 10.1359/jbmr.090305. [DOI] [PubMed] [Google Scholar]
- 9.Greenstein S, Krett NL, Kurosawa Y, Ma C, Chauhan D, Hideshima T, et al. Characterization of the MM.1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol. 2003 Apr;31(4):271–282. doi: 10.1016/s0301-472x(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 10.Burger R, Guenther A, Bakker F, Schmalzing M, Bernand S, Baum W, et al. Gp130 and ras mediated signaling in human plasma cell line INA-6: a cytokine-regulated tumor model for plasmacytoma. Hematol J. 2001;2(1):42–53. doi: 10.1038/sj.thj.6200075. [DOI] [PubMed] [Google Scholar]
- 11.Vallet S, Mukherjee S, Vaghela N, Hideshima T, Fulciniti M, Pozzi S, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proceedings of the National Academy of Sciences of the United States of America. Mar 16;107(11):5124–5129. doi: 10.1073/pnas.0911929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derkx P, Nigg AL, Bosman FT, Birkenhager-Frenkel DH, Houtsmuller AB, Pols HA, et al. Immunolocalization and quantification of noncollagenous bone matrix proteins in methylmethacrylate-embedded adult human bone in combination with histomorphometry. Bone. 1998 Apr;22(4):367–373. doi: 10.1016/s8756-3282(97)00299-8. [DOI] [PubMed] [Google Scholar]
- 13.Tassone P, Neri P, Carrasco DR, Burger R, Goldmacher VS, Fram R, et al. A clinically relevant SCID-hu in vivo model of human multiple myeloma. Blood. 2005 Jul 15;106(2):713–716. doi: 10.1182/blood-2005-01-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasband WS. In: ImageJ. U. S. National Institutes of Health B; Maryland U, editor. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997-2006. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 15.Stein GS, Lian JB, Owen TA. Relationship of cell growth to the regulation of tissue-specific gene expression during osteoblast differentiation. Faseb J. 1990 Oct;4(13):3111–3123. doi: 10.1096/fasebj.4.13.2210157. [DOI] [PubMed] [Google Scholar]
- 16.Uneda S, Hata H, Matsuno F, Harada N, Mitsuya Y, Kawano F, et al. Macrophage inflammatory protein-1 alpha is produced by human multiple myeloma (MM) cells and its expression correlates with bone lesions in patients with MM. British journal of haematology. 2003 Jan;120(1):53–55. doi: 10.1046/j.1365-2141.2003.04040.x. [DOI] [PubMed] [Google Scholar]
- 17.Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995 Feb 15;85(4):997–1005. [PubMed] [Google Scholar]
- 18.Graf L, Iwata M, Torok-Storb B. Gene expression profiling of the functionally distinct human bone marrow stromal cell lines HS-5 and HS-27a. Blood. 2002 Aug 15;100(4):1509–1511. doi: 10.1182/blood-2002-03-0844. [DOI] [PubMed] [Google Scholar]
- 19.Boskey AL, Gadaleta S, Gundberg C, Doty SB, Ducy P, Karsenty G. Fourier transform infrared microspectroscopic analysis of bones of osteocalcin-deficient mice provides insight into the function of osteocalcin. Bone. 1998 Sep;23(3):187–196. doi: 10.1016/s8756-3282(98)00092-1. [DOI] [PubMed] [Google Scholar]
- 20.Kavukcuoglu NB, Patterson-Buckendahl P, Mann AB. Effect of osteocalcin deficiency on the nanomechanics and chemistry of mouse bones. Journal of the mechanical behavior of biomedical materials. 2009 Aug;2(4):348–354. doi: 10.1016/j.jmbbm.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Terpos E, Politou M, Szydlo R, Goldman JM, Apperley JF, Rahemtulla A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. British journal of haematology. 2003 Oct;123(1):106–109. doi: 10.1046/j.1365-2141.2003.04561.x. [DOI] [PubMed] [Google Scholar]
- 22.Sordi V, Malosio ML, Marchesi F, Mercalli A, Melzi R, Giordano T, et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood. 2005 Jul 15;106(2):419–427. doi: 10.1182/blood-2004-09-3507. [DOI] [PubMed] [Google Scholar]
- 23.Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM, Silberstein LE. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem cells (Dayton, Ohio) 2006 Apr;24(4):1030–1041. doi: 10.1634/stemcells.2005-0319. [DOI] [PubMed] [Google Scholar]
- 24.Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, et al. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005 Oct 1;106(7):2472–2483. doi: 10.1182/blood-2004-12-4986. [DOI] [PubMed] [Google Scholar]
- 25.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. The New England journal of medicine. 2003 Dec 25;349(26):2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 26.Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. The Journal of biological chemistry. 2006 Mar 10;281(10):6297–6306. doi: 10.1074/jbc.M507804200. [DOI] [PubMed] [Google Scholar]
- 27.Roussou M, Tasidou A, Dimopoulos MA, Kastritis E, Migkou M, Christoulas D, et al. Increased expression of macrophage inflammatory protein-1alpha on trephine biopsies correlates with extensive bone disease, increased angiogenesis and advanced stage in newly diagnosed patients with multiple myeloma. Leukemia. 2009 Nov;23(11):2177–2181. doi: 10.1038/leu.2009.130. [DOI] [PubMed] [Google Scholar]
- 28.Hipskind RA, Bilbe G. MAP kinase signaling cascades and gene expression in osteoblasts. Front Biosci. 1998;3:d804–816. doi: 10.2741/a323. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, et al. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res. 2002 Oct;17(10):1785–1794. doi: 10.1359/jbmr.2002.17.10.1785. [DOI] [PubMed] [Google Scholar]
- 30.Kono SJ, Oshima Y, Hoshi K, Bonewald LF, Oda H, Nakamura K, et al. Erk pathways negatively regulate matrix mineralization. Bone. 2007 Jan;40(1):68–74. doi: 10.1016/j.bone.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Raucci A, Bellosta P, Grassi R, Basilico C, Mansukhani A. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. Journal of cellular physiology. 2008 May;215(2):442–451. doi: 10.1002/jcp.21323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.