

Abstract
Many normally cytosolic yeast proteins form insoluble intracellular bodies in response to nutrient depletion, suggesting the potential for widespread protein aggregation in stressed cells. Nearly 200 such bodies have been found in yeast by screening libraries of fluorescently tagged proteins. In order to more broadly characterize the formation of these bodies in response to stress, we employed a proteome-wide shotgun mass spectrometry assay in order to measure shifts in the intracellular solubilities of endogenous proteins following heat stress. As quantified by mass spectrometry, heat stress tended to shift the same proteins into insoluble form as did nutrient depletion; many of these proteins were also known to form foci in response to arsenic stress. Affinity purification of several foci-forming proteins showed enrichment for co-purifying chaperones, including Hsp90 chaperones. Tests of induction conditions and co-localization of metabolic enzymes participating in the same metabolic pathways suggested those foci did not correspond to multi-enzyme organizing centers. Thus, in yeast, the formation of stress bodies appears common across diverse, normally diffuse cytoplasmic proteins and is induced by multiple types of cell stress, including thermal, chemical, and nutrient stress.
Introduction
A multitude of large cytoplasmic structures formed by protein assemblies have recently been discovered in eukaryotic cells. In the budding yeast S. cerevisiae alone, nearly 200 normally diffuse cytoplasmic proteins, including enzymes from nearly all main branches of metabolism, can be induced to form large subcellular bodies, as was shown by assaying ~800 yeast strains, each expressing a green fluorescent protein (GFP)-tagged protein from its native locus in the genome. Normally cytoplasmic proteins are distributed diffusely, but when grown to stationary phase, 180 proteins involved in intermediary metabolism and stress response form cytoplasmic bodies [1].
Such bodies predominantly appear within cells as foci by fluorescence microscopy, though a small percentage may appear as fibers, forming in response to specific nutrient manipulations [1] or stresses [2][3]. The proteins' foci/fiber-forming tendencies are in some cases conserved across large evolutionary distances. For instance, cytidine triphosphate synthase has been observed to form fibers in bacteria, yeast, fly, and human cells; a cytoskeletal role has been proposed for the fiber form of the enzyme in bacteria [4], though this function has not been confirmed nor is it known in any of the other organisms [2][5][6]. For most of these intracellular assemblies, cellular functions remain an open question.
It is possible that in vivo aggregation is a common feature of many cytoplasmic proteins. In support of this, in yeast there are a number of intracellular bodies to which aggregated proteins are shunted. For refolding or degrading, proteins collect in the `juxtanuclear quality control' compartment JUNQ [8], endoplasmic reticulum associated protein degradation (ERAD) machinery [9], or aggresome [10], all of which appear as foci by fluorescence microscopy. Permanently misfolded proteins are sequestered near cells' exterior in distinct foci dubbed `insoluble protein deposits' (IPODs) [8]. Mounting evidence suggests that many types of aggregation are highly specific for similar sequences [11]. Thus, even within the above mentioned bodies, particular aggregated proteins may be associated largely with themselves and chaperones.
In the case of the yeast protein bodies, the foci often appear during the nutrient depletion phase of the post diauxic shift transition to stationary phase [12][13]. Yeast stationary phase culture conditions are inherently stressful due to nutrient depletion and the accumulation of toxic metabolites [14]. Formation in response to stressful conditions is a hallmark of aggregation, raising the possibility that many of these newly discovered protein assemblies represent endemic aggregation of much of the cytoplasmic proteome.
In order to better characterize the formation of these protein bodies, we employed a proteome-wide assay of endogenous protein solubility, which uses shotgun mass spectrometry to quantify the partitioning of each protein in the proteome between clarified cell lysate and pelleted cell debris. In this manner, we assayed proteins that shifted significantly into the pellet following heat stress. We found a significant overlap of these same proteins with those known to form intracellular bodies in response to nutrient stress or following arsenic toxicity. Finally, affinity-purification of foci-forming proteins showed significant association with a variety of chaperones, including Hsp82. Tests of foci induction in response to specific metabolic cues and stresses confirmed their formation by independent methodologies, and generally argued against coordinate formation of multi-enzyme bodies spanning single metabolic pathways. Our data show that many yeast proteins, especially many proteins of central metabolism, aggregate in response to a variety of cell stresses including heat stress, arsenic toxicity, and nutrient starvation.
Results
Foci are generally insoluble
Previous evidence suggested that the GFP-tagged proteins seen as foci are in insoluble protein clusters. In untagged strains, many proteins that become insoluble in stationary phase also form foci in GFP-tagged strains [1], demonstrating the two sets are strongly correlated. We first explicitly tested if the insoluble shift seen for untagged proteins holds for tagged proteins in order to directly link observations of the two phenomena. Using mass spectrometry, we assayed fractionated Gln1-GFP cell lysate from stationary phase cells, choosing Gln1-GFP for the high penetrance of the phenotype (>90% of cells) and its stability through lysis [1] (Figure 1). We found Gln1-GFP foci still visible in the insoluble fraction of the cellular lysate, where the majority of Gln1-GFP was detected by mass spectrometry (Figure 1). Thus we conclude that the observation of foci by microscopy or of a preponderance of insoluble peptides by mass spectrometry are likely orthogonal observations of the same protein assemblies. To ensure that this partitioning was independent of the GFP tag, we repeated the experiment on an untagged strain carrying a high-copy plasmid that constitutively expressed the same variant of GFP (GFPS35T). We found Gln1p was again insoluble in stationary phase cells while the GFP was soluble demonstrating that native Gln1p are still forming large, insoluble protein assemblies, and supporting the observation that insolubility of cytoplasmic proteins was a reasonable proxy for foci formation. We exploit this physical property to broadly assay aggregate body formation by simultaneously measuring insoluble phase shifts across all cytoplasmic proteins (Figure 1).
Figure 1. Foci are cytoplasmic, insoluble protein assemblies that form independent of a GFP-tag, shown here for the example of glutamine synthetase (Gln1).

A) Microscopy of stationary phase yeast cells expressing Gln1-GFP from its native locus or GFP from a plasmid pRS426-GPD-GFPS35T-HIS3. Upon lysis, the Gln1-GFP foci are still visible in the insoluble fraction, in contrast to the GFP only. Scale bars are 10μm. B) Mass spectrometry of the fractionated cell lysate shows the majority of Gln1-GFP is in the insoluble fraction. Similarly untagged, Gln1p is also insoluble whereas the plasmid expressed GFP is soluble. Grey lines indicate the threshold for the 95% confidence interval (|Z| > 1.96).
Heat stress induces intracellular protein foci formation
If some of the protein bodies observed in stationary phase were formed by aggregation in response to stress, it stands to reason an orthogonal stress should also induce those proteins. It is well-known that heat stress in vitro can lead to protein unfolding and aggregation [15]. In order to connect this phenomenon to formation of protein foci we systematically screened for changes in protein aggregation in response to heat stress.
By comparing the relative changes in the solubility of cytoplasmic proteins between heat-shocked and normal log-phase cells, we explicitly controlled for possible biases introduced by the lysis or quantification methodology. We were able to observe 395 proteins in both conditions, roughly half of the ~800 proteins annotated as normally exclusively cytoplasmic (Figure 2A). Following heat stress, 117 proteins became significantly more insoluble (Z-score ≥ 1.96, Supplemental Table 1) relative to the normal control.
Figure 2. A proteome-wide mass spectrometry survey reveals that foci-forming proteins are significantly more likely to precipitate in vivo following heat stress, suggesting they are assembling into stress bodies.

A) Experimental design for measuring solubility changes of endogenous, untagged proteins during heat stress as changes in the relative partitioning between the clarified lysate and cell pellet compared to controls. B) The set of proteins that become insoluble during heat stress significantly overlapped those that formed foci in stationary phase (when GFP-tagged) and those previously identified by mass spectrometry as becoming insoluble in stationary phase. The probability of overlaps was calculated as the iterated cumulative hypergeometric probability of all three sets. The 184 cytoplasmic proteins detected by all three assays were used as background set. Proteins seen in all three sets are shown in the inset table. C) Proteins from the intersection of the GFP-tagged protein screen and those that were insoluble following heat shock were tested and found to form foci in response to heat stress. Scale bars are 10μm. D) Quantifying the change in GFP-tagged protein distribution shows a specific and distinct shift toward foci formation following heat stress, as defined by manually scoring the localization of GFP-tagged proteins within cells as diffuse (green), structured but not foci (blue), or foci (red). Bar height represents the mean of 3 biological replicates, error bars show standard deviation (n ≥ 200 cells/replicate).
A three-way hypergeometric analysis found significant overlap (p ≤ 2.2×10−5) between proteins were more insoluble in heat stress (Figure 2B) or were more insoluble in stationary phase [1] or that formed foci in stationary phase [1]. This suggested the three experiments had assayed the phase change of a common set of proteins under stress. We hypothesized that the phase change produced by heat stress should predict heat stress-induced foci formation for proteins shared between the two sets. To test this we selected four proteins (Gln1, Gpm1, Ssa1, and Ura7) from the intersection of proteins that were more insoluble in heat stress and proteins that formed foci in stationary phase based on their diverse functions [1] (Figure 2B). Additionally, two proteins that have not been observed to form foci were selected as negative controls: Rpl24a, which became insoluble following heat stress, and Yef3, which remained soluble. We repeated the heat stress experiment with GFP-tagged strains of these proteins and assayed for foci formation. The four proteins that formed foci in stationary phase also formed foci following heat stress, whereas the two negative control proteins from outside the intersection did not form foci (Figure 2C).
Interestingly, we observed a difference in the penetrance of the foci phenotype following heat stress compared to stationary phase, with fewer cells showing single, distinct foci and more cells showing a number of smaller foci (Figure 2D). This difference is particularly stark for Gln1-GFP, where 90% of cells have a single clear protein aggregate in stationary phase compared to only 25% in heat stress.
Chemical stress also induces in vivo protein insolubility
Proteins can be chemically as well as thermally denatured. Arsenic is thought to be toxic to cells in multiple ways. It can damage cytoplasmic proteins directly by reacting with reduced thiol groups [16], ultimately destabilizing and unfolding them. It can also inhibit chaperones that would normally help fold unfolded, nascent chains and degrade misfolding proteins [17]. Jackobson and colleagues reported widespread protein aggregation in yeast after treating cells with 1.5 mM arsenite, As(III), for one hour as measured by shotgun mass spectrometry of fractionated cell lysate. Some 143 proteins were significantly shifted toward the insoluble fraction following arsenic treatment, 59 of which were cytoplasmic proteins [17]. Comparing this set of 59 proteins to the set of 114 cytoplasmic proteins that were more insoluble in stationary phase [1], we observed a significant overlap of 22 proteins (p ≤ 10−6, Figure 3A).
Figure 3. A similar set of cytoplasmic proteins aggregate and form foci in response to arsenic treatment and stationary phase nutrient depletion.
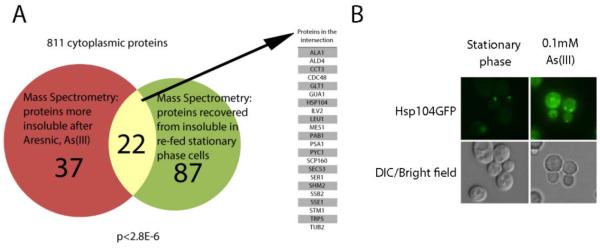
A) A Venn diagram illustrating the statistically significant overlap between proteins that become more insoluble following arsenic treatment of yeast cells and proteins more insoluble in stationary phase cells. The set of 811 cytoplasmic proteins screened in both assays was used for the background. B) Log-phase cells stressed with 0.1 mM arsenic and stationary phase cells both harbor aggregate bodies, which appear as foci in GFP-tagged strains, such as Hsp104-GFP shown here.
In agreement with this data, both the presence of arsenic and the onset of stationary phase induced foci formation, as seen with the AAA chaperone Hsp104-GFP (Figure 3B) and the stress granule marker Pab1-CFP [1][17].
Foci formation is accompanied by interactions with chaperones
We took a more targeted approach to characterizing the components of a subset of protein bodies by immunoprecipitating them and identifying co-purifying proteins by shotgun mass-spectrometry. We initially began by purifying protein bodies formed by the fluorescently-tagged glutamine synthetase Gln1-GFP in stationary phase cells. These Gln1-GFP protein bodies survive cell lysis ostensibly intact, even apart from cellular debris as foci (Figure 4A) and are strongly and specifically purified by antibodies targeting the GFP fusion partner (Figure 4B) – making Gln1-GFP the ideal test candidate. We found that Gln1-GFP foci are composed almost exclusively of Gln1-GFP and the hsp90 chaperones Hsp82p and Hsc82p (Figure 4C, Spreadsheet S1 for full results). This differs markedly from the co-purifying partners seen in log-phase cells, where tandem affinity purification (TAP) tagged Gln1 was associated with trehalose synthase (Tps1p) and the small heat shock protein Hsp42p [18].
Figure 4. Immunopurification reveals a diversity of foci composition, with a common theme of protein quality control.

A) Gln1-GFP, while diffuse in log-phase cells, localized predominantly into foci in stationary-phase cells, which persisted in lysate (scale bars are 10μm) and B) could be selectively purified by immunoprecipitation with goat anti-GFP antibodies and visualized by western blot with mouse anti-GFP antibodies. Proteins that co-immunopurified with various GFP-tagged, foci-forming proteins were assayed by shotgun mass spectrometry. Identified proteins are arranged by significance of enrichment relative to the untagged control strain, BY4741, on the X-axis and the fold change of enrichment on the Y-axis for each bait protein tested. C) Immunoprecipitation of Gln1-GFP from stationary phase cells co-immunoprecipitated the cytoplasmic hsp90 class heat shock proteins Hsp82 and Hsc82. D) Ils1-GFP also co-purified with chaperones, but instead with the hsp70 class chaperones Ssb1 and Ssb2. Several other tRNA synthetases also co-purified with Ils1-GFP. E) Cdc19-GFP also co-purified with hsp70 class chaperones as fellow members of glycolysis, notably Pdh1p.
Given these results, we were curious whether chaperones could also be seen to co-immunoprecipitate with other proteins observed to form foci. Interestingly, isoleucine tRNA synthetase, Ils1-GFP, was found to primarily co-purify with the ribosome-associated hsp70 chaperones, Ssb1p and Ssb2p (Figure 4D). The other members of the ribosome-associated complex (RAC), Ssz1p and Zuo1p, also co-purified; RAC members collectively chaperone nascent polypeptide chains during translation. Finally, the tRNA synthetases for methionine, alanine, valine, and threonine as well as the tRNA structural protein, Arc1p, co-purified with Ils1-GFP. Of these, all but Mes1-GFP formed foci in stationary-phase cells [1]. The protein interactions we observed in stationary phase differ from the results of log-phase protein interaction studies, where no chaperone or RAC interactions have been reported for Ils1p, although multiple tRNA synthetases are known to associate in a multiprotein tRNA synthetase complex.
Pyruvate kinase (Cdc19-GFP) similarly showed enrichment for hsp70 class chaperones: Ssz1, Ssa2, and Ssb2 (Figure 4E). Hsp82 and Hsp104 were also detected as significantly enriched, though at lower Z-scores and fold enrichments. Interestingly, several translation-associated proteins as well as fellow members of the glycolysis pathway co-purify with Cdc19 and are significantly enriched in stationary vs. log-phase. Among these is Acs1, whose paralog (Acs2) was detected as an interaction partner in a log-phase protein-protein interaction screen [18].
Many Hsp82 clients also form foci
After finding that several chaperones co-precipitated with foci forming proteins, we decided to reverse the previous experiments and immunoprecipitate a chaperone itself and identify its partners. A set of 50 cytoplasmic proteins co-immunoprecipitated with Hsp82-GFP in stationary phase cells. Comparing this set to the previously established set of foci forming proteins [1] we found a significant overlap (hypergeometric, p ≤ 10−3, Figure 5A), with 21 of the Hsp82-GFP co-immunoprecipitation partners also forming foci in GFP-tagged strains (Supplemental Table 2). Of particular note, Gln1p co-immunoprecipitated when Hsp82-GFP was pulled down, a reciprocal enrichment (Figure 5B). We constructed a dually fluorescent protein-tagged yeast strain in order to confirm the association and verified that Gln1-GFP foci co-localized with Hsc82-TagRFP foci in yeast grown to stationary phase in synthetic complete medium (Figure 5C). This would suggest that cytoplasmic Hsp82p chaperones a subset of foci-forming proteins in subcellular structures, which is reminiscent of similar findings for IPOD and JUNQ forming proteins [8]. Moreover, the fact that the same proteins formed foci with Hsp82 in stationary phase in their endogenous, untagged forms helped alleviate the concern that the observed interactions might be the result of tag artifacts.
Figure 5. Immunopurification of Hsp82-GFP from stationary phase cells co-precipitated a significant number of foci forming proteins.
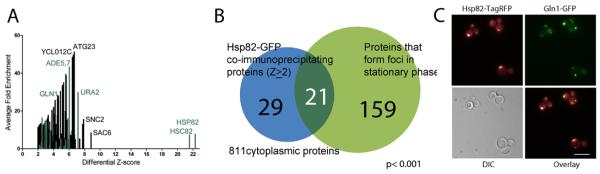
A) Among the many interaction partners identified with Hsp82-GFP, Gln1 was reciprocally enriched (combined Z-score > 4.3, fold enrichment >3.7), as were other foci forming proteins (green bars). B) Of the 50 cytoplasmic proteins that co-purify with Hsp82-GFP, a significant number (21) have been observed to form foci (p ≤ 10−3, Table S3, 811 cytoplasmic proteins as background set), suggesting that a subset of foci-forming proteins are Hsp90 clients in their foci-forming state. C) Partial co-localization of Gln1-GFP with Hsp82-TagRFP in dually fluorescent protein tagged yeast cells grown to stationary phase in synthetic complete medium (Spearman rank correlation r = 0.50 across GFP and TagRFP image fluorescent pixel intensities. Scale bar is 10 μm).
A lack of correspondence between foci and functional multienzyme complexes
In order to test whether the foci observed were also multienzyme assemblies of metabolic pathways, we focused on proteins that both i) were observed in our previous non-directed screen to form foci [1] and ii) were known to participate in related metabolic complexes in yeast or other organisms. If proteins in foci were in fact part of a multienzyme complex dedicated to a particular metabolic function, then we would expect co-localized proteins to share similar induction conditions for foci formation. To test this, we measured the induction responses of members of metabolic pathways to both specific and general nutrient depletion. We assayed the foci formation of 75 genomically GFP-tagged proteins from 8 major cytoplasmic processes selected from pathways that were either up-regulated in stationary phase [19] or that were found to contain multiple foci forming proteins [1]. Genomically GFP-tagged strains were grown in each of 5 metabolically-limiting conditions, including two general and three nutrient-specific depletions [20]. Using fluorescence, strains were scored for the presence or absence of fluorescent foci as a binary score for a total of 370 strain/condition pairs. Figure 6A plots these binary scores as a similarity matrix. The matrix yielded two key results. First, the less nutrient rich the medium was, the more foci formed as a result. Second and more surprisingly, the specific nutrient depletions showed that, for the most part, proteins of a given cellular process showed little correlation in their foci induction patterns.
Figure 6. Disparate induction patterns suggest that metabolic enzymes in the same pathway do not form foci in a coordinated fashion.

A) A test of 75 GFP-tagged proteins spanning 8 major cellular processes for their tendency to form foci in each of 5 metabolically limiting conditions found little concerted foci induction. B) The de novo purine biosynthesis pathway illustrates to case; only the enzyme responsible for the first step, Ade4-GFP, forms foci in response to adenine depletion. C) Even under conditions where other members of the purine biosynthesis pathway formed foci (stationary phase, SC-uracil), Ade4 did not co-localize with any other pathway members. The absence of coordinated induction, a hallmark of co-complex members, argues that many foci do not represent pathway-specific multi-enzyme complexes.
In fact, only one purine biosynthesis enzyme in our induction screen (Ade4p) formed foci in response to end-product depletion (Figure 6B), in contrast to observations in transiently transfected human cells [21]. Follow-up studies have since suggested that the reported foci formation of transiently expressed human purine biosynthesis enzymes was independent of purines and thus may explain this discrepancy [22]. In order to more carefully assay for a potential purinosome, we additionally tested for the co-localization of yeast purine biosynthesis enzymes in foci. We transfected a plasmid expressing Ade4-mCherry into strains where a member of the purine biosynthesis pathway was genomically GFP-tagged and induced foci formation by growth to stationary phase. Of all the pairs tested, only Ade4-GFP co-localized with Ade4-mCherry (Figure 6C). This argues against i) the tag being essential to formation of the foci and ii) the foci of purine biosynthetic enzymes being a functional multienzyme organizing centers for nucleotide production.
Discussion
General, widespread aggregation of cytoplasmic proteins
There are several lines of evidence indicating that many of the foci we observed represent aggregates. We previously showed that foci-forming proteins are computationally predicted to have an intrinsically higher propensity to aggregate than non-foci forming proteins using the TANGO algorithm [23] (with a caveat being that TANGO is trained on in vitro peptide data.). Stressing cells with either heat stress or arsenic, which are known to cause aggregation, drives many of the same proteins that form foci to the insoluble phase. Indeed, insoluble shifts were predictive of foci formation in heat stress and additionally served to control for the possibility that the GFP tag itself was driving the aggregation [24]. This effect is important to control for, as many fluorescent proteins were originally multimers that required sequence optimization to function as monomers [58], and many cytoplasmic proteins are innately homo-oligomers. Proteins whose native form is oligomeric may thus stabilize residual interaction tendencies of these proteins through avidity. Thus any novel body observed by fluorescent tagging, particularly those of natural oligomers, requires additional verification. Indeed, independent assays using alternate tags (e.g., TAP [59][1], HA [2], and RFP [this work]) additionally support many of these foci. Noree et al. also verified several other proteins still formed fibers when C-terminally tagged with a human influenza hemagglutinin (HA)-tag [2]. Further solubility shifts of many native, untagged proteins as measured by mass spectrometry correlated well with observations of foci formation in stationary phase cells [1], suggesting protein body formation is largely a property of the native protein.
Of the foci-forming proteins we studied in detail, Gln1 formed protein bodies in response to nearly every stress tested and co-immunoprecipitated with chaperones. Gln1-GFP protein bodies are unlikely to represent storage bodies because foci formed by nutrient depletion did not disperse when new medium with cycloheximide was added [1], indicating that recovery of diffuse Gln1-GFP requires new protein synthesis. We speculate that the set of proteins that, like Gln1, shift to the insoluble phase in response to various stresses, form protein bodies composed of aggregates. Top candidates would be foci-forming proteins that show novel chaperone associations in stationary phase or under stress, such as the foci-forming proteins that co-immunoprecipitate with Hsp82-GFP.
Given that the fitness cost of clearing aggregates formed by the expression of a non-functional gene can be a decrease in growth rate [25], why do cells allow up to 20% of cytoplasmic proteins to crash out in response to common, even routine stresses such as nutrient depletion or thermal stress? One possibility is that many evolutionarily ancient proteins are trapped in a local minimum in sequence space, with limited options to explore while maintaining essential protein functions. These proteins may be expressed near the limits of solubility or beyond to maximize growth rate in stable conditions; cells might occasionally accept the costs of aggregated proteins in order to achieve higher enzyme concentrations. Alternatively, such aggregation might serve a function by rapidly altering protein activity, as we discuss below.
Foci rarely represent consecutive metabolic enzymes
Massive rearrangements of cytoplasmic structures occur in response to a variety of stresses. Notably, many proteins condense into concentrated pockets [26][27][1], effectively decreasing the concentration of normally diffuse proteins. These changes alter translation, chaperone capacity, and likely metabolism in the cytoplasm, both within and outside the foci. There are many examples of metabolically active, heterogeneous protein megacomplexes whose formation and activity is regulated in response to nutrient availability. Notable examples include carboxysomes [28], cellulosomes [29], and pyruvate dehydrogenase complexes [30]. Most of the observed yeast protein bodies are inconsistent with such structures; we observed that proteins within the same metabolic pathway do not generally form foci under the same induction conditions. If proteins do not form assemblies at the same time, it precludes the possibility of their co-assembly into the same structures. Thus, in general, most yeast foci do not appear consistent with multi-enzyme factories catalyzing consecutive metabolic reactions. It remains possible that some foci could represent homo-oligomeric assemblies which cooperatively regulate enzymatic activities or flux through specific branch points in the metabolic network. Future work could attempt to systematically measure dissociation rates for aggregated proteins in vitro and in vivo as well as to measure enzymatic activity for the subset of aggregates stable enough to survive cell lysis, native-state purification, and specific activity measurements. It is also worth noting that several of the enzymes studied, among them Ade4p, Ura2p, and Pfk1p/Pfk2p, catalyze rate limiting steps in their respective metabolic pathways. The failure to assemble into multi-enzyme factories does not rule out the potential for aggregation to serve a regulatory function in these pathways.
Adaptation to aggregation
While some proteins, particularly RNA-binding proteins and chaperones, are known to aggregate in response to nutrient depletion or heat stress, the scope of additional proteins from diverse pathways added to the list in this study is extensive. Many proteins spanning central metabolism, translation, and chaperones collapse into insoluble bodies in response to a variety of stresses. Recent work on RNA granules suggests they form by β-amyloid aggregation of low-complexity regions within partially unfolded RNA-binding proteins [31]. The granules appear as foci within cells and are insoluble upon lysis. However, the proteins retain their RNA-binding capacity, and unlike amyloid aggregates associated with prions or neural plaques, are highly dynamic and specific only to the type rather than the sequence of amino acid side chains. Thus the hypotheses for storage body and aggregate are not be mutually exclusive, and future work may try to more precisely define roles, if present, for these bodies.
Conclusions
In summary, we have used a combination of mass spectrometry and fluorescence microscopy to assay yeast cells for protein aggregation into insoluble bodies, which are formed by an extensive assortment of normally cytoplasmic proteins in response to a variety of stresses. We observe a correspondence between foci formation and the tendency for proteins to show a shift in lysed cells from the clarified lysate to the cell pellet. Based on this, we employed mass spectrometry to survey protein aggregation in response to heat stress, and find that many of the same proteins form protein bodies in response to thermal, nutrient, and chemical stresses. Many of these proteins tend strongly to associate with chaperones, including Hsp90 chaperones, and, do not appear to represent multi-enzyme complexes. Instead, they appear in general to represent widespread intracellular protein aggregation. Thus, yeast cells exhibit dramatic alterations in their cytoplasmic architecture in response to a variety of stresses, and the soluble phase assumptions for metabolism may no longer be valid in these cases.
Methods
Media and yeast strains
Yeast strains had a genetic background of BY4741 (genotype: MATa his3_1 leu2_0 met15_0 ura3_0). Strains expressing natively-regulated, genomically-tagged C-terminal GFP-fusion proteins were obtained from the OpenBiosystems GFP collection. Rich (YPD) medium containing yeast extract (1%), peptone (2%), and glucose (2%) was purchased from Sunrise Sciences. Synthetic complete medium (SC) was purchased premixed from Sunrise Biosciences or made locally. Local SC contained 1x yeast nitrogen base (BD Biosciences/Difco) without amino acids, synthetic drop-out medium supplement mix (Sigma), with or without glucose (2%), as noted. Frozen yeast stocks were inoculated into YPD and grown overnight before subculturing to new medium for assay. All cultures were maintained by shaking at 30 °C unless otherwise specified.
Solubility of Gln1-GFP Foci
We measured the cytoplasmic solubility of Gln1 with and without a GFP-tag by shotgun mass spectrometry and microscopy of fractionated whole cell lysate. Cultures of Gln1-GFP were grown in SC-His medium to stationary phase (48 hours) at 30 °C. Cells were first imaged, then lysed vortexing with glass beads in 50 mM Tris, 50 mM NaCl, 5 mM MgCl2, 1x protease inhibitor cocktail I (CalBiosciences), and 1 mM DTT. Whole cell lysate was partitioned into soluble and insoluble fractions by centrifugation at 10,000xg for 10 minutes, and resulting fractions were imaged as described below.
Heat stress experiments
Cultures were inoculated from frozen stocks into SC and grown in triplicate for each strain. Overnight cultures were subcultured once to an initial OD660nm of 0.2 OD/ml, regrown to approximately 1 OD660nm/ml, and then transferred to shaking incubators at either 30 °C or 42 °C for 2 hours. For proteomic analysis, cells were lysed and separated into soluble and an insoluble fractions as described above. To measure foci induction, GFP-tagged strains were grown under identical heat stress or control conditions, fixed with formaldehyde, and imaged as described below.
Proteomics sample preparation and analysis
Insoluble fractions were resuspended in denaturing buffer consisting of 50% TFE in lysis buffer (50 mM Tris, 50 mM NaCl, 5 mM MgCl2). Soluble protein fractions were reduced to near-dry (<10μ l) by speedvac and resuspended in denaturing buffer (50% TFE in lysis buffer). All samples were then subjected to reduction, alkylation, and digestion with trypsin as previously described [32]. Following digestion, trypsin activity was halted by the addition of 1% formic acid. Sample volume was reduced to ~100 μl by SpeedVac centrifugation and the volume adjusted to 150 μl with Buffer C (95% H2O, 5% acetonitrile (ACN), 0.1% formic acid). Tryptic peptides were bound and washed on Hypersep C-18 SpinTips (Thermo), eluted with 60% acetonitrile, 0.1% formic acid, reduced to near-dry by speedvac and resuspended in Buffer C. Soluble and insoluble fractions from heat-shock experiments were additionally filtered through Microcon 10,000 NMWL Centrifugal Filters (Millipore) to remove larger contaminants and undigested proteins.
Soluble and insoluble fractions and Gln1-GFP immunoprecipitations were analyzed by nano LC-MS/MS using a Thermo Surveyor Plus HPLC coupled to an LTQ-Orbitrap Classic hybrid mass spectrometer (Thermo Scientific). Analyses the of remaining immunoprecipitations were carried out on a Dionex Ultimate 3000 nanoRSLC system coupled to an LTQ-Orbitrap Velos Pro hybrid mass spectrometer (Thermo Scientific). Data-dependent ion selection was activated, with parent ion scans (MS1) collected at high resolution (60,000 for Classic, 100,000 for Velos Pro). Ions with charge >+1 were selected for collision-induced dissociation fragmentation, with fragment spectra (MS2) collected by LTQ (12 MS2 per MS1 for Classic, 20 MS2 per MS1 for Velos Pro). Dynamic exclusion was activated, with an exclusion time of 45 seconds for ions selected more than twice in a 30 second window. Four injections (technical replicates) were performed for each biological replicate.
With a reference database of non-redundant yeast protein-coding sequences downloaded from SGD, mass spectra were interpreted using the MSBlender search algorithm [33], which employed an ensemble of Tide [34], MS-GFDB [35], and InsPecT [36] search algorithms. Results were filtered to achieve a 1% false discovery rate for peptide spectrum matches (PSMs), using a reverse-sequence decoy database. As an additional confidence filter, peptides observed in only one injection were removed.
LC-MS/MS injections were analyzed independently for each biological replicate. Data from multiple injections per biological replicate were combined by adding the total count of peptide mass spectra for each protein across injections. Proteins observed only once in a biological replicate were subsequently omitted from further analyses. For heat-shock experiments, the data sets were curated to assign peptides to a single proteins or protein groups to account for the occurrence of degenerate peptides assigned to multiple proteins (e.g., paralogs). For IP samples, peptide spectral counts were divided evenly among all protein group members sharing that common peptide.
Relative protein quantification by mass spectrometry
Proteins were quantified by comparison of peptide spectral counts as described [37]. To estimate the significance of relative protein abundance changes between two samples, a Z-score was calculated for each protein as in [37] by comparing the protein's spectral count frequency in the sample to a matched control (e.g., GFP strain vs. untagged control; soluble fraction vs. insoluble fraction). Where Z-scores are combined, e.g. as for calculating significance across biological replicates, composite Z-scores were calculated as the sum of individual Z-scores per protein for each biological replicate divided by the square root of the number of Z-scores combined (e.g., Eq. 1):
| Eq. 1 |
A score of |Z| ≥ 1.96 was considered significant for all IP samples, corresponding to a 95% confidence level. For the heat stress experiments, a score of |Z| ≥ 1.64 was considered significant, corresponding to 90% confidence. Additionally for the heat-shock experiments, pairwise differences in protein abundance between samples were estimated as fold-change, calculated as the ratio between normalized frequencies of spectral counts.
Immunoprecipitation of GFP-tagged proteins
Strains from the GFP collection or the parental strain BY4741 were cultured in SC medium to stationary phase (48 hours) at 30 °C to reach 100 OD per sample. Foci formation was verified by microscopy before lysing cells by vortexing with glass beads in 50 mM Tris, 50 mM NaCl, 5 mM MgCl2, 1x protease inhibitor cocktail I (CalBiosciences), and 1 mM DTT. After verifying by microscopy that foci were retained in the lysate, GFP-tagged proteins were immunoprecipitated with 4 μg rabbit anti-GFP antibodies (Sigma) and bound to 200 μl Protein A-conjugated Dynabeads (Immunoprecipitation Kit, Invitrogen) according to the manufacturer's instructions. Immunoprecipitated proteins were washed three times with PBS (pH 7.4) and eluted by incubating beads in 120 μl 50% trifluoroethanol at 70 °C for 10 minutes. For western blot verification, samples were first eluted by boiling protein-A beads in loading buffer for 5 minutes and then detected with mouse anti-GFP primary antibodies (Covance) and horseradish-peroxidase conjugated goat anti-mouse secondary antibodies (Santa Cruz Biotechnology). Samples were visualized using luminol (Santa Cruz Biotechnology).
To measure the relative enrichment of proteins between the GFP-tagged strains and the untagged parental strain, we compared the total counts between samples of MS/MS peptide mass spectra for all peptides attributed to a given protein, calculating the significance of enrichment as a Z-score as described above [32]. Proteins with Z ≥ 2 were considered significantly enriched (97.7% confidence level) by immunoprecipitation.
Foci induction by metabolite depletion
Select GFP-tagged strains were picked into 96-well plates and grown overnight in YPD. Cultures were divided into a dozen plates for storage as single-use glycerol stocks. For each experiment, a plate was thawed and used to inoculate a YPD plate for overnight growth. Overnight cultures were used to inoculate a new plate to 0.2 OD/ml in YPD or SC for nutrient dropout and SC growth to stationary phase. For medium exchange experiments, cells were regrown to approximately 1 OD/ml in SC, washed once with the destination medium, and then resuspended in SC minus the specified metabolite for 2 hours to induce foci. Cells were fixed with 4% formaldehyde (SPI-CHEM) at room temperature for 60 min, and washed with PBS before storing in PBS at 4 °C until imaging.
Cells were imaged on either a Nikon E800 fluorescence microscope with a Photometrix Coolsnap CCD camera under oil immersion at 100× magnification or a Nikon TE2000-E with a Photometrics Cascade II camera under oil immersion at 60× magnification. Differential interference contrast and widefield fluorescent images were collected using standard filter sets and processed using a Nikon Elements AR. Cell images were manually scored for the presence or absence of foci to build an array of foci formation per condition. Proteins were hierarchically clustered based on their foci formation patterns using the hclust function in R/Bioconducter and employing Euclidian distance as the measure of similarity.
Co-localization assays
Dual-tagged yeast strains for testing co-localization of purine biosynthesis pathway enzymes were created by transforming genomically GFP-tagged strains with a high copy plasmid (pRS426) expressing Ade4-mCherry under the control of the GPD promoter with a URA3 selection marker. A dual-tagged yeast strain for testing the co-localization of Gln1p and Hsp82p was created by using homologous recombination to chromosomally insert TagRFP and the URA3 selectable marker at the C-terminus of HSP82 in the Gln1-GFP strain. Cells were grown for 48 hours in SC-uracil to stationary phase to induce foci and were then fixed with formaldehyde and imaged as above. Spearman's rank correlations were measured in ImageJ using the PSC Colocalization Plugin [39].
Supplementary Material
Acknowledgments
We would like to acknowledgement the generous support of the ICMB core facilities, particularly Dr. Richard Salinas and Dr. Angela Bardo. We are deeply indebted to Caitlin Sanford for her efforts in editing this manuscript. This work was supported by grants from the National Institutes of Health, National Science Foundation, Cancer Prevention Research Institute of Texas, and the Welch (F-1515) Foundation to EMM. MT acknowledges funding and support from Michele Vendruscolo and the University of Cambridge. All raw mass spectrometry datasets are available for download at: http://marcottelab.org/index.php/Widespreadaggregation.2013
Footnotes
The authors declare that they have no conflict of interest.
References
- [1].Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O'Connell JD, Mirrielees J, Ellington AD, Marcotte EM. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. U. S. A. 2009 Jun;106(25):10147–52. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Noree C, Sato BK, Broyer RM, Wilhelm JE. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 2010 Aug;190(4):541–51. doi: 10.1083/jcb.201003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bagola K, Sommer T. Protein quality control: on IPODs and other JUNQ. Curr. Biol. 2008;18(21):R1019–21. doi: 10.1016/j.cub.2008.09.036. [DOI] [PubMed] [Google Scholar]
- [4].Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat. Cell Biol. 2010;12(8):739–746. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu J-L. Intracellular compartmentation of CTP synthase in Drosophila. J. Genet. genomics. 2010 May;37(5):281–96. doi: 10.1016/S1673-8527(09)60046-1. [DOI] [PubMed] [Google Scholar]
- [6].Chen K, Zhang J, Tastan OY, Deussen ZA, Siswick MY-Y, Liu J-L. Glutamine analogs promote cytoophidium assembly in human and Drosophila cells. J. Genet. genomics. 2011 Sep;38(9):391–402. doi: 10.1016/j.jgg.2011.08.004. [DOI] [PubMed] [Google Scholar]
- [7].O'Connell JD, Zhao A, Ellington AD, Marcotte EM. Dynamic Reorganization of Metabolic Enzymes into Intracellular Bodies. Annu. Rev. Cell Dev. Biol. 2012 Oct;28:89–111. doi: 10.1146/annurev-cellbio-101011-155841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008 Aug;454(7208):1088–95. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat. Cell Biol. 2005 Aug;7(8):766–72. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- [10].Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 1998 Dec;143(7):1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A. 2010 Feb;107(8):3487–92. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005 Oct;310(5747):486–9. doi: 10.1126/science.1115791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sagot I, Pinson B, Salin B, Daignan-Fornier B. Actin bodies in yeast quiescent cells: an immediately available actin reserve? Mol. Biol. Cell. 2006 Nov;17(11):4645–55. doi: 10.1091/mbc.E06-04-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. Sleeping beauty': quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004 Jun;68(2):187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chernoff YO. Stress and prions: lessons from the yeast model. FEBS Lett. 2007 Jul;581(19):3695–701. doi: 10.1016/j.febslet.2007.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Beyersmann D, Hartwig A. Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch. Toxicol. 2008 Aug;82(8):493–512. doi: 10.1007/s00204-008-0313-y. [DOI] [PubMed] [Google Scholar]
- [17].Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, Grant CM, Christen P, Goloubinoff P, Tamás MJ. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci. 2012 Nov;125(Pt 21):5073–83. doi: 10.1242/jcs.107029. [DOI] [PubMed] [Google Scholar]
- [18].Gavin A-C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier M-A, Hoffman V, Hoefert C, Klein K, Hudak M, Michon A-M, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006 Mar;440(7084):631–6. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- [19].Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11(12):4241–57. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003 Oct;425(6959):686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- [21].An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008 Apr;320(5872):103–6. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- [22].Zhao A, Tsechansky M, Swaminathan J, Cook L, Ellington AD, Marcotte EM. Transiently transfected purine biosynthetic enzymes form stress bodies. PLoS One. 2013 Jan;8(2):e56203. doi: 10.1371/journal.pone.0056203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004 Oct;22(10):1302–6. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- [24].Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 2012 Apr;9(5):480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc. Natl. Acad. Sci. U. S. A. 2011 Jan;108(2):680–5. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Williams R. mRNAs sit out the stress in EGP bodies. J. Cell Biol. 2007 Oct;179(1):2–2. [Google Scholar]
- [27].van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Séraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002 Dec;21(24):6915–24. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shively JM, Ball F, Brown DH, Saunders RE. Functional Organelles in Prokaryotes: Polyhedral Inclusions (Carboxysomes) of Thiobacillus neapolitanus. Science (80-.) 1973 Nov;182(4112):584–586. doi: 10.1126/science.182.4112.584. [DOI] [PubMed] [Google Scholar]
- [29].Doi RH, Kosugi A. Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat. Rev. Microbiol. 2004 Jul;2(7):541–51. doi: 10.1038/nrmicro925. [DOI] [PubMed] [Google Scholar]
- [30].Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006 Apr;34(Pt 2):217–22. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- [31].Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell. 2012 May;149(4):753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Orr SJ, Boutz DR, Wang R, Chronis C, Lea NC, Thayaparan T, Hamilton E, Milewicz H, Blanc E, Mufti GJ, Marcotte EM, Thomas NSB. Proteomic and protein interaction network analysis of human T lymphocytes during cell-cycle entry. Mol. Syst. Biol. 2012 Jan;8:573. doi: 10.1038/msb.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kwon T, Choi H, Vogel C, Nesvizhskii AI, Marcotte EM. MSblender: A probabilistic approach for integrating peptide identifications from multiple database search engines. J. Proteome Res. 2011 Jul;10(7):2949–58. doi: 10.1021/pr2002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Diament BJ, Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. J. Proteome Res. 2011 Sep;10(9):3871–9. doi: 10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJR, Pevzner PA. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics. 2010 Dec;9(12):2840–52. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tanner S, Shu H, Frank A, Wang L-C, Zandi E, Mumby M, Pevzner PA, Bafna V. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005 Jul;77(14):4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- [37].Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007 Jan;25(1):117–24. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- [38].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003 Nov;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP. Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat. Protoc. 2008 Jan;3(4):619–28. doi: 10.1038/nprot.2008.31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.