

Abstract
Signaling via Programmed Death Ligand (PD-L)-1 and PD-L2 is crucial for maintaining peripheral tolerance. CD90+ myofibroblasts/fibroblasts (CMFs) are major PD-1 ligands -expressing cells in normal human colonic mucosa. CMFs suppress activated CD4+ T cell proliferation via PD-1 ligands. It is not known whether signaling through TLRs contribute to the regulation PD-1 ligands on CMFs upon colonic mucosal tolerance. Herein, we demonstrated that stimulation of TLR4 on human CMFs upregulates PD-L1, but not PD-L2, and reinforces CMF-mediated suppression of CD4+ T cell proliferation and IFN-γ production. TLR4-mediated upregulation of PD-L1 on CMFs involved NF-κB pathways and was JAK2- and MyD88-dependent. MyD88-dependent stimulation of TLR1/2 and TLR 5 also upregulated PD-L1 expression on CMFs in culture. PD-L1 expression was drastically decreased in vivo in the colonic mucosa of mice devoid of MyD88. Induction of MyD88 deficiency in CMFs in fibroblast-specific MyD88 conditional knockout mice resulted in a strong increase in a mucosal IFN-γ expression concomitantly with the abrogation of PD-L1 expression in CMFs under homeostasis and epithelial injury induced by dextran sodium sulfate. Together these data suggest that MyD88-dependent TLR stimulation of CMFs in the normal colonic mucosa may reinforce these cells' anti-inflammatory capacity, and thus contribute to the maintenance of mucosal tolerance.
Keywords: Colon, fibroblasts, tolerance, PD-L1, MyD88
Introduction
The gastrointestinal (GI) mucosa plays a fundamental role in the immunoregulatory response to pathogens, sensitization/desensitization to dietary antigens, and tolerance to self antigens and commensal flora in the GI system (1). The mammalian colon is highly populated by variety of different microorganisms. Using standard culture methods it was estimated three decades ago that at least 400 species of bacteria are present in fecal samples (2), while current metagenomic methodology has suggested that those numbers may have been an underestimate, representing only 30-40% of the total microbial diversity in colon (3-5). In spite of this high level of exposure to microbial antigens, the normal colonic mucosa is protected by innate and adaptive pro-tolerogenic mechanisms that homeostatically control inflammatory responses. Disruption of mucosal tolerance in the colon is associated with several incurable and chronic diseases, such as inflammatory bowel disease (IBD). Therefore, understanding the underpinning mechanisms responsible for colonic mucosal tolerance has potential translational value for IBD therapy and management.
Despite significant progress in understanding gastrointestinal mucosal tolerance, the cells and pathways that determine the establishment and maintenance of tolerance in the colon remain poorly understood. A prominent role in peripheral tolerance has been proposed for the inhibitory molecules Programmed Death-1 ligand (PD-L)-1 (a.k.a B7-H1, CD274) and PD-L2 (a.k.a B7-DC, CD273). These B7 family molecules are mainly expressed on antigen-presenting cells (APCs), and appear to have dual roles as both negative modulators of activated T cell responses and as co-stimulators of early T cell priming and differentiation (6-10). Signaling through these ligands limits the proliferation of activated effector T cells, and regulates the balance among Th1, Th2, Th17 and CD4+ regulatory T responses (10-13). The best-characterized tolerogenic receptor is PD-1 (CD279), which negatively regulates TCR signals and controls the balance of T-cell activation, immune-mediated tissue damage and provides multiple tolerance checkpoints that prevent autoimmunity (7).
Despite extensive investigation of the B7 family of co-stimulators in various tissues, their involvement in the regulation of mucosal immune responses in the GI tract remains poorly understood. The crucial role of PD-L1-mediated signaling in mucosal tolerance in the small intestine has recently been highlighted by the report that loss of PD-L1:PD-1 signaling leads to a development of an autoimmune enteritis and destruction of the small intestinal epithelium (6). PD-L1-/- animals have elevated systemic IFN-γ production (14). PD-L1-mediated control of inflammatory IFN-γ-producing effector T cells in the GI mucosa has particular importance, as immune surveillance by these cells is critical for the host responses to tumors and infection, and contributes to autoimmune responses (13). In particular, altered PD-L1-mediated signaling/expression have been reported to contribute to chronic inflammation during IBD (15, 16).
Limited data are available on cells expressing PD-L1 and PD-L2 in the GI tract. CD90+ stromal cells (a.k.a. myofibroblasts/fibroblasts or CMFs) are located just beneath the epithelial basement membrane and represent a major cell population expressing both PD-L1 and PD-L2 in the normal colonic mucosa (17). We and others previously reported that CMFs represent a distinct class of local non-professional APCs and are negative for the markers of professional APCs such as macrophages and dendritic cells (DCs) (18) (19). CMFs express MHC class II molecules. Under homeostasis, CMFs do not express CD80 (18) (a.k.a. B7-1), which is also capable of signaling via PD-L1 (20). CMFs limit activated CD4+ T cell responses via signaling initiated by PD-L1 and PD-L2. Taken together (17-19, 21), these recently published data demonstrate that normal human colonic mucosa colonic stromal cells, although capable of serving as APCs, play an immunosuppressive role instead.
Toll-like receptors (TLRs) recognize conservative molecular motifs of commensal and pathogenic microorganisms. Activation of TLRs results in the transduction of innate pro- and anti- inflammatory gene expression programs through distinct intracellular signaling pathways, controlled by signaling adapter complexes. MyD88 is a shared cytoplasmic TLR adapter, which plays a critical role in innate signaling through the NF-κB and IRF transcription factors (22-24). Signaling through TLRs on innate and adaptive immune cells plays a critical role in the maintenance of mucosal homeostasis. These interactions contribute to the maintenance of tolerance to commensal microorganisms and the induction of the inflammatory responses against pathogens (23, 25, 26). While the colonic epithelial layer is a dynamic structure undergoing constant renewal, CMFs located just beneath the epithelium and with a slower turnover rate are constantly exposed to a vast array of colonic microbiota products that might stimulate innate immune receptor mediated signaling (19, 27). Under conditions of homeostasis these interactions do not result in overt inflammatory responses, presumably due to mechanisms of mucosal tolerance. A critical contribution of PD-1 ligand mediated signaling to peripheral tolerance in vivo has been demonstrated (6, 14). Recent studies showed that stimulation of TLRs induce PD-L1 expression on DCs, macrophages and cancer cells (28-30). However, it is not known whether signaling via TLRs contributes to the regulation of PD-1 ligands in the GI tract. Thus, the objective of the study presented herein was to investigate the role of MyD88-mediated TLR signaling in the modulation of the PD-L1expression by CMFs.
Herein we demonstrate that stimulation of TLR4 on CMFs derived from the human normal mucosa leads to the NF-κB-dependent upregulation of PD-L1, but not PD-L2 with reinforced CMF-mediated suppression of CD4+ effector T cell responses. Lack of intact MyD88 signaling specifically within the fibroblast compartment in vivo resulted in an increased expression of the inflammatory cytokine IFN-γ concomitantly with the decrease in PD-L1 expression by CMFs. Taken together with previous observations by ourselves and others (14, 17, 31) these new data suggest that upregulation of PD-L1 expression by CMFs via MyD88-dependent TLR signaling is among the critical processes contributing to the suppression of inflammation under mucosal tolerance and disruption of this mechanism might be an important factor involved in acute and chronic colonic inflammatory conditions.
Materials and Methods
Antibodies
Fluorochrome-conjugated and unconjugated murine anti-α-smooth muscle actin (α-SMA, clone 1A4) monoclonal and anti-MyD88 goat polyclonal Abs were purchased from Sigma (St. Louis, MO). Fluorochrome-conjugated forms of IgG1κ, IgG2a, isotype controls and monoclonal mAbs directed against human CD90 (clone 5E10) were from BD PharMingen and eBioscience (San Diego, CA). Fluorochrome-conjugated mAbs against human and murine CD4 (clone RPA-T4 and RM4-5, respectively), T-bet (clone eBIo4B10), isotype controls as well as mAbs against human PD-L1 (clone M1H1), PD-L2 (clone M1H18) and murine PD-L1 (clone M1H5), murine and human Fc receptor blocker were from eBioscience (San Diego, CA). Alexa Fluor® (AF®) 488- and AF®633-labeled donkey anti-mouse IgG2a and IgG1κ (respectively), Zenon Mouse IgG and Apex™ Ab labeling kits were purchased from Invitrogen Inc. (CA). Anti-total IkBα murine mAbs (clone L35A5) and anti-biotin HRP-conjugated Abs were from Cell Signaling Technology, Inc. (Danvers, MA). Goat anti-human PD-L1 polyclonal biotinylated Abs were purchased from R&D Systems, Inc. (Minneapolis, MN). Goat anti-murine IgG HRP-conjugated Abs were from Bio-Rad Life Science (Hercules, CA).
Human tissue & CMF isolation
For CMF isolation, full-thickness fresh human mucosal samples were obtained from discarded surgical resection material of colons in compliance with protocols approved by the University of Texas Medical Branch Institutional Review Board. Areas of uninvolved colon tissue from patients undergoing colectomy for colon cancer were studied. Total mucosal cell preparation was done as described previously (18). CMFs were isolated according to the protocol of Mahida et al. (32), which is routinely used in our laboratory (18). The purity of isolated CD90+ CMFs (98-99%) was confirmed by flow cytometry, as previously described (18). Studies were performed with primary CMF isolates at passages 4-10. Cells were cultured as described previously (18).
Animals
C57BL/6 mice were purchased from the Jackson Laboratories (Bar Harbor, ME). MyD88-deficient mice (MyD88-/-) on a C57BL/6 background were obtained from the University of California at San Diego. MyD88flox mice were obtained from Jackson laboratory and were crossed with fibroblast specific Cre mice (TgCol1α2-CreER™) in order to generate fibroblast specific Tamoxifen-inducible conditional knockout mice under fibroblast specific Col1α2 promoter selective for MyD88 (Myd88floxTgCol1α2-CreER™ or Fib-Myd88 KO). The fibroblast specific Cre mice (TgCol1α2-CreER™) mice were provided by Dr Andrew Leask at University of Western Ontario (London, Ontario, Canada) and have been described previously (33). Deletion of myd88 was induced with the intraperitoneal (i.p.) injection of tamoxifen (TM, 1 mg/mice for four days. total injection volume/animal is 100 μL) one week prior to the experimental procedure. The presence of MyD88 floxed and Cre recombinase under Col1α2 promoter transgene was identified by using standard PCR protocols suggested by Jackson Laboratory. For the detection of MyD88 floxed trasgene the following primer pair was used: oIMR9481fw 5′GTT GTG TGT GTC CGA CCG T 3′ and oIMR9481rev 5′GTC AGA AAC AAC CAC CAC CAT GC 3′. Cre recobinase trasgene was detected using primers: CreSfw 5′ AGG TTC GTT CAC TCA TGG A 3′ and CreASrev 5′ TCG ACC AGT TTA GTT ACC C 3′. Absence of MyD88 expression within the CMF compartment was controlled using immunostaining followed by confocal microscopy and flow cytometry. All mice (female, 6-12 weeks of age) were housed under pathogen-free, but not germ-free conditions. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of California at San Diego and University of Texas Medical Branch at Galveston.
Dextran sodium sulfate (DSS) treatment
The DSS model of colitis was used to induce epithelial injury using the standard protocol (34). On day 1, animals received DSS solution (2% DSS in drinking water). Control animals received regular drinking water. On days 3 and 5 water supplies were refilled with fresh DSS and control solutions, respectively. On day 8 the DSS solution was replaced by regular water. Colitic changes and immune responses were analyzed at day 10. For this purpose mice were anesthetized (80 μl of ketamine at 80 mg/Kg and xylazine 10 mg/Kg, i.p.) and euthanized by cervical dislocation and samples taken for analysis enlisted below. Total mucosal cell preparation was done as described previously for human tissue (18).
Transfection of siRNA into CMFs
Primary human CMFs with repressed expression of RelA, RelB or MyD88 were generated in our lab using Stealth™ siRNA probes (Invitrogen, CA). Negative siRNA controls with appropriate GC content were included in each experiment. An optimal concentration of each siRNA (0.3 nM) was used for each transfection. Transfection of primary cells was performed using Nucleofector™ technology (Amaxa Biosystems, Gaithersburg, MD) according to the manufacturer's instructions as previously described (10).
Costimulation of T cell responses
Assays were performed as previously described using allogeneic CMF: T cell co-cultures (17). Briefly, human CD4+ CD45RA+ T cells were purified from PBMCs of healthy donors by negative selection using Naïve CD4+ T Cell Isolation magnetic bead Kit II (Miltenyi Biotec., Auburn, CA). T cells were CFSE labeled as previously described (35) and were pre-activated with anti-human CD3 and CD28 microbeads (Myltenyi Biotec, per manufacturer's instructions) for 1 h prior to co-culture initiation. A ratio of 4:1 (T cells to CMFs) was used in these experiments. In some assays CMFs were pre-stimulated with LPS (1 μg/mL) for 24h. LPS was removed and CMFs were washed three times with MEM containing polymyxin B (10 μg/mL) to remove residual LPS. Pre-activated T cells were then added and the co-cultures were maintained in polymyxin-containing media. T cells were then recovered from co-cultures on day 4, immunostained for the markers of interest, and analyzed by flow cytometry. Conditioned media from mono- and co-cultures of T cells and CMFs were collected and analyzed for IFN-γ using single-plex analysis (Millipore, CA) per the manufacturer's instructions.
Flow cytometry
CMFs were detached from the culture flasks by treatment with cell dissociation buffer (Sigma) at 37°C for 15-30 min, followed by two washes with cold PBS. Single- and multi-color immunostaining were performed according to standard eBioscience surface and intracellular FACS staining protocols. Cells were analyzed by flow cytometry using LSRII cytometers (BD Biosciences) per the manufacturer's procedure. Area, height and width parameters for forward and side scatters (FSC and SSC, respectively) were used to discriminate single live cells. An additional gate was set up based on the negativity for the fixable viability dye eFluor® 780 (eBIoscience), which was added during the surface marker staining to exclude dead cells from the analysis. Flow cytometry data were analyzed using FACSDiva 6.2 (Becton Dickinson) and FlowJo (Tree Star, USA) software.
Confocal microscopy
Frozen murine colon tissue sections were fixed in 1% paraformaldehyde for 20 minutes at room temperature, blocked with normal rabbit serum (1:10 in PBS) for 15 min at room temperature, and then incubated with anti-PD-L1 (1 μg/mL) mouse monoclonal Abs (clone M1H5) overnight at 4°C. The sections were then stained with AF®594-conjugated rabbit anti-rat IgG Abs (1:300) and AF®488-conjugated anti-α-SMA mAbs for 30 min at room temperature. Each staining step was followed by six washes with PBS with Ca++/Mg++. Secondary mAbs and isotype controls were included in the analyses. The sections were then mounted in SlowFade® Gold antifade reagent with DAPI (Invitrogen). Confocal microscopy was performed with a Zeiss LSM510 META laser-scanning confocal microscope (Carl Zeiss, Thornwood, NY) as previously described (17).
Western blot analysis
This analysis was performed on 10 μg of protein as previously described (18).
Real-time RT-PCR
Analysis was performed as previously described (17) according to the Applied Biosystems's two-step RT real-time PCR protocol (Applied Biosystems, Foster City, CA). Briefly, all reagents were purchased from Applied Biosystems. The appropriate assays-on-demand™ gene expression assay mix (Applied Biosystems) for human 18S RNA and the gene of interest (a 20X mix of unlabeled PCR primers and TaqMan® MGB probe, FAM™ dye-labeled) and 2 μL of cDNA were added to the PCR reaction step. The reactions were carried out in a 20 μL final volume using a BioRad Q5 real-time PCR machine with the protocol 2 min at 50°C, 10 min at 95°C (1 cycle) and 15 sec at 95°C and one min at 60°C (40 cycles).
Statistical analysis
Unless otherwise indicated, the results were expressed as the mean ± SE of data obtained from at least three independent experiments each performed in triplicate. Differences between means were evaluated by ANOVA using Student's t-test for multiple comparisons. Values of P <0.05 were considered statistically significant.
Results
Stimulation of TLR4 on colonic CD90+ myofibroblasts/fibroblasts (CMFs) upregulates their PD-L1 expression
Because abnormal TLR4-mediated signaling is involved in chronic inflammation associated with IBD (36-38) and TLR4 stimulation of the resident peritoneal macrophages upregulates PD-L1 (11), we first determined how LPS stimulation affected PD-L1 expression on CMFs in freshly digested normal colonic mucosal cell (MC) preparations. Stimulation of normal colonic MCs with LPS (1μg/mL) led to strong upregulation of PD-L1 on CD90+ CMFs, which were the major population expressing PD-L1 in LPS-stimulated MC preparations (Fig. 1A). Minor increases in PD-L1 expression were also noted on non-stromal cells in the MC preparations. To discern whether this process is due to direct stimulation of TLR4 on CMFs, we analyzed pure primary cultures of human CMFs isolated from normal mucosa. By flow cytometry analysis we confirmed that normal CMFs express surface TLR4 (Fig. 1B), as was previously reported (27). Stimulation of cultured CMFs with LPS also resulted in significant upregulation of PD-L1 surface protein (Fig. 1C) and mRNA (Fig. 1D). Analysis of the geometric size of CMFs (FSC) vs the fluorescence intensity of the anti-PD-L1 mAb-labeled cells confirmed that the observed LPS-mediated increase in PD-L1+ CMFs was due to surface expression of PD-L1 and not to the increase in the size of cells (data not shown). Further, the observed level of the LPS-mediated upregulation of PD-L1 was comparable to those reported earlier for macrophages and mucosal Langerhans cells (11, 28). The kinetics of PD-L1 mRNA expression by LPS increased significantly between 2 and 4 h of LPS exposure, and returned to baseline levels after 24h (Fig. 1D). Since PD-L2 and CD80 may serve as a ligand/receptor for PD-1 and PD-L1, respectively, we also analyzed whether TLR4 stimulation modulates their expression on normal CMFs. No detectable CD80 expression was observed upon TLR4 stimulation of the CMFs. There was a minor increase in surface PD-L2 expression by isolated normal CMF upon TLR4 stimulation. However, this increase did not reach statistical significance (Supplement, online Fig. S1). These data suggest that in normal human colonic mucosa, CMFs respond to TLR4 stimulation with the upregulation of the T cell negative co-stimulator PD-L1.
Figure 1.
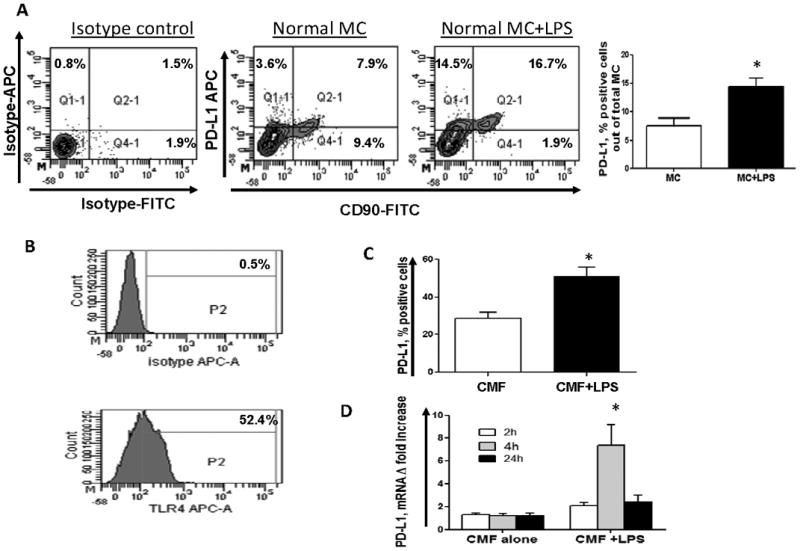
(A) Freshly digested normal colonic mucosal cell (MC) preparations were stimulated with 1 μg/mL of LPS for 16 h and stained with CD90, PD-L1 or isotype controls and analyzed by flow cytometry. One representative experiment and the collective results of 5 separate experiments are shown, * = p < 0.05. (B) Primary normal CMF cultures express TLR4 as detected by flow cytometry. One representative experiments of ten is shown. (C) Stimulation of cultured CMFs with 1 μg/mL of LPS (24h) upregulates PD-L1 on the cell surface (flow cytometry analysis); values are expressed as percentage of positive cells ± SE of triplicate CMF cultures isolated (n= 9 donors,). * = p < 0.05. (D) Stimulation of cultured CMFs with 1 μg/mL of LPS upregulates PD-L1 mRNA levels (real time RT-PCR analysis). The means ± SE are shown as the results of duplicates of three representative experiments,* = p<0.05.
TLR4-mediated upregulation of PD-L1 reinforces CMF-mediated suppression of activated CD4+ T effector cell responses
To assess the functional significance of the TLR4-mediated PD-L1 upregulation on CMFs, we analyzed their ability to suppress proliferation of activated CD4+ effector T cells. LPS pre-stimulation of CMFs led to an increase in the CMF-mediated suppression of activated CD4+ effector T cell proliferation. This process involved PD-L1 since its neutralization with anti-PD-L1 blocking mAbs, but not an isotype control, partially restored T cell proliferation (Fig. 2A-B). Upregulated PD-L1 expression is reported on activated T cells and this may inhibit T cell proliferation in an endogenous manner (39, 40). In our experiments, addition of PD-L1 blocking mAbs did not alter proliferation of the activated T cell monocultures, suggesting that CMF-mediated PD-L1 signaling and not intrinsic T cell PD-L1 signaling is important in alteration of the T cell proliferation. Interestingly, we observed that the major reduction of CFSElow population of CD4+ T cells co-cultured with CMFs was mostly in P10 (later) phase (Fig. 2A-B), and much less in earlier P5-P9 phases of proliferation. Because PD-1 ligand mediated signaling may render PD-1+ effector T cells more susceptible to apoptosis (41, 42), our data may indicate that the apparent decline in T cell proliferation in the in vitro CMF:T cell co-cultures may be due to increases in PD-L1 mediated cell death.
Figure 2.
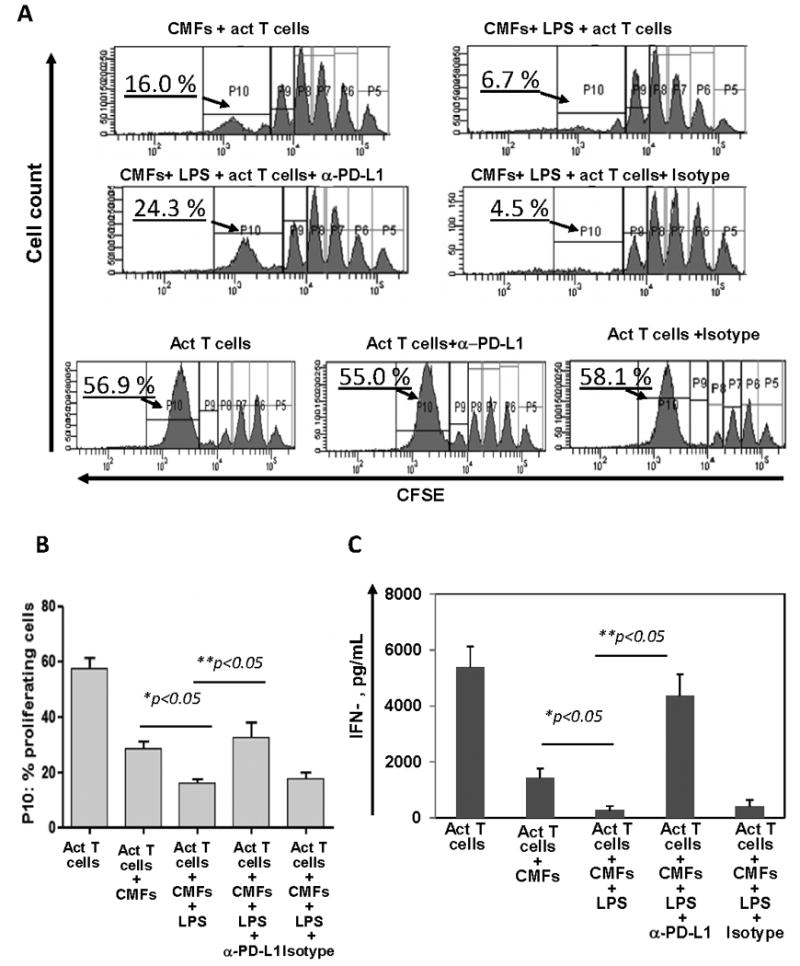
LPS pre-stimulation of normal CMFs reinforces their capacity to suppress activity of the CD4+ T effector cells. CMFs were pre-stimulated with 1 μg/mL of LPS for 24h, the remaining LPS was removed by extensive washes, and the CMFs co-cultured with allogeneic CFSE-labeled, CD2/CD3/CD28-preactivated naive CD4+ T cells at a ratio 1:4 for 4 days in 24-well plates. T cell monocultures were included as experimental controls. In some experiments co-cultures were performed in the presence of anti-PD-L1 blocking mAbs (clone M1H1) or isotype controls. (A-B) Proliferation of the CD4+ T cell was analyzed based on the dilution CFSE dye, and (C) IFN-γ production in conditioned medium was analyzed using singleplex cytokine analysis, n=5 allogeneic donor pairs, two experimental replicates each.
Since PD-L1-mediated signaling is involved in down-regulation of IFN-γ production by activated T cells, we then analyzed production of IFN-γ in these co-cultures. LPS-pre-stimulated CMFs co-cultured with activated CD4+ T cells were found to have an increased capacity to suppress T cell IFN-γ production in a PD-L1 dependent manner (Fig. 2C). Thus, our data suggest that upon exposure to the bacterial ligands that are recognized by TLR4 CMFs may contribute to mucosal tolerance via an increased capacity to suppress IFN-γ which is involved in pro-inflammatory CD4+ T effector cell responses.
TLR4-mediated upregulation of PD-L1 on CMFs from normal colonic mucosa is NF-κB- dependent
Exposure to LPS induces activation of NF-κB in murine and human intestinal stromal cells (24, 37). We evaluated whether NF-κB is involved in LPS-mediated upregulation of PD-L1 on CMFs. Using Western blot analysis we demonstrated that stimulation of CMFs with LPS resulted in a rapid degradation of IκBα, which was maximal at 1 hour and returned to the basal level by 24 h post-LPS exposure (Fig. 3A). Moreover, LPS-induced upregulation of total (Fig. 3B) and surface PD-L1 (Fig. 4A-B) on CMFs was significantly inhibited by the presence of the NF-κB translocation inhibitor triptolide (20 ng/mL). Interestingly, we noted that basal expression of both PD-L1 and PD-L2 on N-CMFs was slightly decreased by treatment with triptolide (Figure 4, Figure S1). Additional studies are currently in progress to understand the role of the NF-κB activity in the basal expression levels of B7 molecules. A previous report suggested that the PI3K/mTOR pathway may also enhance PD-L1 expression (43). Thus, we tested the effect of the P70S6/mTOR kinase inhibitor rapamycin on CMFs, and this did not affect LPS-induced PD-L1 upregulation (data not shown). LPS-induced activation of the NF-κB-mediated response is reported to involve activation of protein tyrosine kinase JAK2 (44). We then analyzed whether inhibition of JAK2 activation decreased the LPS-induced PD-L1 expression by CMFs. AG490 specifically blocks JAK2 activation at the concentration range of 5-20 μM. Its application at 10 μM concentrations significant decreased the LPS-induced upregulation of PD-L1 (Fig. 4C). A previous study (45-47) demonstrated that AG490 may have off-target effects (blocking of the constitutive activation of Stat3) when used at higher concentration 60-100 μM. This seems unlikely in our study since we used AG490 at a final concentration of 10 μM. Thus, activation of JAK2 is involved in LPS-mediated induction of PD-L1 on CMFs.
Figure 3.
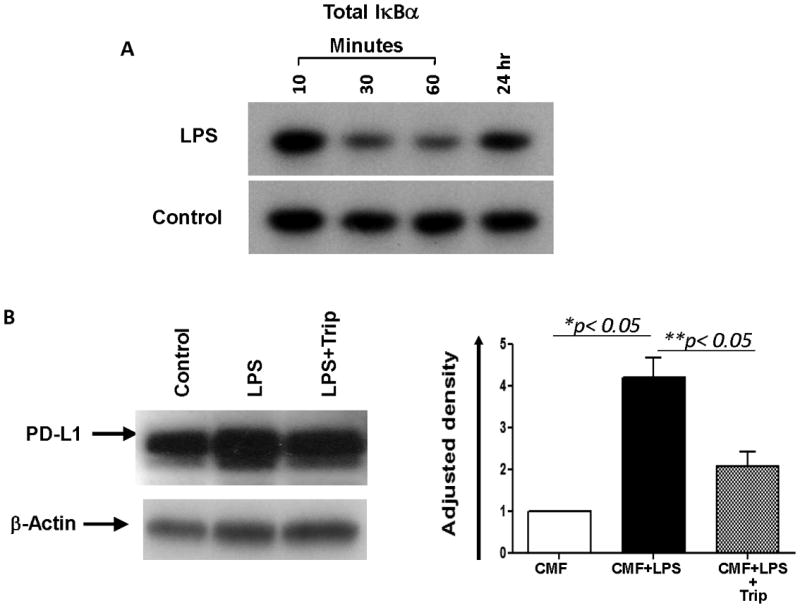
The NF-κB pathway is involved in the TLR4-mediated upregulation of PD-L1. (A) Western blotting using Abs specific for total IκBα protein demonstrated that the total IκBα level rapidly decreases in LPS-treated (1 μg/mL) CMFs cultures compared with the untreated controls. (B) LPS-induced total cell PD-L1 upregulation was inhibited in the presence of triptolide (Trip, a NF-κB translocation inhibitor). Under denaturing conditions, the PD-L1 protein migrates as subunits of ∼50 kDa. Image J software available through NIH website was used to calculate Relative Density. Adjusted density values for tested samples were calculated by dividing the Relative Density of each Sample lane obtained with anti-PD-L1 Abs by the Relative Density of the loading-control for the same lane (stain with anti-β-actin Abs), n=3, *,** = p < 0.05.
Figure 4.
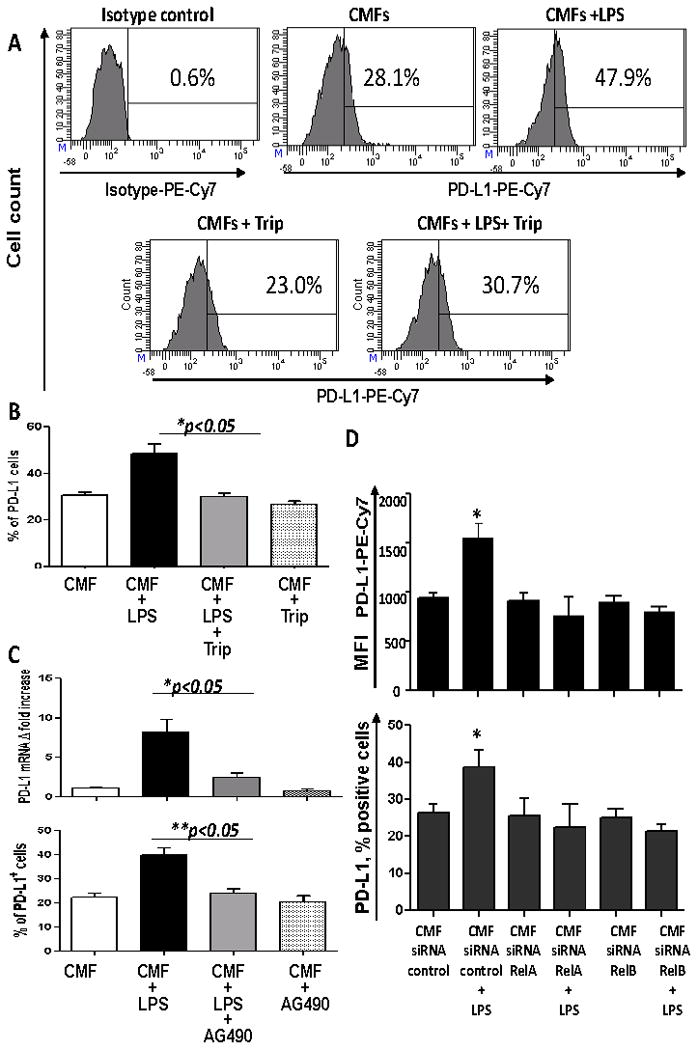
TLR4 stimulation of CMFs increases surface PD-L1 and depends on NF-κB and JAK2 activation. (A) Immunostaining followed by flow cytometry analysis demonstrated that LPS -induced PD-L1 upregulation was inhibited in the presence of the NF-κB translocation inhibitor triptolide (Trip). (B) Summary of PD-L1 surface expression modulation by LPS in presence/absence of Trip by CMF primary isolates. Values are expressed as a % of PD-L1+ cells ± SE of triplicate CMF cultures isolated from each donor, n=10. (C) JAK2 is involved in the LPS-mediated upregulation of PD-L1 levels on CMFs. Real time RT-PCR and flow cytometry was used to analyze expression of the PD-L1 on N-CMFs treated with LPS in presence/absence of JAK2 inhibitor AG490, 10 μM, n=4, * = p < 0.05). (D) CMFs isolated from normal colonic mucosa were transected with RelA- or RelB-specific siRNA or negative control siRNA, allowed to recover for 7 days, and stimulated with LPS (1 μg/mL) for 24 h. Immunostaining followed by multi-color flow cytometry demonstrated that silencing of either relA or relB gene expression abrogated LPS-induced surface upregulation of PD-L1 (n=4 donors, two experimental replicates each), * = p < 0.05.
Because both canonical (RelA-dependent) and non-canonical (RelB-dependent) NF-κB pathways can be active in fibroblasts (48, 49), we determined which of these two NF-κB pathways is implicated in TLR4-mediated upregulation of PD-L1 expression on CMFs. We also demonstrated that silencing the expression of RelA or RelB with specific siRNAs, but not control siRNA, abrogated LPS-induced PD-L1 upregulation on CMFs (Fig. 4D). These data suggest that both pathways contribute to the TLR4-mediated upregulation of PD-L1.
The adaptor MyD88 is required for the TLR4-mediated upregulation of PD-L1 on human CMFs
Since stimulation of TLR4 on APCs may activate both MyD88-dependent and -independent pathways (50), we analyzed whether the adaptor MyD88 is required for the TLR4-mediated PD-L1 upregulation on CMFs. Using a siRNA approach, we demonstrated that silencing MyD88 expression in CMFs abrogated the LPS-induced upregulation of PD-L1 (Fig. 5A).
Figure 5.
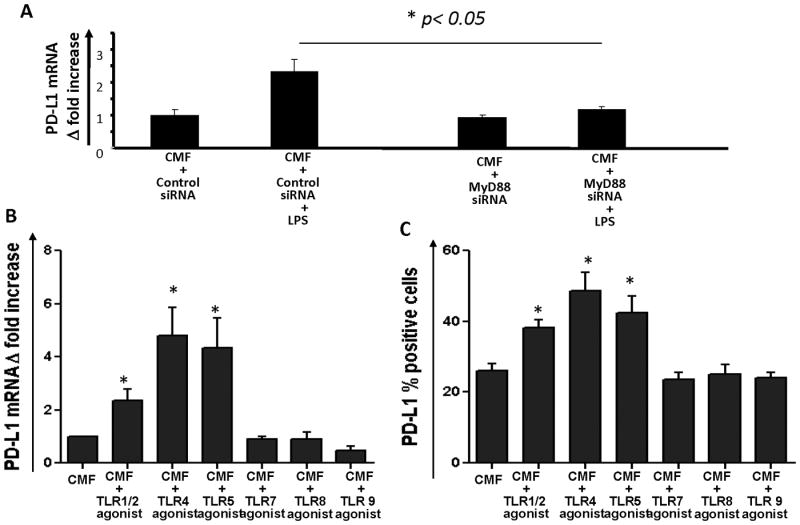
MyD88-dependent TLR signaling increases PD-L1 expression by human CMFs. (A) (A) Silencing of the myd88 gene in CMFs abrogates TLR4-mediated increases in PD-L1 mRNA expression. Primary CMFs isolated from normal colonic mucosa were transfected with MyD88 siRNA or control siRNA seven days prior exposure to the TLR4 agonist LPS (100 ng/mL) for 4 h. PD-L1 expression was analyzed using real-time RT-PCR. The data are shown as means ± SE, n=3; * = p<0.05.CMFs were exposed to TLR agonists and PD-L1 mRNA (B) and surface levels of PD-L1 (C) and were analyzed using real time RT-PCR (4h treatment) and flow cytometry (24 h treatment), respectively. TLR agonists were used at following concentrations: TLR1/2 agonist (Pam3CSK4) -100 ng/mL; TLR4 agonist (E.coli K12 LPS) – 1 μg/mL; TLR5 agonist (S. typhimurium flagellin) – 100 ng/mL; TLR 7 agonist (Imiquinod) – 100 ng/mL; TLR8 agonist (ssRNA40) – 100 ng/mL; TLR 9 agonist (ODN 2006) – 0.5 mM. The results are shown as means ± SE, n=4 donors,*p<0.05.
Signaling through other innate immunity receptors has been reported to activate PD-L1 expression on the professional immune cells (51, 52). We tested whether stimulation of other MyD88-dependent TLRs modulates PD-L1 expression by CMFs. Stimulation of CMFs with the bacterial agonists for TLR1/2 (Pam3CSK4, 100 ng/mL) and TLR5 agonist (S. typhimurium flagellin, 100 ng/mL) also resulted in upregulation of PD-L1 (Fig. 5B-C). Similar to the TLR4-mediated PD-L1 upregulation, the TLR1/2 and TLR5 mediate increase in PD-L1 expression by CMFs was MyD88 dependent. Silencing of the MyD88 mRNA expression using specific siRNA abrogated this PD-L1 increase (Supplement online, Fig. S2). By contrast, tested agonists to TLR7 (Imiquinod 100 ng/mL), TLR8 (ssRNA40, 100 ng/mL) and TLR 9 (ODN 2006, 0.5 mM) did not increased PD-L1 expression by CMFs. Importantly, bacteria whose products are recognized by those TLR1/2, 4 and 5 are highly represented within the normal colon (2, 4). Because tissue expression of PD-L1 has been demonstrated to be among the key factors mediating peripheral T cell tolerance (31), our data suggest that CMFs are important contributors to mucosal tolerance via upregulation of PD-L1 in response to stimulation by colonic bacterial products.
MyD88 dependent signaling is required for the expression of PD-L1 on murine CMFs both at homeostasis and upon epithelial injury
Our in vitro experiments demonstrated that MyD88-dependent TLRs stimulation results in the PD-L1 upregulation by CMFs. This observation raised the question whether MyD88 is required for the PD-L1 expression on CMFs in vivo. Using immunostaining followed by confocal microscopy, we demonstrated that CMFs (α-SMA+ subepithelial cells) in C57Bl/6 wild-type mice express a strong basal level of PD-L1 in normal colonic mucosa (Fig. 6A). The level of total mucosal PD-L1 expression was vastly reduced in MyD88-/- mice on the same genetic background. We also noted that the number of the α-SMA+ CMFs was reduced in the colonic mucosa of these knockout (KO) mice. This may be due to the reduced MyD88-dependent fibroblast migration and differentiation as has been noted in the small intestine and other organs (22, 53). However, the α–SMA+ positive cells (a.k.a. activated CMFs) that remain were essentially devoid of PD-L1. Further, the number of α-SMA negative cells expressing PD-L1 was also strongly reduced in the colonic mucosa from MyD88-/- mice (Fig. 6B).
Figure 6.
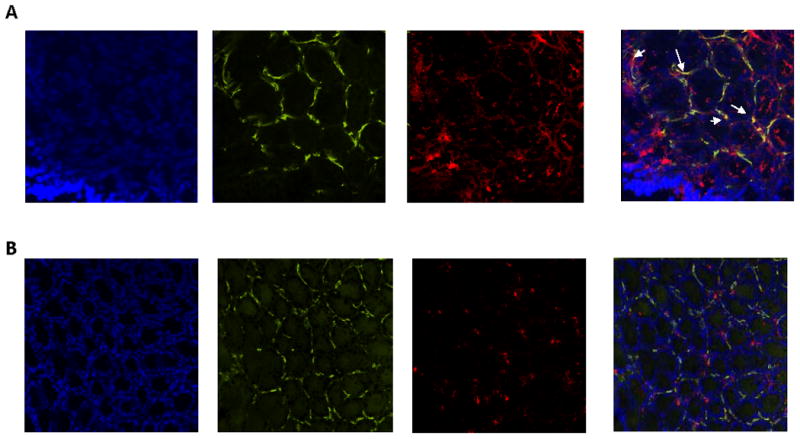
Expression of PD-L1 is reduced in colonic mucosa from mice with impaired MyD88 signaling. Immunostaining of frozen mucosal colonic tissue cross-sections from (A) wild-type C57B6 and (B) MyD88-/- mice was performed, followed by confocal microscopy. DAPI was used to stain cell nuclei (blue); CMFs were detected by anti-α-SMA mAb (green, clone A4) and stained for PD-L1 with mAb (red, clone M1H5). A yellow-orange color on merged images indicates co-localization of α-SMA and PD-L1, and thus the expression of PD-L1 by CMFs (indicated by arrows). Cross-sections are representative of data from six wild-type and six MyD88-/- mice.
We then tested how CMF specific activation of MyD88 regulates the expression of PD-L1 at homeostasis and upon epithelial injury. For this purpose we generated fibroblast-specific tamoxifen-inducible conditional knockout mice selective for MyD88 (Fib-MyD88 KO). The specificity of MyD88 deletion within the CMF, as well as retained MyD88 expression in other relevant innate immunity cells (CD11c+DC, F4/80+ macrophages and EpCAM+ epithelial cells) was confirmed by immunostaining followed by confocal microscopy analysis (Supplement online, Figure S3). Similar to the MyD88-/- mice we observed a decrease in a PD-L1 expression within the colonic mucosa (real-time RT-PCR analysis, Supplement online, Fig. S4A). Using confocal microscopy and flow cytometry we demonstrated that CMF intrinsic MyD88 deficiency resulted in the abrogation of PD-L1 expression by CMFs in Fib-MyD88 KO control group (Fig 7A, C, see also Supplement online Figure S4B). Similar observations were made in the DSS acute inflammation model, in which the exposure of colonic mucosal lamina propria cells to commensal microbiota is increased due to the epithelial damage induced by DSS (Fig. 7B,C and Supplement online Figure S4B).
Figure 7.
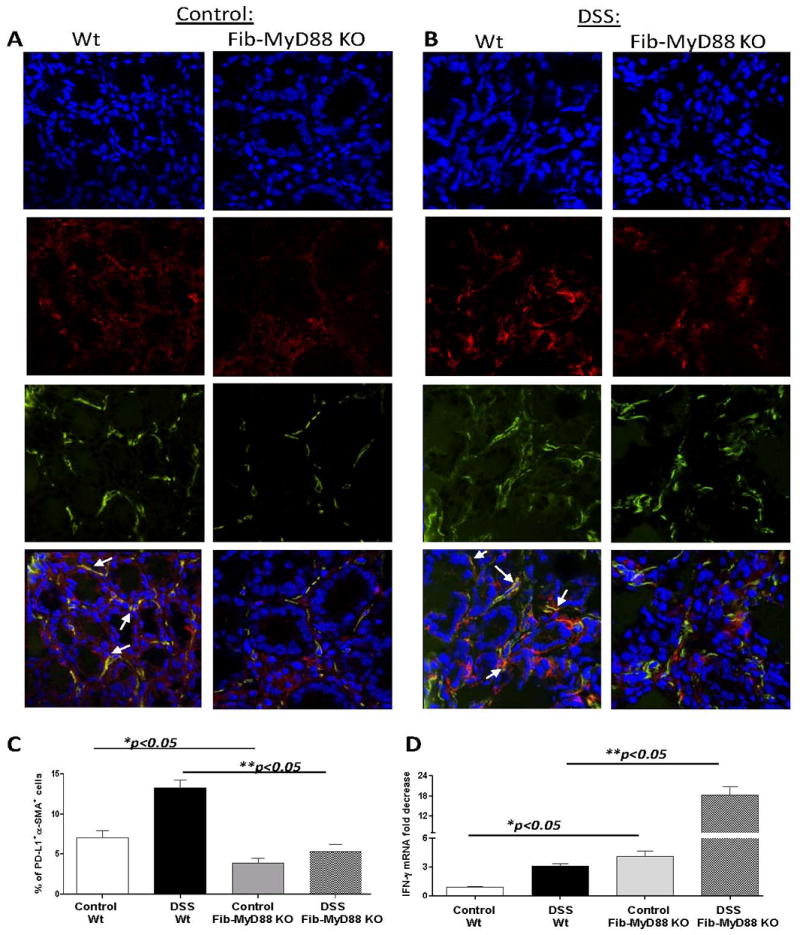
Fibroblast-intrinsic MyD88 signaling is required for the PD-L1 expression by CMFs and controls IFN-γ expression in colonic mucosa. PD-L1 expression colonic mucosa upon (A) homeostasis and in (B) DSS-colitis (day 10) in wild-type C57B6 and Fibroblast specific Fib-MyD88fl/fl mice was analyzed by confocal microscopy. DAPI was used to stain cell nuclei (blue); activated CMFs were detected by anti-α-SMA mAb (green, clone A4) and stained for PD-L1 with mAb (red, clone M1H5). A yellow-orange color on merged images indicates co-localization of α-SMA and PD-L1, and thus the expression of PD-L1 by CMFs (indicated by arrows). Cross-sections are representative of a two independent experiments including four – five animals per group. (C) Freshly digested murine colonic mucosal cell preparations were stained with mAbs against α-SMA and PD-L1 and analyzed by flow cytometry. Isotype controls were included for each staining. Five animals per group were analyzed. (D) IFN-γ mRNA level in colonic mucosa was determined using real time RT-PCR. The level of the IFN-γ mRNA was normalized to the level of murine β-actin mRNA. The results are shown as means ± SE from two independent experiments. Four and five animals per group per experiment were analyzed.
Using PD-L1 knockout animals, Keir et al (14) demonstrated that animals lacking intact PD-L1 signaling have elevated production of the inflammatory cytokine IFN-γ. Our in vitro experiments suggest that stimulation of MyD88-dependent TLRs potentiate CMF-mediated suppression of the IFN-γ production by preactivated T cells in a PD-L1 dependent manner. Thus, we analyzed IFN-γ levels in the mice lacking an intact MyD88-PD-L1 immunoregulatory axis. We observed that in the absence of MyD88-PD-L1 signaling (in Fib-MyD88 KO mice), there was a significant increase in mucosal IFN-γ expression at homeostasis (Fig 7D). Similar observations were made in colonic mucosa from MyD88-/-: a decrease in mucosal IFN-γ was observed (Supplement online, Fig. S4C). Further, production of an IFN-γ inflammatory response was even stronger in Fib-MyD88 KO mice upon intestinal injury induced by DSS (Fig 7D).
Discussion
Tolerance is defined as the immune system's endogenous regulatory mechanisms that prevent potentially injurious inflammatory immune responses by producing a systemic state of antigen-specific non-responsiveness to antigens previously encountered (54, 55). A similar but more localized process regulates responses to commensal bacteria in the intestinal mucosa, and is known as mucosal tolerance (56, 57, 58). However, physiological pathways and cells responsible for the induction and maintenance of tolerance in the colonic mucosa are not well characterized. Taken together with our previous publications (17, 35), the present study provides additional evidence supporting the importance of human colonic CD90+ stromal cells (CMFs) in the maintenance of local mucosal tolerance.
Many mechanisms have been proposed to explain the defects in the activation of T cell-dependent immune responses that occur during tolerance, including clonal anergy and deletion, as well active regulatory processes (3, 56). Immunosupression due to the upregulation of the B7 negative co-stimulatory molecules, as well as induction of regulatory T cells, are important mechanisms contributing to the induction of the tolerogenic (homeostatic) state (30, 32). During mucosal tolerance, acute inflammatory effector T cell responses are regulated by their interactions with APCs. We and others recently reported that CMFs may act as non-professional APCs in normal colonic mucosa (18, 19), and so may be involved in the regulation of T cell responses. It has been suggested that, in contrast to parenchymal cells, a “tolerogenic” anti-proliferative effect is a fundamental characteristic of all mesenchymal stromal cells (59). Our recently published data are consistent with this idea. CMFs isolated from normal human mucosa suppress proliferation of activated CD4+ T cells (17).The mechanisms responsible for CMF's suppressive effect on activated T effector cell responses involve cell contact-mediated interactions and/or the production of soluble factors. We previously demonstrated that direct CMF-mediated suppression of activated T effector cell responses involves signaling through PD-L1 and PD-L2 (17). However, in colonic mucosal sites in vivo, these interactions occur within an extremely dynamic milieu containing abundant cytokines, chemokines, growth factors and TLR ligands.
The continuous presence of physiological microflora in the colonic lumen provides a significant source for TLR ligands; thus, it is likely that TLR signaling plays a major role in orchestrating mucosal tolerogenic responses. Therefore, the immunosuppressive capacity of CMFs is likely to be modulated by signaling through TLRs. We have confirmed expression of TLR4 on normal CMFs, and demonstrated that signaling through TLR4 upregulates PD-L1, but not PD-L2 on CMFs. TLR4-mediated upregulation of PD-L1 results in greater CMF-mediated suppression of CD4+ effector T cell proliferation. Although further investigation is required to understand the mechanism responsible for CMF-mediated suppression of proliferating T cells, our data suggest that this inhibitory effect may be caused by an increase in PD-L1 mediated T cell death. Such an APC-mediated PD-L1 dependent inhibitory action on T cell growth/survival was previously described in the other organs (41, 42)
CMF:T cell co-culture experiments also demonstrated that signaling through TLR4 enhanced CMF mediated suppression of IFN-γ production by activated CD4+ effector T cells and this process was PD-L1 dependent. CMFs isolated from normal colonic tissue respond to exogenous IFN-γ by increased PD-L1 expression (66). This suggests that reduced production of IFN-γ by CMF-derived PD-L1 engagement of T cells may be an important physiological negative feedback mechanism to control the inflammation in the GI track and mucosal homeostasis.
The intracellular mechanisms responsible for TLR-mediated upregulation of PD-L1 on APCs are not well characterized. IFN-γ- and LPS-induced PD-L1 upregulation on tumor cells was reported to be mediated by MEK and PI3K/AKT/mTOR, but not NF-κB pathways (30, 51). However, IFN-γ regulation of PD-L1 expression on dermal fibroblasts was demonstrated to be NF-κB- mediated (67). NF-κB pathways are activated in the intestinal mucosal stromal cells upon stimulation with LPS (37), and we have made the novel observation that TLR4-mediated upregulation of PD-L1 on CMFs is NF-κB-dependent, involving both the canonical and non-canonical NF-κB pathways. Further, our data suggest that activation of JAK2 is involved in the NF-κB-mediated upregulation of PD-L1 on CMFs in response to LPS. A similar mechanism was described for the LPS-induced production of nitric oxide in macrophages (44). The question that needs further investigation is whether activation of JAK2 is directly involved in the TLR4-mediated increase of PD-L1 or it is due to the LPS-mediated increase in the inflammatory cytokines and growth factors on CMFs. Recent data by Wolfe at al (68) suggest that IL-6 and IL-10 via STAT3 activation may contribute to the increase in PD-L1 expression on professional APCs. LPS treatment may induce minor increases in IL-6, but not IL-10 production by CMFs (69). However, we did not observe significant upregulation of PD-L1 expression by CMFs after 24h of treatment with IL-6 (10 ng/mL; data not shown). This suggests that JAK2 activation might be directly involved in the TLR4-mediated regulation of PD-L1 expression by CMFs, as reported previously in B cell lymphomas (70). However we do not exclude the possibility that exogenous IL-6 and IL-10 produced by other cells in the normal colonic mucosa may contribute to the regulation of the PD-L1 expression in vivo. In contrast to previous findings with tumor-derived cells (30, 51), inhibition of mTOR with rapamycin did not affect TLR4-mediated upregulation of PD-L1 on CMFs. In summary, TLR-mediated upregulation of PD-L1 on different cell type may involve different intracellular pathways in health and disease, and thus may have different pathophysiological effects.
TLR4-mediated signaling may activate two downstream pathways, Myd88-dependent and -independent, each able to directly activate NF-κB. The adaptor MyD88 is also required for the TLR1, 2 and TLR5-9 mediated signaling. This adaptor has been reported to be essential for the TLR4- and TLR7/8-medated upregulation of PD-L1 on plasma cells and DCs, respectively (51, 52). We observed that signaling through MyD88-dependent TLRs (1/2, 4 and 5) upregulates PD-L1. In contrast to professional APCs, stimulation of CMFs via TLR6-9 agonists did not result in PD-L1 upregulation. This may be due either to limited availability of these receptors on CMFs or different regulatory functions of these TLRs on CMFs as contrasted to professional APCs, as has previously been reported for gingival fibroblasts (71).
In the GI tract, MyD88 signaling is critical for host protection against DSS-induced colitis by limiting mucosal damage and acute inflammatory responses (58, 72). MyD88-dependent signaling has also been shown to limit the systemic enteropathogen burden in the model of Salmonella-induced colitis (22). At the same time, MyD88 signaling is critical in spontaneous and inflammation-induced intestinal tumorogenesis (73-74). Because of the intrinsic complexity of MyD88 signaling in various type of cells (74,58), the mechanisms underlying MyD88-protective vs pathological effects are unclear. PD-L1 signaling is known to play a dual role: it is critical for the maintenance of mucosal tolerance (6-7, 10, 14), but its persistent, increased expression within a tumor promotes cancer-associated immunosuppression (10, 75-77). The normal colonic mucosa is highly colonized by large variety of bacterial species that are a source of multiple ligands for MyD88 dependent TLRs (4, 78). However, the role of MyD88 signaling in the in vivo regulation of the PD-L1 in GI tract has not been investigated. Our in vivo data suggest that total lack of intact MyD88 signaling in colonic mucosa (Myd88-/- mice) drastically decreases PD-L1 expression especially by CMFs. We have demonstrated that fibroblast-restricted MyD88 signaling was required for the PD-L1 expression by CMFs in vivo. Using the DSS acute colitis-conditional MyD88 knockout model, we demonstrated that despite intact MyD88 signaling on other immune cells that accumulate in the mucosa of this model there does not appear to be any compensatory mechanism present to restore the expression of PD-L1 on CMFs during acute intestinal epithelial injury.
Our in vitro data in human cells and our in vivo observations in mice suggest that direct interactions between CMFs, stromal cells located just beneath the epithelial basement membrane, and colonic microbial products is critical for PD-L1 expression by CMFs. Further, our in vitro culture experiments suggest that activation of MyD88-dependent TLR4 signaling potentiates PD-L1 dependent CMF-mediated suppression of IFN-γ production by T cells. We observed also that impairment of MyD88/PD-L1 regulation in CMFs in vivo concomitantly increased the expression of IFN-γ in the colonic mucosa. Thus, taking into consideration the key role of PD-L1 in the negative regulation of the IFN-γ production in vivo (14), the shift in PD-L1 expression on CMFs even by the 10-20 % observed by us in culture might be relevant to the TLR-mediated tuning of the immune balance in the colonic mucosa. This might be particularly relevant when the epithelial layer is disrupted and PD-L1 upregulation on CMFs might help to maintain the equilibrium between tolerance and immunity in order to protect the colonic mucosa against overt inflammatory responses toward innocuous microflora.
Data from these studies point to mechanisms that deserve further detailed investigation such as the contribution of MyD88 signaling to the basal levels of PD-L1 expression on CMFs in vivo and whether it affords a protective function in human IBD. Further in vivo investigation of the role of PD-L1 expression by CMFs in health and diseases is also needed. Finally, our data strongly suggest that modulation of PD-L1 expression by CMFs via MyD88-dependent TLR stimulation might be among the main processes contributing to the control of colonic inflammation during mucosal tolerance and its disruption may be among the factors involved in several acute and chronic gastrointestinal inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Dr Andrew Leask at University of Western Ontario (London, Ontario, Canada) for providing us with the TgCol1α2-CreER™ mice. We also thank Dr David Konkel, PhD (UTMB) for critical revision of the manuscript and our technical assistant Ms. Yu Lin for her excellent assistance in performing the animal work.
Footnotes
Supported by grants from the NIAID (1R56AI085474), NCATS (KL2TR000072), NCATS (TR000071), American Gastroenterology Association, American Cancer Society (RSG-10-159-01-LIB), CTSA Multi-Institutional Pilot Fund, and Crohn's & Colitis Foundation of America. The content is solely the responsibility of the authors and does not necessarily represent the views of the supporting agencies listed above.
The authors have declared that no conflicts of interest exist.
References
- 1.Untersmayr E, Jensen-Jarolim E. Mechanisms of type I food allergy. Pharmacol Ther. 2006;112:787–798. doi: 10.1016/j.pharmthera.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Tannock GW. Probiotics : a critical review. Wymondham: Horizon Scientific; 1999. [Google Scholar]
- 3.Novak N, Haberstok J, Bieber T, Allam JP. The immune privilege of the oral mucosa. Trends Mol Med. 2008;14:191–198. doi: 10.1016/j.molmed.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Ricanek P, Lothe SM, Frye SA, Rydning A, Vatn MH, Tonjum T. Gut bacterial profile in patients newly diagnosed with treatment-naive Crohn's disease. Clin Exp Gastroenterol. 2012;5:173–186. doi: 10.2147/CEG.S33858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Geng J, Tang, Fan H, Xu J, Wen X, Ma ZS, Shi P. Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 2013 Oct 17; doi: 10.1038/ismej.2013.185. 2013. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, Freeman GJ, Turley SJ. Intestinal tolerance is converted to autoimmune enteritis upon PD-1 ligand blockade. J Immunol. 2009;182:2102–2112. doi: 10.4049/jimmunol.0802769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 9.Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer-induced immune suppression. Clin Dev Immunol. 2012;2012:656340. doi: 10.1155/2012/656340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Podojil JR, Miller SD. Targeting the B7 family of co-stimulatory molecules: successes and challenges. BioDrugs. 2013;27:1–13. doi: 10.1007/s40259-012-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Addio F, Riella LV, Mfarrej BG, Chabtini L, Adams LT, Yeung M, Yagita H, Azuma M, Sayegh MH, Guleria I. The link between the PDL1 costimulatory pathway and Th17 in fetomaternal tolerance. J Immunol. 2011;187:4530–4541. doi: 10.4049/jimmunol.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, Levine BL, June CH, Medin JA, Fowler DH. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011;3:111ra20. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, Azuma M, Mayer L. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology. 2004;126:1347–1357. doi: 10.1053/j.gastro.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, Machida U, Iwai H, Azuma M, Chen L, Watanabe M. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171:4156–4163. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- 17.Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, Raju GS, Reyes VE, Powell DW. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology. 2008;135:1228–1237. doi: 10.1053/j.gastro.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saada JI, Pinchuk IV, Barrera CA, Adegboyega PA, Suarez G, Mifflin RC, Di Mari JF, Reyes VE, Powell DW. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J Immunol. 2006;177:5968–5979. doi: 10.4049/jimmunol.177.9.5968. [DOI] [PubMed] [Google Scholar]
- 19.Owens BM, Steevels TA, Dudek M, Walcott D, Sun MY, Mayer A, Allan P, Simmons A. CD90+ Stromal Cells are Non-Professional Innate Immune Effectors of the Human Colonic Mucosa. Front Immunol. 2013;4:307. doi: 10.3389/fimmu.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, Liu Y, Strome SE, Chen L, Tamada K. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116:1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. 2011;73:213–237. doi: 10.1146/annurev.physiol.70.113006.100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansson LE, Montero M, Zarepour M, Bergstrom KS, Ma C, Huang T, Man C, Grassl GA, Vallance BA. MyD88 signaling promotes both mucosal homeostatic and fibrotic responses during Salmonella-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2012;303:G311–G323. doi: 10.1152/ajpgi.00038.2012. [DOI] [PubMed] [Google Scholar]
- 23.Fukata M, Arditi M. The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol. 2013;6:451–463. doi: 10.1038/mi.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke JP, Cunningham MF, Watson RW, Docherty NG, Coffey JC, O'Connell PR. Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br J Surg. 2010;97:1126–1134. doi: 10.1002/bjs.7045. [DOI] [PubMed] [Google Scholar]
- 25.Kamdar K, Nguyen V, DePaolo RW. Toll-like receptor signaling and regulation of intestinal immunity. Virulence. 2013;4:207–212. doi: 10.4161/viru.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 28.Allam JP, Peng WM, Appel T, Wenghoefer M, Niederhagen B, Bieber T, Berge S, Novak N. Toll-like receptor 4 ligation enforces tolerogenic properties of oral mucosal Langerhans cells. J Allergy Clin Immunol. 2008;121:368–374. doi: 10.1016/j.jaci.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 29.Bao H, Lu P, Li Y, Wang L, Li H, He D, Yang Y, Zhao Y, Yang L, Wang M, Yi Q, Cai Z. Triggering of toll-like receptor-4 in human multiple myeloma cells promotes proliferation and alters cell responses to immune and chemotherapy drug attack. Cancer Biol Ther. 2011;11:58–67. doi: 10.4161/cbt.11.1.13878. [DOI] [PubMed] [Google Scholar]
- 30.Berthon C, Driss V, Liu J, Kuranda K, Leleu X, Jouy N, Hetuin D, Quesnel B. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol Immunother. 2010;59:1839–1849. doi: 10.1007/s00262-010-0909-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owens BM, Simmons A. Intestinal stromal cells in mucosal immunity and homeostasis. Mucosal Immunol. 2013;6:224–234. doi: 10.1038/mi.2012.125. [DOI] [PubMed] [Google Scholar]
- 32.Mahida YR, Beltinger J, Makh S, Goke M, Gray T, Podolsky DK, Hawkey CJ. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase-1 and -2. Am J Physiol. 1997;273:G1341–G1348. doi: 10.1152/ajpgi.1997.273.6.G1341. [DOI] [PubMed] [Google Scholar]
- 33.Zheng B, Zhang Z, Black CM, de Crombrugghe B, Denton CP. Ligand-dependent genetic recombination in fibroblasts : a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol. 2002;160:1609–1617. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 35.Pinchuk IV, Beswick EJ, Saada JI, Boya G, Schmitt D, Raju GS, Brenmoehl J, Rogler G, Reyes VE, Powell DW. Human Colonic Myofibroblasts Promote Expansion of CD4+ CD25high Foxp3+ Regulatory T Cells. Gastroenterology. 2011;140:2019–2030. doi: 10.1053/j.gastro.2011.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 37.Walton KL, Holt L, Sartor RB. Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2009;296:G601–G611. doi: 10.1152/ajpgi.00022.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590:447–458. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor- associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 42.Venkatachari NJ, Buchanan WG, Ayyavoo V. Human immunodeficiency virus (HIV-1) infection selectively downregulates PD-1 expression in infected cells and protects the cells from early apoptosis in vitro and in vivo. Virolog y. 2008;376:140–153. doi: 10.1016/j.virol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008;68:5432–5438. doi: 10.1158/0008-5472.CAN-07-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones E, Adcock IM, Ahmed BY, Punchard NA. Modulation of LPS stimulated NF-kappaB mediated Nitric Oxide production by PKCepsilon and JAK2 in RAW macrophages. J Inflamm (Lond) 2007;4:23. doi: 10.1186/1476-9255-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burdelya L, Catlett-Falcone R, Levitzki A, Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton WS, Jove R, Yu H. Combination therapy with AG-490 and interleukin 12 achieves greater antitumor effects than either agent alone. Mol Cancer Ther. 2002;1:893–899. [PubMed] [Google Scholar]
- 46.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman CM. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 47.Abe M, Funakoshi-Tago M, Tago K, amishimoto JK, Aizu-Yokota E, Sonoda Y, Kasahara T. The polycythemia vera-associated Jak2 V617F mutant induces tumorigenesis in nude mice. Int Immunopharmacol. 2009;9:870–877. doi: 10.1016/j.intimp.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 48.Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem. 2003;278:19852–19860. doi: 10.1074/jbc.M301945200. [DOI] [PubMed] [Google Scholar]
- 49.Jacque E, Tchenio T, Piton G, Romeo PH, Baud V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc Natl Acad Sci U S A. 2005;102:14635–14640. doi: 10.1073/pnas.0507342102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukherjee S, Chen LY, Papadimos TJ, Huang S, Zuraw BL, Pan ZK. Lipopolysaccharide-driven Th2 cytokine production in macrophages is regulated by both MyD88 and TRAM. J Biol Chem. 2009;284:29391–8. doi: 10.1074/jbc.M109.005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J, Aamrouni H, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, Saudemont A, Quesnel B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- 52.Meier A, Bagchi A, Sidhu HK, Alter G, Suscovich TJ, Kavanagh DG, Streeck H, Brockman MA, LeGall S, Hellman J, Altfeld M. Upregulation of PD-L1 on monocytes and dendritic cells by HIV-1 derived TLR ligands. AIDS. 2008;22:655–658. doi: 10.1097/QAD.0b013e3282f4de23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li G, Liu D, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, Hisamitsu T, Liu Y. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-kappaB-mediated matrix metalloproteinase-9 expression. PLoS One. 2013;8:e68905. doi: 10.1371/journal.pone.0068905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothstein DM. Immunosuppression and regulation: Cast in new light? J Am Society Nephrol. 2006;17:2644–2646. doi: 10.1681/ASN.2006080844. [DOI] [PubMed] [Google Scholar]
- 55.Halloran PF, Bromberg J, Kaplan B, Vincenti F. Tolerance versus immunosuppression: a perspective. Am J Transplant. 2008;8:1365–1366. doi: 10.1111/j.1600-6143.2008.02289.x. [DOI] [PubMed] [Google Scholar]
- 56.Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol. 2012;5:232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuhn KA, Stappenbeck TS. Peripheral education of the immune system by the colonic microbiota. Semin Immunol. 2013;25:364–369. doi: 10.1016/j.smim.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kirkland D, Benson A, Mirpuri J, Pifer R, Hou B, DeFranco AL, Yarovinsky F. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity. 2012;36:228–238. doi: 10.1016/j.immuni.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones S, Horwood N, Cope A, Dazzi F. The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol. 2007;179:2824–2831. doi: 10.4049/jimmunol.179.5.2824. [DOI] [PubMed] [Google Scholar]
- 60.Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, Lu L, Qian S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004;40:1312–1321. doi: 10.1002/hep.20488. [DOI] [PubMed] [Google Scholar]
- 61.Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, Pennesi G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482–1490. doi: 10.1002/eji.200425405. [DOI] [PubMed] [Google Scholar]
- 62.Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, Hardy W, Devine S, Ucker D, Deans R, Moseley A, Hoffman R. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 63.Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Jr, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol. 2007;178:5552–5562. doi: 10.4049/jimmunol.178.9.5552. [DOI] [PubMed] [Google Scholar]
- 64.Nagamatsu T, Schust DJ, Sugimoto J, Barrier BF. Human decidual stromal cells suppress cytokine secretion by allogenic CD4+ T cells via PD-1 ligand interactions. Hum Reprod. 2009;24:3160–3471. doi: 10.1093/humrep/dep308. [DOI] [PubMed] [Google Scholar]
- 65.Ghahary A, Li Y, Tredget EE, Kilani RT, Iwashina T, Karami A, Lin X. Expression of indoleamine 2,3-dioxygenase in dermal fibroblasts functions as a local immunosuppressive factor. J Invest Dermatol. 2004;122:953–964. doi: 10.1111/j.0022-202X.2004.22409.x. [DOI] [PubMed] [Google Scholar]
- 66.Saada JI, Barrera CA, Reyes VE, Adegboyega PA, Suarez G, Tamerisa RA, Pang KF, Bland DA, Mifflin RC, DiMari JF, Powell DW. Intestinal myofibroblasts and immune tolerance. Ann N Y Acad Sci. 2004;1029:379–381. doi: 10.1196/annals.1309.023. [DOI] [PubMed] [Google Scholar]
- 67.Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, Maeng PJ, Lim JS. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40:95–103. doi: 10.1016/j.jdermsci.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 68.Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, Dalpke AH, Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–24. doi: 10.1002/eji.201040979. [DOI] [PubMed] [Google Scholar]
- 69.Rogler G, Gelbmann CM, Vogl D, Brunner M, Scholmerich J, Falk W, Andus T, Brand K. Differential activation of cytokine secretion in primary human colonic fibroblast/myofibroblast cultures. Scand J Gastroenterol. 2001;36:389–398. doi: 10.1080/003655201300051216. [DOI] [PubMed] [Google Scholar]
- 70.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahanonda R, Sa-Ard-Iam N, Montreekachon P, Pimkhaokham A, Yongvanichit K, Fukuda MM, Pichyangkul S. IL-8 and IDO expression by human gingival fibroblasts via TLRs. J Immunol. 2007;178:1151–1157. doi: 10.4049/jimmunol.178.2.1151. [DOI] [PubMed] [Google Scholar]
- 72.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 73.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 74.Schiechl G, Bauer B, Fuss I, Lang SA, Moser C, Ruemmele P, Rose-John S, Neurath MF, Geissler EK, Schlitt HJ, Strober W, Fichtner-Feigl S. Tumor development in murine ulcerative colitis depends on MyD88 signaling of colonic F4/80+CD11bhighGr1low macrophages. J Clin Invest. 2011;121:1692–16708. doi: 10.1172/JCI42540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brahmer JR. PD-1-targeted immunotherapy: recent clinical findings. Clin Adv Hematol Oncol. 2012;10:674–675. [PubMed] [Google Scholar]
- 77.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian L. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:31673175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.